
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative process leading to irreversible dementia. The main hallmark of AD is the extracellular accumulation of amyloid β-peptide (Aβ) to form oligomers and fibrils, which are synaptotoxic [1–3] and induce neuronal death [4–6].
Aβ is produced by all the cells of the organism as a result of amyloid precursor protein (APP) proteolytic processing, when it is sequentially cleaved by beta- and gamma-secretase complex [7–10]. Under physiological conditions, Aβ is produced within the brain at a low rate. Aβ length varies depending on its cleaving sites: Aβ1-40 is the most abundant, followed by Aβ1-42. Additionally, there are larger (1-43) and N-terminal truncated (X-40 and X-42) Aβ species [11]. Once Aβ is released into the brain parenchyma it can be degraded, mostly by neprilysin [12] and insulin-degrading enzyme (IDE)[13]. Nevertheless, there are other proteolytic enzymes contributing to its degradation, such as plasmin [14], the angiotensin converting enzyme (ACE) [15] and endothelin converting enzyme (ECE) [16]. Monomeric Aβ that is not degraded is drained into the blood through the Low-Density Lipoprotein Receptor-Related Protein (LRP), which is in the basal membrane of the endothelium and releases Aβ to plasma by transcytosis [17]. The beta-secretase (BACE1) activity increases with age [18], which causes an increase in Aβ production. Particularly, Aβ1-42 levels are increased in the elder, probably due to the nitrotyrosination of gamma-secretase complex [10]. Aβ1-42 misfolding into β-sheet favours its aggregation and the formation of oligomers [6], preventing its drainage through the blood brain barrier (BBB). This aggregation also produces cerebral amyloid angiopathy, due to the accumulation of Aβ in brain vessels [19, 20].
Currently, there is no effective treatment for AD. However, different approaches to reduce Aβ deposition are being studied, including the modulation, clearance or inhibition of Aβ aggregation [21]. There are strong evidences showing that the reduction of Aβ could be helpful for treating AD [22, 23]. In this context, there are reports on immunotherapy, which relies on evidences showing that Aβ antibodies not only inhibit fibril formation, but also disassemble pre-formed aggregates [24, 25]. Gamma immunoglobulins (IgG) can cross the BBB to act in the central nervous system [26]. Different groups have studied the mechanism by which antibodies targeting Aβ N-terminal region inhibit its aggregation [24, 27, 28]. In addition, it has been shown that peripheral administration of anti-Aβ antibodies could reduce brain plaques in an AD mouse model [26]. Several clinical trials using humanized monoclonal antibodies against Aβ, showed promising results regarding safety and tolerance [21, 29–33]. Although none of them has overcome phase III for mild to moderate AD until now [30, 31], some trials are still ongoing because of their potential [32, 34, 35]. The administration of an intravenous human serum IgG preparation was reported to be safe, and the treatment was confirmed to be present in the cerebrospinal fluid (CSF). Even though the treatment was effective among APOE-e4 carriers, it was not able to overcome phase III [36, 37]. Recently, the humanized IgG aducanumab has obtained very promising results, demonstrating a decrease in brain Aβ that correlates with slow clinical decline [32].
Here, we report the role of the human pool of IgG in the inhibition of Aβ aggregation and neurotoxicity, independently of IgG antigen-specificity. Particularly, we show experimentally for the first time the importance of the antigen-binding fragment (Fab) in the inhibition of Aβ aggregation.
RESULTS
IgG prevents amyloid toxicity in cortical neurons
Ultimately, the deposition of Aβ in AD brain leads to neuronal death. Therefore, we have tested IgG effect on Aβ-mediated neurotoxicity. Aβ oligomers, resulting from the aggregation of Aβ1-42, were prepared in the presence or absence of IgG, either at plasmatic physiological concentration (7 mg/mL) or at 0.04 mg/mL because it has been reported as the non-pathological IgG concentration in the central nervous system (CNS) [38]. We used Aβ1-42 because it is the most aggregation-prone species and, in consequence, the most neurotoxic amyloid form [39]. The different treatments were administered to cortical primary neurons to further evaluate cell viability. Aβ1-42 oligomers-mediated neurotoxicity was significantly reduced in the presence of 7 mg/mL of IgG, but not with IgG at 0.04 mg/mL (Figure 1A). To confirm this observation, we analyzed apoptosis performing an immunofluorescence staining against active caspase 3 and β3-tubulin (Figure 1B). We observed higher levels of active caspase 3 and an altered neuronal morphology, characterized by less neurites in neurons treated with Aβ1-42 oligomers prepared in the absence of IgG. Conversely, Aβ1-42 oligomers prepared in the presence of IgG at 7 mg/mL prevented the increase in caspase 3 and preserved the morphology. No protective effect was observed when IgG at 0.04 mg/mL was co-incubated with Aβ1-42. Controls with different concentrations of IgG showed no effects on the viability of cortical neurons (Figure 1C).
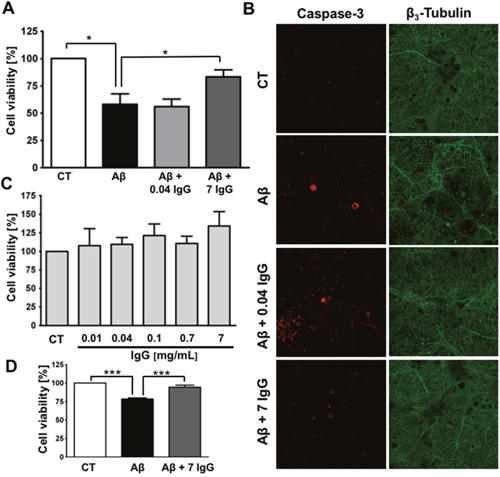
Figure 1: IgG protects cortical neurons against amyloid toxicity. (A) Cell viability was assayed by MTT reduction in cortical neurons treated with 10 μM Aβ1-42 or 10 μM Aβ1-42 + IgG at either 0.04 or 7 mg/mL for 24 h. Data are the mean ± SEM of 6 independent experiments. *p < 0.05 by one-way ANOVA using Newman-Keuls post-test. (B) Immunofluorescence study of cortical neurons treated with 10 μM Aβ1-42, 10 μM Aβ1-42 + 0.04 mg/mL of IgG or 10 μM Aβ1-42 + 7 mg/mL of IgG for 24 h. Staining for active caspase-3 is shown in red and for β3-tubulin in green. β3-tubulin is an overlap of images of the same area (C) Cell viability was assayed in cortical neurons by MTT reduction treated with increasing concentrations of IgG for 24 h. Data are the mean ± SEM of 4 independent experiments. (D) Cell viability was assayed by MTT reduction in endothelial cells treated with 0.1 μM Aβ1-40 or 0.1 μM Aβ1-40 + IgG 7 mg/mL for 24 h. Data are the mean ± SEM of 5 independent experiments. *** p < 0.001 by one-way ANOVA using Newman-Keuls post-test.
Considering that cerebral amyloid angiopathy is a concomitant pathology in most of AD cases [40, 41], we assayed the effect of IgG on endothelial protection against amyloid challenge (Figure 1D). Brain vascular amyloid deposits are mainly composed of Aβ1-40 [42], in consequence, we used this species to perform the experiment. The treatment was performed using a final concentration of 0.1 μM because it produces a significant decrease in cell viability. As expected, simultaneous incubation of Aβ1-40 with 7 mg/mL of IgG protects endothelial cells against oligomers cytotoxicity.
IgG inhibits Aβ aggregation
After demonstrating that IgG prevents Aβ-mediated neurotoxicity, we were interested in elucidating the mechanism by which IgG mediates its protective effect. Therefore, Aβ1-42 was set at aggregation conditions in the presence or absence of IgG (Figure 2A). We used the plasmatic physiological concentration of IgG (7 mg/mL) and a subphysiological concentration (0.7 mg/mL). Thioflavin T (ThT) fluorescence was analyzed after 96 h of aggregation assay to allow fibril formation. We observed a significant reduction of Aβ1-42 fibril formation in the presence of 7 mg/mL of IgG (p<0.05). To confirm these results, Aβ1-42 was visualized on freshly cleaved highly oriented pyrolytic graphite (HOPG) with atomic force microscopy (AFM; Figure 2B). In the untreated Aβ1-42 controls after 96 h of aggregation mature fibers could be clearly identified, as well as clusters of oligomers (inset in Figure 2B), whereas after co-incubation with 7 mg/mL of IgG, there were mostly amorphous globular structures concomitant with a significant reduction of mature fibrils. Overall, these results are in agreement with the data obtained in ThT assays.
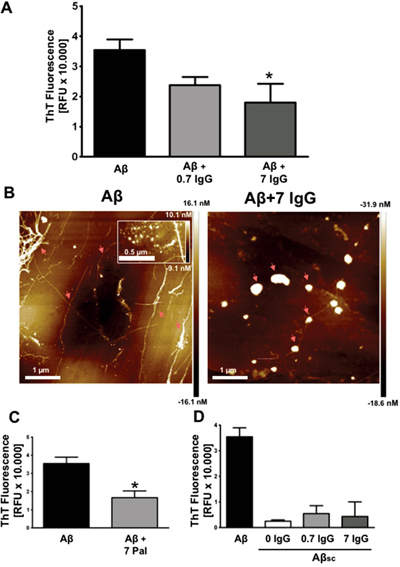
Figure 2: IgG inhibits amyloid aggregation. (A) Aggregation assay of 27 μM Aβ1-42 with or without IgG at 0.7 and 7 mg/mL during 96 h. ThT fluorescence was measured at 96 h. Data are the mean ± SEM of 3 independent experiments. * p < 0.05 compared to control by one-way ANOVA using Newman-Keuls post-test. (B) AFM images of 88,6 μM Aβ1-42 (left) and 88,6 μM Aβ1-42 treated with 7 mg/mL of IgG (right) for 96 h. The inset in the left image shows a cluster of oligomers. Arrows indicate mature fibers (left) and amorphous globular structures (right) (C) Aggregation assay of 27 μM Aβ1-42 with or without Palivizumab at 7 mg/mL during 96 h. ThT fluorescence was measured at 96 h. Data are the mean ± SEM of 3 independent experiments. * p < 0.05 by t-student test. (D) Aggregation assay of 27 μM Aβ1-42, 27 μM Aβ1-42 scramble (Aβsc) and 27 μM Aβsc co-incubated with IgG at 0.7 mg/mL and 7 mg/mL. ThT fluorescence was measured at 96 h. Data are the mean ± SEM of 3 independent experiments.
To study the importance of antibody’s specificity we used Palivizumab (Pal), a humanized IgG against respiratory syncytial virus [43] (Figure 2C). Aβ1-42 was aggregated in the presence of Pal (7 mg/mL). The aggregation process was analyzed by ThT fluorescence after 96 h. Our results show a significant decrease in Aβ1-42 fibril formation similar to the observed with the pool of human IgG (Figure 2A). Thus, the inhibition of amyloid aggregation in vitro by IgG seems to be independent of IgG specificity.
In order to rule out an unspecific effect of IgG, we measured ThT fluorescence in an aggregation assay of scramble Aβ1-42 (Aβsc; which is not able to aggregate) and Aβsc co-incubated with IgG at 7 mg/mL and 0.7 mg/mL (Figure 2D). Data showed the specificity of ThT fluorescence on fiber formation and the absence of inherent fluorescence of IgG.
Finally, the presence of different amyloid aggregates populations was analyzed by dynamic light scattering (DLS, Figure 3A and 3B) and the corresponding secondary structure was characterized using attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR, Figure 3C and 3D). Aβ1-42 oligomers measurement by DLS showed a detectable population of high molecular weight (particles > 1 μm) and even larger aggregates, which could not be detected and that were over the device detection limit (sharp decay at sizes > 5 μm, top panel Figure 3A). For IgG, there is a major population (ca. 17%) centered at around 13 nm, corresponding to the size of antibodies, and two less populated (<5%) fractions 500 nm and 5 μm (middle panel Figure 3A). Aβ1-42 oligomers incubated with 7 mg/ml of IgG for 24h show two populations, one centered at 13 nm corresponding to IgG and a second one widely centered at 1 μm (lower panel Figure 3A). In Figure 3B, the size distribution as a function of particle volume shows that the sample containing Aβ1-42 oligomers has a dominant population at > 1 μm, whereas in the Aβ1-42 oligomers incubated with 7 mg/ml of IgG sample two populations appear, the IgG at 13 nm and a minor population at 2 μm. The concentration of IgG (7 mg/ml) compared to Aβ1-42 oligomers (0.4 mg/ml) may mask the observation of aggregation in DLS, thus we performed ATR-FTIR in thin hydrated films, normalizing the signal for the protein concentration present in the film (Figure 3C). The Aβ1-42 oligomers spectrum shows a distinctive amyloid β-sheet band at 1628 cm-1, which is absent in the IgG containing samples. To confirm that amyloid β-sheet bands were absent in the IgG containing samples, band narrowing using the spectra second derivative is shown in Figure 3D. None of the β-sheet bands is present in sample Aβ1-42 oligomers co-incubated with 7 mg/ml of IgG. The results obtained by DLS and ATR-FTIR further support that IgG prevents Aβ1-42 aggregation in vitro. Overall, these experiments suggest a protective effect of IgG against Aβ1-42 aggregation and toxicity.
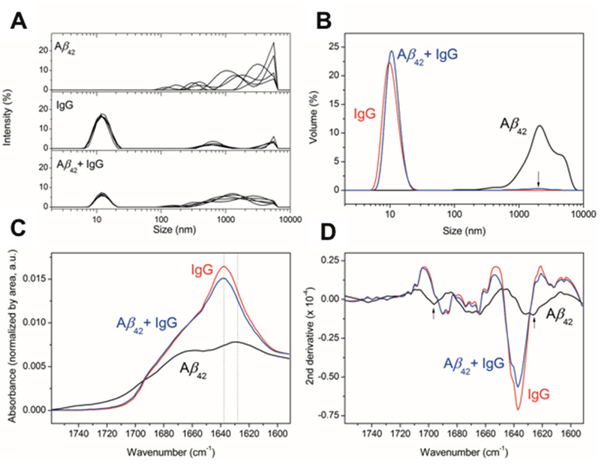
Figure 3: Effect of IgG on Aβ oligomerization. (A) DLS. Size distribution measured by DLS as a function of signal intensity of particles for 5 DLS measurements of 10 scans each. (B) Size distribution as a function of volume of the measured particles for the averaged measurements. The arrow indicates the presence of a low percentage population at the specified size for the mixture of Aβ plus IgG. (C) ATR-FTIR spectroscopy of the amide I region for the indicated samples in a thin hydrated film. All spectra are normalized by the area under the curve, representative of the protein amount measured in each thin hydrated film. The dotted lines indicate the wavenumber position of the highest intensity peaks for the samples analyzed. (D) Second derivative of the ATR-FTIR to highlight different secondary structures due to mathematical band narrowing [59]. The arrows indicate the position of the bands characteristic for β-sheet amyloid aggregation.
Fab fragment is involved in the inhibitory effects in Aβ aggregation
The IgG is formed by the Fab, which contains the antigen-binding site and it is responsible for IgG specificity and the crystallizable fragment (Fc). Our next step was to study whether IgG inhibitory effect in Aβ1-42 aggregation and neurotoxicity was a property of the whole IgG or it relies on one of these fragments. Therefore, Aβ1-42 was co-incubated, separately, with the Fab and with the Fc for 96 h and ThT fluorescence was measured. Fab and Fc concentrations were calculated based on IgG stoichiometry. We observed that Fab inhibits Aβ1-42 aggregation in a concentration-depending manner (Figure 4A), suggesting that it is the region that binds to amyloid. On the other hand, Fc did not show any effect in Aβ1-42 aggregation (Figure 4B).
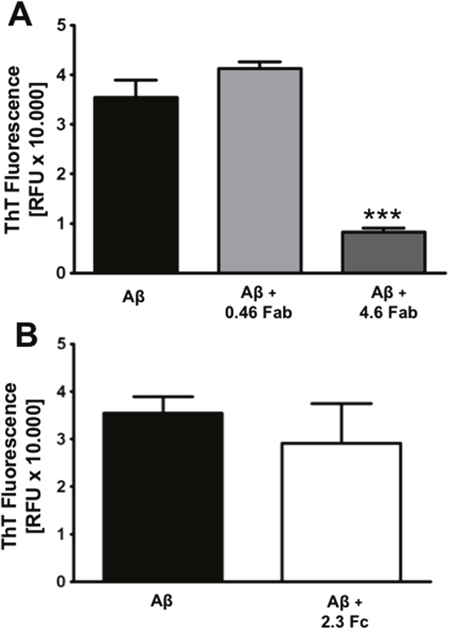
Figure 4: Fab is the region that inhibits amyloid aggregation. Aggregation assay of 27 μM Aβ1-42 co-incubated with Fab fragment at 0.46 and 4.6 mg/mL (A) or Fc fragment at 2.3 mg/mL (B) during 96 h. ThT fluorescence was measured at 96 h. Mean ± SEM of 3 independent experiments *** p < 0.001 by one-way ANOVA using Newman-Keuls post-test.
To explore the mechanism for non-specific binding of Fab-Aβ1-42, a rigid-body docking analysis was performed between a selected group of non-redundant human Fabs with known structure and the structure of Aβ1-42 [44]. The top scored decoys show the Aβ1-42 peptide in complex with the human myeloma IgG [45] (Figure 5A) and the human HIV-neutralizing antibody 2F5 [46] (Figure 5B). In both cases, Aβ1-42 bound through a non-specific β–β ladder strands-pairing, like the intra-strand interactions of Aβ1-42 peptides in fibrillation. This configuration seems favorable, especially in the case of human myeloma IgG, because the stacked β–β pairing with IgG has a Cα-Cα distance of 10.8±0.4Å like the Aβ1-42 stacking distance in fibrillation but higher (with more distance between the stacked β–strands) than other exposed β–strands of IgG (8.5±1Å). Combined with experimental data, this model suggests that IgG domains can affect fibrillation by binding Aβ1-42 peptides like a new monomer of the fiber, blocking its progression. Considering that fibrillation of Aβ1-42 progresses from both strand sides, this mechanism can also explain the concentration-dependence of the process, requiring two IgG to block each side of fiber progression (see Figure 5 and Video: https://drive.google.com/drive/folders/0B_99pS9URra_U2dZS2dxRUJFWnM?usp=sharing).
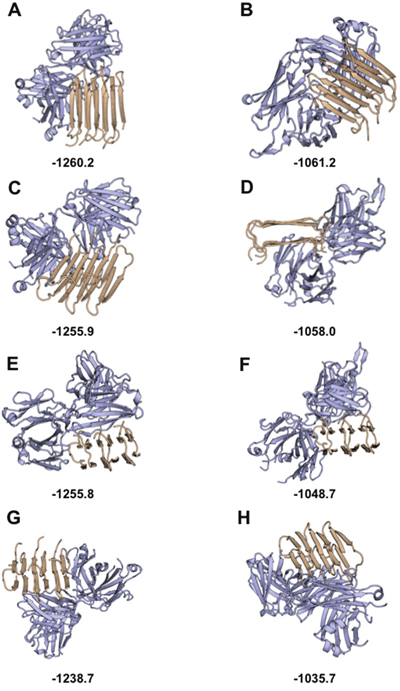
Figure 5: In silico study of Aβ1-42 binding the IgG Fab region: Top scored decoys for antibody-Aβ1-42 docking. In silico structural modeling of the interaction between amyloid (light brown) and the functional region of the Fab domain (light blue). Top scored decoys for the interaction with the structure of the Fab region of myeloma IgG are shown in (A, C, E, and G,) with best to worst decoys, respectively. Top scored decoys for the interaction with the structure of the Fab region of HIV IgG are shown in (B, D, F, and H,) with best to worst decoys, respectively. Scores obtained with Rosetta are shown at the bottom of each decoy (low score Rosetta-energies indicate the most stable decoys).
DISCUSSION
Different strategies have been used to control Aβ levels in the brain, although none of them has been successful. Nowadays, there are a few drugs approved for AD treatment. Among them, most of the approved drugs for AD are cholinesterase inhibitors (tacrine, donezepil, galantamine, rivastigmine) and also, a NMDA receptor antagonists (memantine) [47]. However, currently there are not drugs capable of modulating the mechanisms of Aβ oligomerization for therapeutic purposes.
The present work shows that the pool of human IgG protects cortical neurons and endothelial cells against Aß cytotoxicity, and inhibits Aβ aggregation at 7 mg/mL. The inhibitory effect on Aß aggregation had already been described for some anti-Aβ antibodies [24]. The authors attribute this effect to its specific binding to Aβ, however it was not shown in which IgG fragment it resides. Here, we identify for the first time Fab as the fragment responsible for inhibition Aβ aggregation, and that this effect is independent of IgG specificity. Over last years, immunotherapy has been considered a promising approach to prevent amyloid plaque formation, and therefore to ameliorate cognitive decline in AD patients. Different clinical trials have been performed, but none of them have been able to overcome phase III [30, 31]. Nonetheless, there are still some anti-Aβ antibodies ongoing phase III trials due to their potential therapeutic effect [32, 34, 35]. The vast majority of treatments that have been tested following this strategy are antibodies targeting Aβ, our results using a pool of human IgG and Palivizumab (Pal) suggest that the specificity of the antibody is not especially relevant for inhibiting in vitro amyloid aggregation. However, our results were obtained by IgG or Pal co-incubation with soluble Aβ, and therefore recognizing its native non-folded sequence. Thus, we cannot discard that human anti-Aβ antibodies recognizing oligomers or fibrils folded domains could produce beneficial results in AD treatment.
Clinical trials have shown that immunotherapy reduce Aβ amyloid deposits in the brain, besides showing safety and tolerance [21, 29–33]. However, they also present several significant side effects that should be considered. These side effects, such as cerebral amyloid angiopathy (CAA)-associated cerebral hemorrhage [48] or vascular edema [49], are primarily related to inflammation [50, 51]. Taking into consideration that Fc is the fragment responsible for triggering the immune response, administration of IgG could be a method to overcome inflammatory problems. Moreover, reducing IgG size could be a manner to potentiate their pass through BBB. In addition, intravenous IgG therapy has also been proposed as a treatment for AD. It has been shown to reduce Aβ levels in the CSF, increase its levels in serum and improve cognitive function, but it has not been able to conclude any clinical trial [36, 37, 52]. Importantly, adverse reactions to this treatment are not common and they are usually minor, including mild to moderate headache, fever, chills and myalgia [53]. Nevertheless, a quite promising study on a human anti-Aβ antibody able to recognize oligomers of Aβ has been recently published [32].
On the other hand, amyloid is produced by all the cells of the body [54], but paradoxically, Aß aggregation is just observed inside the brain. These evidences suggest that amyloid aggregation would be physiologically inhibited in systemic tissues and in brain, until the amount of amyloid exceeds a concentration threshold. Is in this scenario that IgG would be acting as a physiological inhibitor of amyloid aggregation, and therefore ameliorating AD condition.
In conclusion, the data presented here contribute to the understanding of the role of IgG regarding Aβ aggregation. Further studies on Fab fragment may lead to the development of new approaches able to prevent Aβ oligomerization without leading to inflammation or other side-effect, hence being a promising treatment for AD.
MATERIALS AND METHODS
Mouse cortical primary cultures
Mouse cortical neurons were isolated from 18-day-old CD1 embryos. The procedure was approved by the Ethics Committee of the Institut Municipal d’Investigacions Mèdiques-Universitat Pompeu Fabra (EC-IMIM-UPF). Cortex was aseptically dissected in ice-cold HBSS (Life Technologies) supplemented with 4.5 g/L glucose (Sigma-Aldrich) and trypsinized for 17 min at 37ºC. After 3 washes in HBSS+glucose and mechanical dissociation, cells were seeded on DMEM (Life Technologies) plus 10% horse serum (Life Technologies) onto 1% poly-D-Lysine (Sigma-Aldrich, USA) coated plates. After 2 h, medium was removed and Neurobasal medium (Life Technologies) was added containing 2% B27 supplement (Gibco BRL), 1% GlutaMAX (Life Technologies) and 1% Penicillin/Streptomycin. On day 3 of culture (DIV), cells were treated with 2 μM 1-β-D-arabinofuranosylcytosine (AraC; Sigma) for 24 h to eliminate proliferating non-neuronal cells. Primary cortical neurons were used at 10 DIV.
Porcine aortic endothelial cultures
Endothelial cells were isolated from porcine aortas provided by FRISELVA (Girona, Spain). Endothelial cells were obtained following Epithelial Cell Culture Protocols of Methods in Molecular Biology. Briefly, porcine aortas were cleaned in washing solution (HBSS with 0.002 mM HEPES and antibiotics) and incubated in collagenase solution (50 mg/mL collagenase type II, 0.1 g/L MgCl2·6H2O, 7 g/L NaCl, 0.1 g/L Na2HPO4·7H2O, 0.18 g/L NaH2PO4·H2O, 0.54 g/L D-Glucose and 0.2 % FBS) for 15 minutes. Aortas were scratched into Endothelial Medium (DMEM F12K, 0.1 mg/mL heparin, 10% fetal bovine serum (FBS) and 2 mM of L-Glutamine plus antibiotics). Cells were collected by centrifugation and seeded in T-75 flasks. Cells were grown in Endothelial Medium and were used up to passage number 8.
Aβ1-42 and Aβ1-40 preparation
Lyophilized Aβ1-40 or Aβ1-42 (Anaspec) were solubilized as previously described [55]. Briefly, 1 mg of Aβ was dissolved in 250 μL of MilliQ water and pH was adjusted to ≥ 10.5 using 1 M NaOH solution to avoid the isoelectric point of Aβ. Then, 250 μL of 20 mM Phosphate Buffer (pH 7.4) were added and the preparation was placed for 1 min in a bath-type sonicator (Bioruptor, Diagenode). Aβ preparation was immediately used for oligomer preparation or stored in 25 μL aliquots at -20ºC until used for ThT experiments. For oligomer preparation, Aβ was dissolved to 0.4 mg/mL in DMEM/F12 without phenol red with or without IgG (Grifols) or 6E10 (Covance).
IgG preparation
The pool of human IgG was provided by Grifols (Flebogamma® 5% DIF; 100 mL).
Cell viability assay by MTT reduction
Cortical primary neurons and endothelial cells were seeded in a 96-well plate at a density of 1x104 cells/well. The treatment (100 μL/well) was added to Neurobasal without phenol red supplemented with 1% GlutaMAX (Life Technologies) for cortical neurons and Endothelial Medium without FBS for endothelial cells. Cells were treated for 24 h at 37ºC. Cell viability was tested by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. Briefly, 11 μL of MTT stock solution (Sigma Aldrich; 5 mg/mL) were added. After 2 h the media was replaced with 100 μL of DMSO. MTT absorbance was determined in an Infinite 200 multiplate reader (Tecan) at A540 nm and corrected by A650 nm. Untreated cells were taken as 100%.
Immunofluorescence
Cortical primary neurons (1x105 cells/well) were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 for 10 min at RT. Samples were blocked with 5% Fetal Bovine Serum, 1% albumine from bovine serum and 0.02% sodium azide overnight at 4ºC. Subsequently, coverslips were incubated for 2 h at RT in a hydration chamber with the following primary antibodies: anti-β3 tubulin (1:200; Cell Signalling) and anti-cleaved Caspase-3 (1:100; Abcam). Then, samples were washed thrice with PBS and incubated with Alexa Fluor 488 goat anti-mouse and Alexa Fluor 555 goat anti-rabbit antibodies (1:1000; Life Technologies) for 1 h at RT. Coverslips were mounted and analyzed using a Leica TCS SP5 confocal microscope (63x objective) and analyzed with Leica confocal software.
Thioflavin T binding assay
A 1 mM stock ThT (Sigma Aldrich) solution was prepared dissolving the dye in PBS. The solution was filtered using a 0.22 μm syringe filter. ThT solutions were stored at -20ºC in the dark. Aβ42 peptide (27.5 μM) was incubated with or without IgG (0.7 and 7 mg/mL; Grifols), Palivizumab (7 mg/mL; Synagis), Fab (4.6 mg/mL; Grifols) and Fc (2.3 mg/mL; Grifols) and 10 μM ThT in a Nunc-96-well flat bottom black polystrol microplate (Thermo Scientific) at 37ºC. ThT fluorescence was measured at 96 h using excitation and emission wavelengths of 430 and 470 nm, respectively, using a multiplate reader fluorimeter (FLUOstar optima, BMG labtech).
Atomic force microscopy
A MultiMode atomic force microscope (Veeco Instruments Inc., Santa Barbara, CA) equipped with a 12 mm scanner (E-scanner) was used to analyze Aβ samples aggregated with or without IgG for 96 h. Sample aliquots were frozen and were thawed at RT just before the AFM experiments. The images were taken in liquid with a liquid cell without the O-ring seal. 5-10 mL of sample were deposited on cleaved mica substrates. After an adsorption time of 5 min, 40 mL of double deionised water were added to form a drop suitable for the imaging procedure. The system was left for equilibration for at least 10 min before carrying out the experiments. NP-S (Veeco) probes were used to scan the samples in tapping mode at 0.5 Hz scan rate. Height and amplitude images were recorded simultaneously, although only the latter are presented in this paper.
DLS measurements
Samples were prepared as indicated above and kept for 24 h at 4ºC. DLS measurements were recorded on a Zetasizer instrument (Nano ZS; Malvern Instruments) at 20º C using a small volume (40 μL) quartz cuvette of 1 cm path length. Scattering data were collected as an average of 5 measurements with 10 scans for each measurement. Refractive indices were 1.330 (water) and 1.45 (protein) for polydispersant and material, respectively. Data were processed with the Malvern Zetasizer Software (Malvern Instruments).
ATR-FTIR spectroscopy
Thin hydrated films of samples placed on top of the ATR diamond crystal were prepared under a N2 stream gentle drying. Spectra were obtained at RT by the average of 250 scans at 2cm-1 resolution in a VARIAN FTS-7000 infrared spectrometer. The second derivative was performed with ten smoothing points.
Size exclusion chromatography
Size exclusion chromatography was performed in an AKTA purifier FPLC system. All chromatograms were run at 4ºC using a flow of 0.4 mL/min in 10 mM Tris·HCl buffer pH 7.4. An initial run in a Superdex 75 10/30 column was performed to separate HMW and LMW aggregates. HMW aggregates were injected in a Superose 6 10/30 column to analyze the size exclusion profile of the non-resolved species.
Fab and Fc fragments purification
To obtain Fab and Fc fragments of IGIV Flebogamma® DIF 5% 100 mL, the commercial Fab preparation kit (Pierce) was used following manufacturer instructions and the molecular distribution analysis method MA_IG-000158A was used. Briefly, 500 μL of IGIV (8 mg/mL) were desalted by Zeba™ Spin Desalting Column following the kit instructions, while 250 μL agarose resin with immobilized papaine, previously packed in a 0.8 mL column, were equilibrated with a digestion buffer (cysteine·HCl 3.5 mg/mL pH 7). Once equilibrated, 500 μL of desalted IGIV were added and maintained in agitation at 900 rpm for 10 h at 37ºC. Then, the microcolumn was centrifuged and washed with PBS at 5000 g for 1 min to separate completely the digest and the agarose resin.
To purify the Fab fragment from de digest, it was added to a NAb™ Protein A Spin Column, previously equilibrated, and incubated for 10 min at RT. After the incubation, Fc fragments and undigested IgGs were retained at the column, while Fab purified fragments were collected with flow through. To recover Fc fragments and undigested IgGs, NAb™ Protein A Spin Column was eluted. The eluted was concentrated to 6 mg/mL of protein using Ultracel YM-3 filters 3 kDa NMWL (Millipore). Then, it was injected to an exclusion molecular column TSKgel G3000 SW 60 cm X 7.5 mm. I.D. (Teknokroma) in order to be fractioned with a high-performance liquid chromatography (HPLC) Waters 2695 Separations Module (Alliance) with UV detector diodo-array Waters 996. After 16.5 min, Fc purified fraction was recollected for 1 min. Finally, Fab and Fc purified fragments were concentrated using Amicon Ultra-15 filters 3 kDa NMWL (Millipore).
Computational analysis of the IgG-amyloid interaction
All human IgGs with less than 70% sequence identity and known structure were retrieved with the Protein Data Bank REST service [56]. From this search, two non-redundant representatives of the IgG Fab region were found: the human myeloma IgG (PDB: 8FAB) [45] and the human HIV-neutralizing antibody 2F5 (PDB:2F5B) [46]. These two structures were tested individually by performing rigid-body docking with the structure of Aβ1-42 obtained by solid-state NMR (PDB: 2LNQ) [44].
Around 19000 decoys of the complex IgG-Aβ1-42 were obtained for each IgG (2LNQ-8FAB and 2LNQ-2F5B) with PatchDock [57]. We selected the top 100 decoys of PatchDock to calculate the binding ΔG with Rosetta [58]. We selected the top 4 decoys with higher binding affinity for each tested IgG.
The final selected models depict the conformations of Aβ1-42 interacting with the human myeloma IgG (Figures 5A, 5C, 5E, 5G) and the human HIV-neutralizing antibody (Figures 5B, 5D, 5F, 5H). The best scored decoys (Figures 5A, 5B) show the interaction between Aβ1-42 and the IgG Fab region through the pairing of β-strands, extending the Ig-like fold core β-sheet. The non-specific pairing of β-strands can be seen in other top scored decoys (Figures 5C, 5F, 5G, 5H), although in these decoys only one β-strand of Aβ1-42 binds the β-sheet of the IgG fold, reinforced with other side-chain contacts. All decoys show the molecular mechanisms by which the IgG Fab region blocks the progression of the fiber: either playing the role of a new Aβ1-42 monomer, or avoiding the attachment of new Aβ1-42 monomer due to steric hindrances.
Statistical analysis
Data are expressed as mean ± SEM of n experiments as indicated in the corresponding figures. Statistical analyses were performed by one-way ANOVA or t-student using GraphPad software. * p < 0.05; *** p < 0.001.
Abbreviations
Ab, Antibody; Aß, Amyloid-ß peptide; AD, Alzheimer disease; CSF, cerebrospinal fluid; FBS, fetal bovine serum; IgG, gamma-globulin; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Palivizumab, Pal.
ACKNOWLEDGMENTS
This work was supported by the Plan Estatal de I+D+I 2013-2016 and the ISCIII-Subdirección General de Evaluación y Fomento de la Investigación (Grants PI13/00408, PI13/00135, and Miguel Servet Grant CP10/00548 to X.A.) and FEDER Funds; SAF2014-52228-R; BIO2014-57518-R and Fundació La Marató-TV3 (Nº 20140210; Nº 20134030). Victòria Valls-Comamala is a recipient of a predoctoral fellowship from the Spanish Ministry of Education. A.P.-M. is a recipient of the Universitat Autònoma de Barcelona-Programa Banco de Santander Fellowship.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
1. Guivernau B, Bonet J, Valls-Comamala V, Bosch-Morató M, Godoy JA, Inestrosa NC, Perálvarez-Marín A, Fernández-Busquets X, Andreu D, Oliva B, Muñoz FJ. Amyloid-β-peptide nitrotyrosination stabilizes oligomers and enhances NMDAR-mediated toxicity. J Neurosci. 2016; 36:11693–11703.
2. Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, Dziewczapolski G, Nakamura T, Cao G, Pratt AE, Kang YJ, Tu S, Molokanova E, et al. A induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci. 2013; 110:2518–2527.
3. Selkoe DJ, Morris JC, Petersen RC, Lewis J, Davies P, Maloney AJ, Whitehouse PJ, Small DH, Mok SS, Bornstein JC, Davies CA, Mann DM, Sumpter PQ, et al. Alzheimer’s disease is a synaptic failure. Science. 2002; 298:789–791.
4. Tajes M, Eraso-Pichot A, Rubio-Moscardó F, Guivernau B, Ramos-Fernández E, Bosch-Morató M, Guix FX, Clarimón J, Miscione GP, Boada M, Gil-Gómez G, Suzuki T, Molina H, et al. Methylglyoxal produced by amyloid-β peptide-induced nitrotyrosination of triosephosphate isomerase triggers neuronal death in Alzheimer’s disease. J Alzheimers Dis. 2014; 41:273–288.
5. Ramos-Fernández E, Tajes M, Palomer E, Ill-Raga G, Bosch-Morató M, Guivernau B, Román-Dégano I, Eraso-Pichot A, Alcolea D, Fortea J, Nuñez L, Paez A, Alameda F, et al. Posttranslational nitro-glycative modifications of albumin in Alzheimer’s disease: implications in cytotoxicity and amyloid-β peptide aggregation. J Alzheimers Dis. 2014; 40:643–657.
6. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002; 297: 53–56.
7. Ill-Raga G, Tajes M, Busquets-García A, Ramos-Fernández E, Vargas LM, Bosch-Morató M, Guivernau B, Valls-Comamala V, Eraso-Pichot A, Guix FX, Fandos C, Rosen MD, Rabinowitz MH, et al. Physiological control of nitric oxide in neuronal BACE1 translation by heme-regulated eIF2α kinase HRI induces synaptogenesis. Antioxid Redox Signal. 2015; 22:1295–1307.
8. Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz E, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999; 286:735–741.
9. Zhang Y, Thompson R, Zhang H, Xu H. APP processing in Alzheimer’s disease. Mol Brain. 2011; 4:3.
10. Guix FX, Wahle T, Vennekens K, Snellinx A, Chávez-Gutiérrez L, Ill-Raga G, Ramos-Fernandez E, Guardia-Laguarta C, Lleó A, Arimon M, Berezovska O, Muñoz FJ, Dotti CG, et al. Modification of γ-secretase by nitrosative stress links neuronal ageing to sporadic Alzheimer’s disease. EMBO Mol Med. 2012; 4:660–673.
11. Vigo-Pelfrey C, Lee D, Keim P, Lieberburg I, Schenk DB. Characterization of beta-amyloid peptide from human cerebrospinal fluid. J Neurochem. 1993; 61:1965–1968.
12. Howell S, Nalbantoglu J, Crine P. Neutral endopeptidase can hydrolyze beta-amyloid(1-40) but shows no effect on beta-amyloid precursor protein metabolism. Peptides. 1995; 16:647–652.
13. Kurochkin IV, Goto S. Alzheimer’s beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994; 345:33–37.
14. Ledesma MD, Da Silva JS, Crassaerts K, Delacourte A, De Strooper B, Dotti CG. Brain plasmin enhances APP alpha-cleavage and Abeta degradation and is reduced in Alzheimer's disease brains. EMBO Rep. 2000; 1:530–535.
15. Parvathy S, Hussain I, Karran EH, Turner AJ, Hooper NM. Alzheimer’s amyloid precursor protein alpha-secretase is inhibited by hydroxamic acid-based zinc metalloprotease inhibitors: similarities to the angiotensin converting enzyme secretase. Biochemistry. 1998; 37: 1680–1685.
16. Eckman EA, Reed DK, Eckman CB. Degradation of the Alzheimer's amyloid beta peptide by endothelin-converting enzyme. J Biol Chem. 2001; 276: 24540–24548.
17. Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004; 43:333–344.
18. Fukumoto H, Rosene DL, Moss MB, Raju S, Hyman BT, Irizarry MC. Beta-secretase activity increases with aging in human, monkey, and mouse brain. Am J Pathol. 2004; 164:719–725.
19. Muñoz FJ, Opazo C, Gil-Gómez G, Tapia G, Fernández V, Valverde MA, Inestrosa NC. Vitamin E but not 17beta-estradiol protects against vascular toxicity induced by beta-amyloid wild type and the Dutch amyloid variant. J Neurosci. 2002; 22:3081–3089.
20. Coma M, Guix FX, Uribesalgo I, Espuña G, Solé M, Andreu D, Muñoz FJ. Lack of oestrogen protection in amyloid-mediated endothelial damage due to protein nitrotyrosination. Brain. 2005; 128:1613–1621.
21. Citron M. Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9:387-398.
22. Selkoe DJ. The therapeutics of Alzheimer’s disease: where we stand and where we are heading. Ann Neurol. 2013; 74:328–336.
23. Ising C, Stanley M, Holtzman DM. Current thinking on the mechanistic basis of Alzheimer’s and implications for drug development. Clin Pharmacol Ther. 2015; 98:469–471.
24. Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc Natl Acad Sci U S A. 1997; 94:4109–4112.
25. Solomon B, Koppel R, Hanan E, Katzav T. Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer beta-amyloid peptide. Proc Natl Acad Sci U S A. 1996; 93:452–455.
26. Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000; 6:916–919.
27. Frenkel D, Katz O, Solomon B. Immunization against Alzheimer’s beta-amyloid plaques via EFRH phage administration. Proc Natl Acad Sci U S A. 2000; 97:11455–11459.
28. Zhang Y, Yang HQ, Fang F, Song LL, Jiao YY, Wang H, Peng XL, Zheng YP, Wang J, He JS, Hung T. Single chain variable fragment against Aβ expressed in baculovirus inhibits Abeta fibril elongation and promotes its disaggregation. PLoS One. 2015; 10:1–16.
29. Ivanoiu A, Pariente J, Booth K, Lobello K, Luscan G, Hua L, Lucas P, Styren S, Yang L, Li D, Black RS, Brashear HR, McRae T. Long-term safety and tolerability of bapineuzumab in patients with Alzheimer’s disease in two phase 3 extension studies. Alzheimers Res Ther. 2016; 8:24.
30. Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, Reichert M, Ketter N, Nejadnik B, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014; 370:322–333.
31. Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, Siemers E, Liu-Seifert H, Mohs R, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014; 370:311–321.
32. Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016; 537:50–56.
33. Ferrero J, Williams L, Stella H, Leitermann K, Mikulskis A, O’Gorman J, Sevigny J. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimer’s Dement Transl Res Clin Interv. 2016; 2:169–176.
34. Siemers ER, Sundell KL, Carlson C, Case M, Sethuraman G, Liu-Seifert H, Dowsett SA, Pontecorvo MJ, Dean RA, Demattos R. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement. 2016; 12:110–120.
35. Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, Klunk WE, Ashford E, Yoo K, Xu ZX, Loetscher H, Santarelli L. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012; 69:198–207.
36. Dodel R, Rominger A, Bartenstein P, Barkhof F, Blennow K, Förster S, Winter Y, Bach JP, Popp J, Alferink J, Wiltfang J, Buerger K, Otto M, et al. Intravenous immunoglobulin for treatment of mild-to-moderate Alzheimer’s disease: a phase 2, randomised, double-blind, placebo-controlled, dose-finding trial. Lancet Neurol. 2013; 12:233–243.
37. Relkin N. Intravenous immunoglobulin for Alzheimer’s disease. Clin Exp Immunol. 2014; 178:27–29.
38. Bouloukos A, Lekakis J, Michael J, Kalofoutis A. Immunoglobulins in cerebrospinal fluid in various neurologic disorders. Clin Chem. 1980; 26:115–116.
39. Chang YJ, Chen YR. The coexistence of an equal amount of Alzheimer’s amyloid-β 40 and 42 forms structurally stable and toxic oligomers through a distinct pathway. FEBS J. 2014; 281:2674–2687.
40. Vinters HV, Pardridge WM, Secor DL, Ishii N. Immunohistochemical study of cerebral amyloid angiopathy. II. Enhancement of immunostaining using formic acid pretreatment of tissue sections. Am J Pathol. 1988; 133:150–162.
41. Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci U S A. 1999; 96:14088–14093.
42. Castaño EM, Prelli F, Soto C, Beavis R, Matsubara E, Shoji M, Frangione B. The length of amyloid-beta in hereditary cerebral hemorrhage with amyloidosis, Dutch type. Implications for the role of amyloid-beta 1-42 in Alzheimer’s disease. J Biol Chem. 1996; 271:32185–32191.
43. Subramanian KN, Weisman LE, Rhodes T, Ariagno R, Sánchez PJ, Steichen J, Givner LB, Jennings TL, Top FH, Carlin D, Connor E. Safety, tolerance and pharmacokinetics of a humanized monoclonal antibody to respiratory syncytial virus in premature infants and infants with bronchopulmonary dysplasia. MEDI-493 Study Group. Pediatr Infect Dis J. 1998; 17:110–115.
44. Qiang W, Yau WM, Luo Y, Mattson MP, Tycko R. Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc Natl Acad Sci. 2012; 109:4443–4448.
45. Strong RK, Campbell R, Rose DR, Petsko GA, Sharon J, Margolies MN. Three-dimensional structure of murine anti-p-azophenylarsonate Fab 36-71. 1. X-ray crystallography, site-directed mutagenesis, and modeling of the complex with hapten. Biochemistry. 1991; 30:3739–3748.
46. Bryson S, Julien JP, Hynes RC, Pai EF. Crystallographic definition of the epitope promiscuity of the broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2F5: vaccine design implications. J Virol. 2009; 83:11862–11875.
47. Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014; 6:37.
48. Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Aβ immunotherapy. Science. 2002; 298:1379.
49. Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, Sabbagh M, Honig LS, Doody R, Van Dyck CH, Mulnard R, Barakos J, Gregg KM, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009; 73:2061–2070.
50. Check E. Nerve inflammation halts trial for Alzheimer’s drug. Nature. 2002; 415:462.
51. Schenk DB, Yednock T. The role of microglia in Alzheimer’s disease: friend or foe? Neurobiol Aging. 2002; 23:677–679.
52. Dodel RC, Du Y, Depboylu C, Hampel H, Frölich L, Haag A, Hemmeter U, Paulsen S, Teipel SJ, Brettschneider S, Spottke A, Nölker C, Möller HJ, et al. Intravenous immunoglobulins containing antibodies against beta-amyloid for the treatment of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2004; 75:1472–1474.
53. Donofrio PD, Berger A, Brannagan TH, Bromberg MB, Howard JF, Latov N, Quick A, Tandan R. Consensus statement: the use of intravenous immunoglobulin in the treatment of neuromuscular conditions report of the AANEM ad hoc committee. Muscle Nerve. 2009; 40:890–900.
54. Del Toro D, Coma M, Uribesalgo I, Guix F, Muñoz FJ. The amyloid β-protein precursor and Alzheimers disease. Therapeutic approaches. Curr Med Chem Nerv Syst Agents. 2005; 5:271–283.
55. Bitan, G. Teplow B. Preparation of aggregate-free, low molecular weight amyloid-beta for assembly and toxicity assays. Methods Mol Biol. 2005; 299:3-9.
56. Costanzo LD, Ghosh S, Zardecki C, Burley SK. Using the tools and resources of the RCSB Protein Data Bank. Curr Protoc Bioinformatics. 2016; 55: 1.9.1–1.9.35.
57. Mashiach E, Schneidman-Duhovny D, Peri A, Shavit Y, Nussinov R, Wolfson HJ. An integrated suite of fast docking algorithms. Proteins. 2010; 78:3197–3204.
58. Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003; 331:281–299.
59. Perálvarez-Marín A, Barth A, Gräslund A. Time-resolved infrared spectroscopy of pH-induced aggregation of the Alzheimer Aβ1-28 peptide. J Mol Biol. 2008; 379:589–596.