
INTRODUCTION
Glioma is the most common primary central nervous system (CNS) malignancy. It is a major health threat [1–3]. Each year, glioma will cause significant cancer-related death [1–3]. Postoperative irradiation and temozolomide (TMZ) chemotherapy are the standard clinical treatments for glioma [4–6]. Yet, the overall survival has not been significantly improved over the past decades [4–6]. The prognosis of high-grade glioma, including glioblastoma, has been poor [1, 6, 7]. One possible cause is the overwhelming resistance to current irradiation (and chemotherapy) [1, 6, 7].
G protein α inhibitory subunit (Gαi) couples with GPCRs (G-protein coupled receptors) [8] to inhibit adenylate cyclase (AC) [8]. Recently, our group [9–11] and others [12] have discovered an un-anticipated function of Gαi: transducing Akt-mTOR signaling by receptor tyrosine kinases (RTKs). We have previously found that Gαi protein was required for EGFR (epidermal growth factor receptor)- and FGFR (fibroblast growth factor receptor)-induced activation of Akt signaling [9–11]. In our model, Gαi could couple with EGFR/FGFR to activate the adaptor protein (i.e. Gab1), which mediates activation of downstream Akt signaling [9–11].
There are at least three Gαi subunits, including Gαi1, Gαi2 and Gαi3 [8]. Our recent study has shown that Gαi3 is over-expressed in human glioma cells, which is required for Akt activation and cancer cell proliferation [11]. The results of the current study indicate that Gαi protein could also be important for irradiation resistance.
RESULTS
Silencing Gαi3 sensitizes irradiation-induced glioma cell death
In order to study the potential function of Gαi3 in irradiation resistance, shRNA strategy was applied. As described previously [11], two distant lentiviral shRNAs against non-overlapping sequence of Gαi3 were utilized. The two were named as “Gαi3 shRNA-a” and “Gαi3 shRNA-b”. As shown in Figure 1A, the two Gαi3 shRNAs silenced Gαi3 in human glioma A172 cells. These Gαi3-silenced A172 cells and control cells were treated with various degree (0–10 Gy) of irradiation. Trypan blue staining assay results in Figure 1B demonstrated that A172 cells with Gαi3 shRNA were significantly more sensitive to irradiation than the control A172 cells. Irradiation led to significantly more A172 cell death after Gαi3 knockdown (Figure 1B). The IC-50 of irradiation, or the intensity that kills 50% of A172 cells, decreased from over 6 Gy to less than 1.5 Gy after Gαi3 silence (Figure 1B). MTT assay results (Figure 1C) and colony formation assay (Figure 1D) further confirmed that Gαi3 knockdown significantly facilitated irradiation (5 Gy)-induced killing of A172 cells. Notably, Gαi3 shRNA-b was more efficient in silencing Gαi3 (than Gαi3 shRNA-a, Figure 1A), it was also more dramatic in sensitizing irradiation-induced A172 cell death (Figure 1B–1D). Notably, Gαi3 silence alone also induced minor/moderate A172 cell death (Figure 1B–1D), which was also reported early [11].
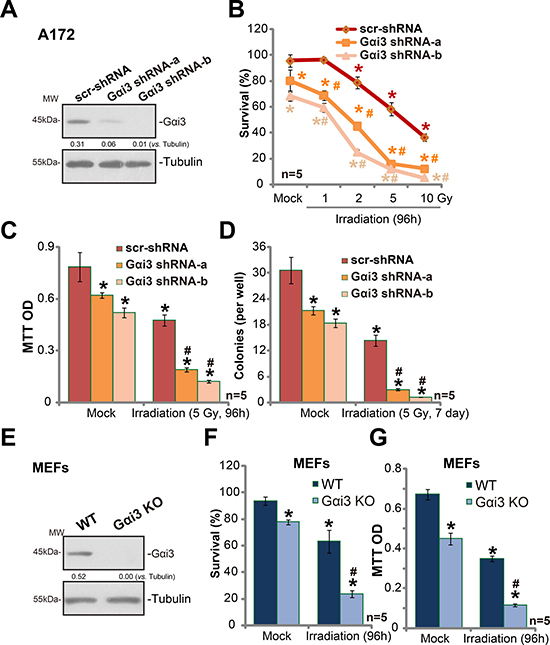
Figure 1: Silencing Gαi3 sensitizes irradiation-induced glioma cell death. Western blotting tested expression of listed proteins in stable A172 cells with Gαi3 shRNA (“-a/-b”) or scramble control shRNA (“scr-shRNA”) (A); A172 cells were also subjected to irradiation (at indicated intensity) and cultured for indicated time, listed assays were performed to test cell survival/death (B–D). Expression of listed proteins in wild-type (WT) and Gαi3 knockout (KO) MEFs was shown (E); MEFs were irradiated (5 Gy) and cultured for additional 96 hours. Afterwards, MEFs were subjected to trypan blue staining assay (F) and MTT assay (G). For all the assays, the exact same number of viable cells of different genetic background was initially plated into each well (Same for all Figures). Same set of lysate samples were run in sister gels (A and E). “Mock” stands for un-irradiated cells (Same for all Figures). “n = 5” means five replicate wells (Same for all Figures). Bars stand for mean ± SD (Same for all Figures). *p < 0.05 vs. “Mock” of “scr-shRNA” A172 cells or WT MEFs. #p < 0.05 vs. “Irradiation” of “scr-shRNA” A172 cells (B–D) or WT MEFs (F and G). Experiments in this figure were repeated three times, with similar results obtained.
The results above suggested that Gαi3 might be important in irradiation resistance. To further support this hypothesis, Gαi3 knockout (“KO”) mouse embryonic fibroblasts (MEFs) [11] were utilized.. Trypan blue assay results in Figure 1F and MTT assay results in Figure 1G confirmed that Gαi3 KO MEFs were significantly more vulnerable to irradiation (5 Gy) than the wild-type (“WT”) MEFs. For instance, 96 hours after irradiation (5 Gy), 63.3 ± 8.6 % of WT MEFs were still alive, yet only 23.6 ± 2.6% of Gαi3 KO MEFs were trypan blue negative (Figure 1F). Together, these results demonstrate that Gαi3 silence or depletion could lead to irradiation-sensitization in glioma cells.
Silencing Gαi3 sensitizes irradiation-induced glioma cell apoptosis
It is known that irradiation kills cancer cells via inducing cell apoptosis [13, 14]. We next wanted to know the potential effect of Gαi3 in the process. In line with our previous studies [10, 11, 15, 16, 17], various apoptosis assays were applied, including Histone DNA apoptosis ELISA assay, TUNEL intensity assay and Annexin V staining assay. As expected, irradiation treatment in A172 cells induced significant apoptosis, which was evidenced by increase of Histone DNA apoptosis ELISA OD (Figure 2A), TUNEL intensity OD (Figure 2B) and percentage of Annexin V positive cells (Figure 2C). Remarkably, Gαi3 silence by targeted shRNA dramatically facilitated irradiation-induced A172 cell apoptosis (Figure 2A–2C). Gαi3 shRNA alone (no irradiation) also induced minor A172 cell apoptosis (Figure 2A–2C). Gαi3 KO MEFs were again utilized. As demonstrated, irradiation (5 Gy) induced significantly more apoptosis in Gαi3 KO MEFs (as compared to WT MEFs, Figure 2D and 2E). For instance, after irritation, 17.6 ± 1.5% of Gαi3 KO MEFs were apoptotic (Annexin V positive), compared to only 6.3 ± 1.9% in WT MEFs (Figure 2E). Basal apoptosis activation was slightly higher in Gαi3 KO MEFs than in the WT MEFs (Figure 2D and 2E) [11].
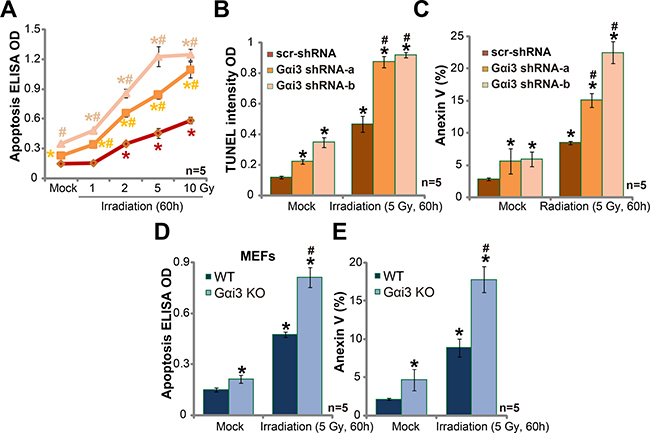
Figure 2: Silencing Gαi3 sensitizes irradiation-induced glioma cell apoptosis. A172 cells with Gαi3 shRNA (“-a/-b”) or scramble control shRNA (“scr-shRNA”) (A–C), as well as wild-type (WT) and Gαi3 knockout (KO) MEFs (D–E) were treated with irradiation (at indicated intensity) and cultured for additional 60 hours; Afterwards, cell apoptosis was tested by the listed assays. *p < 0.05 vs. “Mock” of “scr-shRNA” A172 cells (A-C) or WT MEFs (D–E). #p < 0.05 vs. “Irradiation” of “scr-shRNA” A172 cells (A–C) or WT MEFs (D–E).
Exogenous Gαi3 over-expression in A172 cells cause irradiation resistance
Based on the results above, we would speculate that Gαi3 over-expression shall cause irradiation resistance. Thus, wild-type (“WT”) Gαi3 construct (see our previous study [10, 11]) was introduced to A172 cells. Via puromycin selection, the stable cells with the construct were established. Western blotting assay results in Figure 3A confirmed the expression of exogenous Gαi3 (Flag-tagged) in the stable cells. Significantly, irradiation-induced A172 cell death (MTT OD reduction, Figure 3B) and apoptosis (Histone DNA ELISA OD increase, Figure 3C) were dramatically inhibited in Gαi3-over-expressed A172 cells. Thus, Gαi3 over-expression led to irradiation resistance in glioma cells.
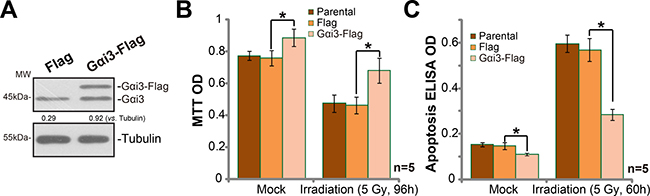
Figure 3: Exogenous Gαi3 over-expression in A172 cells cause irradiation resistance. (A) Western blotting analysis results showed expression of Gαi3 (endogenous and exogenous) in stable A172 cells with the Flag-tagged Gαi3 or empty vector (pSuper-puro-Flag, “Flag”). Cells were treated with irradiation (5 Gy) and cultured for indicated time; Cell death (MTT OD reduction, (B)) and apoptosis (Histone DNA ELISA assay, (C)) were tested. “Parental” stands for control parental A172 cells. *p < 0.05.
Irradiation sensitivity is altered with Gαi3 mutation in A172 cells
Next, mutation strategies were employed to potentially alter the activity of Gαi3 in A172 cells. As discussed in our previous studies [9, 11], the dominant-negative Gαi3 (DN-Gαi3), which has a conserved Gly (G) residue replaced by Thr (T) in the G3 box [9, 10, 11], was introduced to A172 cells (Figure 4A). The DN-Gαi3 shall compete with the wt-Gαi3 for binding with other proteins [18, 19]. Significantly, irradiation-induced A172 cell death (Figure 4B) and apoptosis (Figure 4C) were remarkably potentiated with the Gαi3 DN mutation. On the other hand, a constitutively-active Gαi3 (Q204L, CA-Gαi3) [9] was transfected to A172 cells, and stable cells were again established. Results in Figure 4A confirmed CA-Gαi3 (Flag-tagged) expression in the stable A172 cells (Figure 4A). Remarkably, A172 cells with CA-Gαi3 were protected from irradiation (Figure 4B and 4C). Irradiation-induced A172 cell death (Figure 4B) and apoptosis (Figure 4C) were largely inhibited after CA-Gαi3 expression. These results together indicate that change of Gαi3 activity could alter irradiation sensitivity in A172 cells.
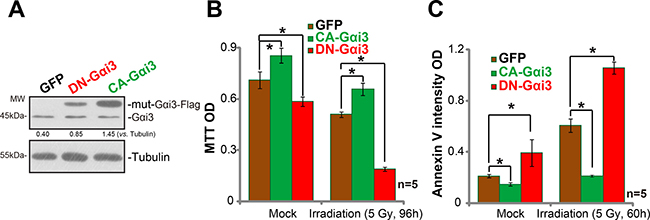
Figure 4: Irradiation sensitivity is altered with Gαi3 mutation in A172 cells. Western blotting assay results showed expression of Gαi3 (endogenous and mutant) in stable A172 cells with the dominant-negative Gαi3 (G202T, “DN-Gαi3”), the constitutively-active Gαi3 (Q204L, “CA-Gαi3”) or the empty vector (pGCL-GFP-puro, “GFP”) (A). Cells were also treated with irradiation (5 Gy) and cultured for indicated time; Cell death (MTT OD reduction, (B)) and apoptosis (Histone DNA ELISA assay, (C)) were tested. *p < 0.05.
Irradiation induces Gαi3 nuclear translocation and association with DNA-PKcs
It is known that irradiation induces DNA damages, which leads to subsequent cell apoptosis [20–22]. DNA repair mechanisms could however repair damaged DNA, causing irradiation resistance [20–22]. One of major protein complex for DNA repair is DNA-dependent protein kinase (DNA-PK). DNA-PK is primarily composed of the 460-kDa catalytic subunit (DNA-PKcs) and the Ku hetero-dimer (Ku-70 and Ku-80) [23, 24]. Intriguingly, we showed that irradiation treatment in A172 cells induced Gαi3 translocation to nuclei (Figure 5A). Basal Gαi3 level in nuclei, as expected, was few (Figure 5A). Following the irradiation, the Gαi3 level in the cell nuclei was significantly increased (Figure 5A), indicating nuclear translocation. Remarkably, the co-immunoprecipitation assay results showed that nuclei-translocated Gαi3 formed a complex with local protein DNA-PKcs (Figure 5B). Considering that DNA-PKcs is critical for DNA damage repair [20–22], we proposed that Gαi3 could also be important for DNA repair. Indeed, we found that irradiation-induced DNA-damage, tested by γ-H2AX increase [25–27], was significantly potentiated with Gαi3 silence (by “Gαi3 shRNA-b”) or DN mutation in A172 cells (Figure 5C). Reversely, expression of CA-Gαi3 inhibited DNA damages by irradiation (Figure 5D). Further, as compared to the WT MEFs, an increase of γ-H2AX staining (indicating DNA damage) was noticed in irradiated Gαi3 KO MEFs (Figure 5E). Notably, basal DNA-damage or γ-H2AX staining was unchanged by above Gαi3 genetic modifications (Figure 5C–5E). Together, our results imply that irradiation induces Gαi3 nuclear translocation and association with DNA-PKcs, which apparently is crucial for DNA-damage repair and irradiation resistance.
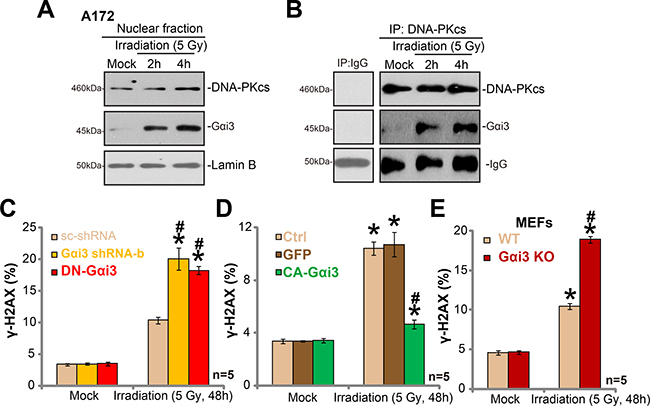
Figure 5: Irradiation induces Gαi3 nuclear translocation and association with DNA-PKcs. A172 cells were treated with irradiation (5 Gy) for 2 and 4 hours, nuclear fractions were isolated, expression of listed protein was tested (A) , Lamin-B is the nuclear marker protein). The association between DNA-PKcs and Gαi3 in cell nuclei was tested by Co-IP assay (B). Stable A172 cells with scramble control shRNA (“scr-shRNA”), Gαi3 shRNA (“-b”), the dominant-negative Gαi3 (G202T, DN-Gαi3) or the constitutively-active Gαi3 (Q204L, CA-Gαi3), as well as wild-type (WT) and Gαi3 knockout (KO) MEFs, were treated with irradiation (5 Gy) and cultured for 48 hours, DNA damage was tested by γ-H2AX FACS assay, and γ-H2AX-postive cell ratio was recorded (C–E). *p < 0.05 vs. “Mock”. # p < 0.05 vs. “Irradiation” of “scr-shRNA” A172 cells (C), “GFP” A172 cells (D) or WT MEFs (E).
DISCUSSION
The results of this study suggest that Gαi3 could be a key resistance factor of irradiation in glioma cells. Gαi3 depletion significantly potentiated irradiation-induced cell apoptosis. On the other hand, forced over-expression of Gαi3 inhibited irradiation-induced A172 cell apoptosis. Meanwhile, irradiation sensitivity in A172 cells was potentiated when expressing DN-Gαi3, but was reduced after CA-Gαi3 expression. Mechanistic study further showed that Gαi3 translocation to nuclei, which was important for DNA damage repair. These results together imply that Gαi3 over-expression in human glioma cells could be a key irradiation-resistance factor.
Irradiation-induced DNA damage will initiate DNA repair pathway [20–22]. There are at least two major signaling pathways that could possibly repair DNA damages, including the non-homologous end joining (NHEJ) pathway and the homologous recombination (HR) pathway [20–22]. In the process of NHEJ, Ku70/80 proteins will sense and bind to ends of the DNA termini in a structure-specific manner, which is followed by the recruitment and activation of DNA-PKcs [28, 29]. Afterwards, DNA ligase IV-XRCC4 complex is recruited to repair damaged DNA [28–30]. HR pathway is the second major pathway for DNA DSB repair [20–22]. After DNA damage, the Mre11/Rad50/Nbs1 (MRN) complex is recruited to the DNA ends, which then activates ATM and other DNA damage response proteins to repair broken DNA [30].
It is known that DNA-PKcs is vital in the repair of damaged DNA by irradiation [21, 22, 24, 31, 23, 24]. DNA-PKcs is a phosphatidylinositol-3-kinase (PI3K)-like protein kinase (PIKK) family kinase protein, which is activated following irradiation-induced DNA double-strand breaks (DSBs) [23, 24]. DNA-PKcs silence, depletion or mutation will disrupt DNA repair mechanism, causing irradiation-sensitization [21, 22, 24, 31]. On the other hand, over-expression and/or constitutive activation of DNA-PKcs could inhibit irradiation-induced DNA damage repair, leading to irradiation resistance [20, 30]. Indeed, DNA-PKcs expression is often elevated in glioma [32, 33] and other malignancies [34], and its upregulation in malignancy often correlates with irradiation resistance. Further studies suggest that DNA-PKcs expression level could be serve as a predictor for irradiation sensitivity in human cancer [35].
In the current study, we discovered an unique function of Gαi3: Irradiation in A172 cells induced Gαi3 translocation to nuclei, where it formed a complex with local protein DNA-PKcs. The complexation between Gαi3 and DNA-PKcs was apparently crucial for DNA repair. Gαi3 silence, depletion or dominant-negative mutation significantly potentiated irradiation-induced DNA damages. Reversely, expression of the constitutively-active Gαi3 inhibited DNA damage by irradiation in A172 cells. Future studies will be needed to further explore the detailed mechanisms of Gαi3’s function in DNA damage repair.
MATERIALS AND METHODS
Reagents
All the antibodies of the current study were described previously [9, 10, 36, 37], and were provided by the Cell Signaling Tech (Shanghai, China) and Santa Cruz Biotech (Shanghai, China). The reagents for cell culture were purchased from Gibco (Shanghai, China). Puromycin was obtained from Sigma (Shanghai, China).
Cell lines
Wild-type (WT) and Gαi3 knockout (KO) mouse embryonic fibroblasts (MEFs) were described previously [9–11]. Human glioma A172 cell line was purchased from the Cell Bank of Fudan University (Shanghai, China). Cells were cultured in routine DMEM medium, with 10% fetal bovine serum (FBS) in the CO2 incubator.
Irradiation
Cells were irradiated with a 137Cs gamma rays source at a dose rate of 1.25 Gy/min (MDS Nordion Gammacell Irradiator).
Western blotting analysis
Following the applied treatment, cells were lysed using the lysis buffer described [9, 10, 36]. Aliquots of 30 μg of protein per treatment were separated by 7.5–10% SDS-PAGE gels, and were transferred to the PVDF membrane (Millipore, Bedford, MA). The membrane was then incubated with indicated primary antibody and corresponding second antibody. Antibody-antigen binding was detected by the ECL reagents (Amersham Biosciences). Each band was quantified through Image J software (NIH). Isolate of nuclei-localized proteins was described previously [15, 16]. For all the Western blotting assay, each lane was loaded with exact same amount of quantified protein lysates (30 μg per sample). Same set of lysate samples were run in sister gels to test different proteins.
Co-immunoprecipitation (Co-IP)
The detailed protocol was described in our previous studies [9, 36]. Briefly, aliquots of 500 μg of nuclei-localized protein lysates from each treatment were pre-cleared with protein A/G beads (30 μL per sample, Sigma). The pre-cleared lysate samples were then incubated with anti-DNA-PKcs antibody [38] overnight. Protein A/G beads (Sigma) were then added again, and the lysates were incubated for 2 hours at 4°C. The beads were washed, and DNA-PKcs-Gαi3 association was then detected by Western blotting assay.
Gαi3 shRNA
The two lentiviral Gαi3 shRNAs (“-a/-b”) were again purchased from Genechem (Shanghai, China), with the targeted sequences 5′-TCAATCATTCTCTTCCTTA-3′ (Gαi3 shRNA-a) and 5′-CCTCAGTGATTATGACCTT-3′ (Gαi3 shRNA-b), respectively. The lentiviral shRNA was added directed to the cells for 24 hours, puromycin (0. 5 μg/mL, 8 days) was added to select the stable cells. Gαi3 knockdown was confirmed by the Western blotting assay. Same amount of lentiviral scramble shRNA (“scr-shRNA”, Santa Cruz, sc-108080) was added to the control cells.
Gαi3 over-expression or mutation
The wild-type Gαi3 (-Flag), the constitutively-active-Gαi3 (CA-Gαi3-GFP-puro, Q204L), the dominant-negative Gαi3 (DN-Gαi3-GFP-puro, G202T), and the empty vector (pGCL-GFP-puro, GeneChem) were described previously [9–11]. The construct was transfected to A172 cells by Lipofectamine 2000 reagents [10]. After 24 hours, cells were subjected to puromycin (0.5 μg/mL, 8 days) selection. Expression of the target protein (Gαi3) in stable cells was always tested by Western blotting assay.
Cell growth, survival and apoptosis assay
MTT assay of cell growth, clonogenicity assay of cell growth, and trypan blue staining of cell death, as well as Histone DNA apoptosis ELISA assay, Annexin V FACS assay of cell apoptosis and TUNEL nuclei staining assay of cell apoptosis were described in detail in our previous studies [9, 10, 16, 17, 36, 37, 39, 40, 41].
γ-H2AX FACS assay of cellular DNA damage.
After irradiation, cells were trypsinized and fixed in ice-cold ethanol. Afterwards, cells were incubated with a mouse monoclonal anti-γ-H2AX antibody (Cellular Signaling Tech, Shanghai, China) for 12 hours, and then incubated with a FITC-conjugated anti-mouse secondary antibody (Cell Signaling Tech). Cells were then subjected to FACS assay to determineγ-H2AX percentage, which indicates DNA damage intensity [27].
Statistical analysis
The data were presented as means ± standard deviation (SD) of one whole set of experiment. All experiments were repeated at least three times, with similar results obtained in each repeat. Statistical differences were analyzed by one-way ANOVA and multiple comparisons with the post hoc Bonferroni test (SPSS version 18.0). Values of p < 0.05 were considered as statistically significant.
CONCLUSIONS
In summary, these results indicate a pivotal function of Gαi3 in irradiation-resistance in human glioma cells. Gαi3 could be a novel oncotarget for irradiation sensitization for glioma.
Authors’ contributions
All the listed authors in the study carried out the experiments, participated in the design of the study and performed the statistical analysis, conceived of the study, and helped to draft the manuscript.
ACKNOWLEDGMENTS AND FUNDING
This work was generously supported by grants from the National Natural Science Foundation of China (Nos. 81302195, 31371139, 81571282, 81502162, 81372411, 81172128); Grants from Natural Science Foundation of Jiangsu Province (BK20130301), and by the Graduate Scientific Research and Innovation Project of Soochow University, KYLX14_1267).
CONFLICTS OF INTEREST
The authors declare no conflicts of interests.
REFERENCES
1. Westphal M, Lamszus K. The neurobiology of gliomas: from cell biology to the development of therapeutic approaches. Nat Rev Neurosci. 2011; 12:495–508.
2. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012; 62:10–29.
3. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64:9–29.
4. Khasraw M, Lassman AB. Neuro-oncology: late neurocognitive decline after radiotherapy for low-grade glioma. Nat Rev Neurol. 2009; 5:646–7.
5. Pollack IF. Neuro-oncology: Therapeutic benefits of reirradiation for recurrent brain tumors. Nat Rev Neurol. 2010; 6:533–5.
6. Wang Y, Jiang T. Understanding high grade glioma: molecular mechanism, therapy and comprehensive management. Cancer Lett. 2013; 331:139–46.
7. Watts C, Price SJ, Santarius T. Current concepts in the surgical management of glioma patients. Clin Oncol (R Coll Radiol). 2014; 26:385–94.
8. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997; 7:261–9.
9. Cao C, Huang X, Han Y, Wan Y, Birnbaumer L, Feng GS, Marshall J, Jiang M, Chu WM. Galpha(i1) and Galpha(i3) are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci Signal. 2009; 2:ra17.
10. Zhang YM, Zhang ZQ, Liu YY, Zhou X, Shi XH, Jiang Q, Fan DL, Cao C. Requirement of Galphai1/3-Gab1 signaling complex for keratinocyte growth factor-induced PI3K-AKT-mTORC1 activation. J Invest Dermatol. 2015; 135:181–91.
11. Li ZW, Cai S, Liu Y, Yang CL, Tian Y, Chen G, Cao C. Over-expression of Galphai3 in human glioma is required for Akt-mTOR activation and cell growth. Oncotarget. 2016 Aug 1. doi: 10.18632/oncotarget.10995. [Epub ahead of print].
12. Wang Z, Dela Cruz R, Ji F, Guo S, Zhang J, Wang Y, Feng GS, Birnbaumer L, Jiang M, Chu WM. G(i)alpha proteins exhibit functional differences in the activation of ERK1/2, Akt and mTORC1 by growth factors in normal and breast cancer cells. Cell Commun Signal. 2014; 12:10.
13. Vink SR, Lagerwerf S, Mesman E, Schellens JH, Begg AC, van Blitterswijk WJ, Verheij M. Radiosensitization of squamous cell carcinoma by the alkylphospholipid perifosine in cell culture and xenografts. Clin Cancer Res. 2006; 12:1615–22.
14. Tu Y, Ji C, Yang B, Yang Z, Gu H, Lu CC, Wang R, Su ZL, Chen B, Sun WL, Xia JP, Bi ZG, He L. DNA-dependent protein kinase catalytic subunit (DNA-PKcs)-SIN1 association mediates ultraviolet B (UVB)-induced Akt Ser-473 phosphorylation and skin cell survival. Mol Cancer. 2013; 12:172.
15. Zhang H, Liu YY, Jiang Q, Li KR, Zhao YX, Cao C, Yao J. Salvianolic acid A protects RPE cells against oxidative stress through activation of Nrf2/HO-1 signaling. Free Radic Biol Med. 2014; 69:219–28.
16. Li KR, Yang SQ, Gong YQ, Yang H, Li XM, Zhao YX, Yao J, Jiang Q, Cao C. 3H-1,2-dithiole-3-thione protects retinal pigment epithelium cells against Ultra-violet radiation via activation of Akt-mTORC1-dependent Nrf2-HO-1 signaling. Sci Rep. 2016; 6:25525.
17. Gong YQ, Huang W, Li KR, Liu YY, Cao GF, Cao C, Jiang Q. SC79 protects retinal pigment epithelium cells from UV radiation via activating Akt-Nrf2 signaling. Oncotarget. 2016; 7:60123–32. doi: 10.18632/oncotarget.11164.
18. Hubbard KB, Hepler JR. Cell signalling diversity of the Gqalpha family of heterotrimeric G proteins. Cell Signal. 2006; 18:135–50.
19. Barren B, Artemyev NO. Mechanisms of dominant negative G-protein alpha subunits. J Neurosci Res. 2007; 85:3505–14.
20. Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2016; 16:20–33.
21. Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer. 2016; 16:35–42.
22. Bunting SF, Nussenzweig A. End-joining, translocations and cancer. Nat Rev Cancer. 2013; 13:443–54.
23. Gao Y, Chaudhuri J, Zhu C, Davidson L, Weaver DT, Alt FW. A targeted DNA-PKcs-null mutation reveals DNA-PK-independent functions for KU in V(D)J recombination. Immunity. 1998; 9:367–76.
24. Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005; 434:605–11.
25. Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004; 64:2390–6.
26. Parsels LA, Morgan MA, Tanska DM, Parsels JD, Palmer BD, Booth RJ, Denny WA, Canman CE, Kraker AJ, Lawrence TS, Maybaum J. Gemcitabine sensitization by checkpoint kinase 1 inhibition correlates with inhibition of a Rad51 DNA damage response in pancreatic cancer cells. Mol Cancer Ther. 2009; 8:45–54.
27. Ewald B, Sampath D, Plunkett W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol Cancer Ther. 2007; 6:1239–48.
28. Dungl DA, Maginn EN, Stronach EA. Preventing Damage Limitation: Targeting DNA-PKcs and DNA Double-Strand Break Repair Pathways for Ovarian Cancer Therapy. Front Oncol. 2015; 5:240.
29. Dobbs TA, Tainer JA, Lees-Miller SP. A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair (Amst). 2010; 9:1307–14.
30. Houtgraaf JH, Versmissen J, van der Giessen WJ. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc Revasc Med. 2006; 7:165–72.
31. Convery E, Shin EK, Ding Q, Wang W, Douglas P, Davis LS, Nickoloff JA, Lees-Miller SP, Meek K. Inhibition of homologous recombination by variants of the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs). Proc Natl Acad Sci U S A. 2005; 102:1345–50.
32. Golding SE, Morgan RN, Adams BR, Hawkins AJ, Povirk LF, Valerie K. Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol Ther. 2009; 8:730–8.
33. Yan D, Ng WL, Zhang X, Wang P, Zhang Z, Mo YY, Mao H, Hao C, Olson JJ, Curran WJ, Wang Y. Targeting DNA-PKcs and ATM with miR-101 sensitizes tumors to radiation. PLoS One. 2010; 5:e11397.
34. Shintani S, Mihara M, Li C, Nakahara Y, Hino S, Nakashiro K, Hamakawa H. Up-regulation of DNA-dependent protein kinase correlates with radiation resistance in oral squamous cell carcinoma. Cancer Sci. 2003; 94:894–900.
35. Noguchi T, Shibata T, Fumoto S, Uchida Y, Mueller W, Takeno S. DNA-PKcs expression in esophageal cancer as a predictor for chemoradiation therapeutic sensitivity. Ann Surg Oncol. 2002; 9:1017–22.
36. Cao C, Rioult-Pedotti MS, Migani P, Yu CJ, Tiwari R, Parang K, Spaller MR, Goebel DJ, Marshall J. Impairment of TrkB-PSD-95 signaling in Angelman syndrome. PLoS Biol. 2013; 11:e1001478.
37. Yang L, Zheng LY, Tian Y, Zhang ZQ, Dong WL, Wang XF, Zhang XY, Cao C. C6 ceramide dramatically enhances docetaxel-induced growth inhibition and apoptosis in cultured breast cancer cells: a mechanism study. Exp Cell Res. 2015; 332:47–59.
38. Zhen YF, Li ST, Zhu YR, Wang XD, Zhou XZ, Zhu LQ. Identification of DNA-PKcs as a primary resistance factor of salinomycin in osteosarcoma cells. Oncotarget. 2016; 7:79417–27. doi: 10.18632/oncotarget.12712.
39. Chen MB, Wei MX, Han JY, Wu XY, Li C, Wang J, Shen W, Lu PH. MicroRNA-451 regulates AMPK/mTORC1 signaling and fascin1 expression in HT-29 colorectal cancer cells. Cell Signal. 2013.
40. Wu CH, Cao C, Kim JH, Hsu CH, Wanebo HJ, Bowen WD, Xu J, Marshall J. Trojan-horse nanotube on-command intracellular drug delivery. Nano Lett. 2012; 12: 5475–80.
41. Yao J, Bi HE, Sheng Y, Cheng LB, Wendu RL, Wang CH, Cao GF, Jiang Q. Ultraviolet (UV) and hydrogen peroxide activate ceramide-ER stress-AMPK signaling axis to promote retinal pigment epithelium (RPE) cell apoptosis. Int J Mol Sci. 2013; 14: 10355–68.