
INTRODUCTION
Primary bone tumors account for 0.2% of all malignancies. Osteosarcoma (OS) is the major tumor type [1]. Sequential neoadjuvant chemotherapy, surgery and adjuvant chemotherapy provide a of 5-year overall survival of 60−70% in patients with localized disease [2-4]. However, 40−50% of patients with initially localized disease develop recurrence during treatment [5,6], and less than 20% of newly diagnosed cases present with metastatic disease [3,7]. Most of these patients will eventually die of the disease due to refractoriness to therapy [2].
Previous studies have revealed several genetic mechanisms in OS tumorigenesis, including dysfunction of tumor suppressor mechanisms, such as cell cycle regulatory genes (e.g., tumor protein 53 [TP53], retinoblastoma 1 [RB1] and mouse double minute 2 homolog [MDM2]) [8-10] as well as novel tumor suppressor genes (e.g., limbic system-associated membrane protein [LSAMP]) [11-13]. In addition, telomere dysfunction [14,15], dysregulation of cell death and cytokine pathways [16,17] and upregulation of ezrin (EZR) have been reported in metastatic OS [18,19]. However, genetic complexity is a hallmark of high-grade OS, and currently, no ideal target has been identified.
Recently, two independent groups have identified polo-like kinase 1 (PLK1), a serine/threonine kinase that regulates many stages of mitosis and maintains genomic stability [20], as a potential target for OS treatment, using short hairpin RNA (shRNA) libraries in lentiviral vectors for screening of protein kinases [21,22]. In addition, previous studies have demonstrated the potential oncogenic role of PLK1 [23-26]. Therefore, inhibition of PLK1 could represent an effective treatment for OS.
15-deoxy-Δ12, 14-prostaglandin J2 (15d-PGJ2) has gained attention as a potential cancer treatment because of its unique anti-tumor activity. Earlier studies showed that it could significantly inhibit cell growth and induce apoptosis in cancer cells through activation of PPARγ [27-29]. However, recent evidence indicates that its cytotoxicity is largely PPARγ-independent, and is closely related to reactive oxygen species (ROS) generation [30-32], with subsequent inhibition of critical pathways, such as v-akt murine thymoma viral oncogene homolog (AKT) [33-37]. Interestingly, in arthritis, 15d-PGJ2 mediated shear-induced chondrocyte apoptosis via protein kinase A (PKA)-dependent regulation of PLK1 [38]. However, the role of 15d-PGJ2 in modulating PLK1 has never been explored in cancer. Since PLK1 has been identified as a potential target of OS, the role of 15d-PGJ2 in the treatment of OS deserves further investigation. In this study, we demonstrated that 15d-PGJ2 could exert a cytotoxic effect on OS cell lines via a ROS-mediated dual inhibition of AKT and the cell cycle.
RESULTS
15d-PGJ2 inhibits growth and induces apoptosis of OS cell lines
To evaluate the effect of 15d-PGJ2 on the growth of OS cells, three OS cell lines (MG63, SaOS2, U2OS) were treated with various concentrations of 15d-PGJ2 for a range of time. Cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich) assay. As shown in Figure 1A and 1B, 15d-PGJ2 significantly inhibited the growth of all three OS cell lines in a dose- and time-dependent manner.
We then investigated whether 15d-PGJ2 induced apoptosis of OS cell lines. After treatment of all three OS cell lines with 15d-PGJ2 at different dose level and durations, cells were co-stained with annexin V and propidium iodide (PI). 15d-PGJ2 significantly induced apoptosis in a dose- and time-dependent fashion (Figure 1C and 1D, respectively). Both these studies indicated that 15d-PGJ2 exerted a cytotoxic effect, inhibiting OS cell growth.
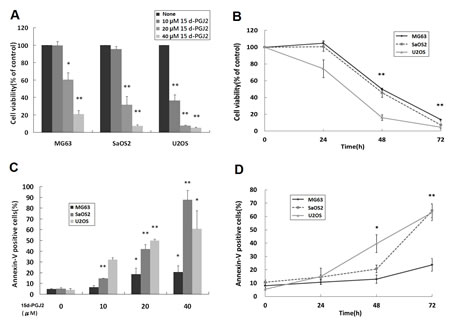
Figure 1: 15d-PGJ2 inhibited the growth and induced apoptosis of OS cell lines. (A, B) MTT assay. MG63, SaOS2 and U2OS cells were seeded 1 day before treatment with (A) various concentrations of 15d-PGJ2 (0, 10, 20, or 40 μmol/L) for 72 h or (B) 15d-PGJ2 (20, 10, 10 μmol/L) for the indicated time. Cell viability was assessed using the MTT assay and expressed as a percentage of viability under controlled culture conditions. (C, D) Apoptosis assay. MG63, SaOS2 and U2OS cells were seeded 1 day before treatment with (C) various concentrations of 15d-PGJ2 (0, 10, 20, or 40 μmol/L) for 72 h or (D) 15d-PGJ2 (20, 10, 10 μmol/L) for the indicated time. The percentage of apoptotic cells was determined using Annexin V-FITC/propidium iodide (PI) staining. All data represent the mean ± SD of three independent experiments. *P < 0.05; **P < 0.01.
15d-PGJ2 induced significant G2/M arrest in OS cell lines
Because PLK1 is a cell cycle regulatory protein, we next examined the effects of 15d-PGJ2 on the cell cycle in OS cells in vitro. After treatment with 15d-PGJ2 for 72 h, increased G2/M DNA content was observed in all three OS cell lines (Figure 2), indicating that 15d-PGJ2 could induce a significant G2/M arrest in OS cells.
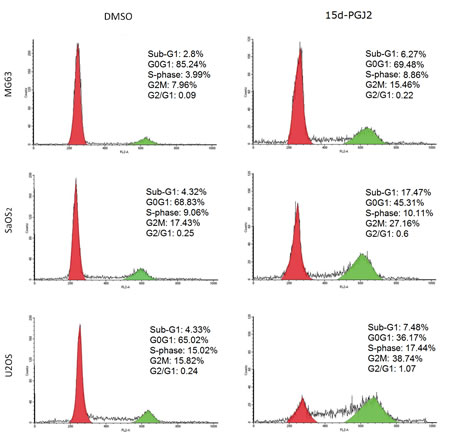
Figure 2: 15d-PGJ2 induced significant G2/M arrest in OS cell lines. DNA profiles of MG63, SaOS2, and U2OS treated with DMSO or 15d-PGJ2 (20, 10, 10 µmol/L) for 72 h were evaluated by flow cytometry. The percentage of cells in sub-G1, G0G1, G2M, and S phases are shown.
15d-PGJ2 activated ERK but down-regulated both AKT and PKA-PLK1 pathways
Since both the PI3K/AKT and PKA-PLK1 signaling pathways have been implicated as targets of 15d-PGJ2, we investigated the effect of 15d-PGJ2 on AKT expression and phosphorylation as well as changes in expression of members of the PKA-PLK1 pathway. As shown in Figure 3, 15d-PGJ2 induced ERK activation, as reported by previous studies [30,39]. On the other hand, 15d-PGJ2 induced a significant time-dependent down-regulation of AKT and p-AKT in U2OS and Saos2 cells, and, to a lesser extent, in MG63 cells. Moreover, 15d-PGJ2 dramatically down-regulated total and phospho- PKA, PLK1, and CDC25 levels in all three cell lines. Cleavage of PARP was also seen after 24–48 h of treatment (Figure 3). Thus, 15d-PGJ2 repressed both the AKT and PKA-PLK1 pathways, thereby inducing apoptosis.
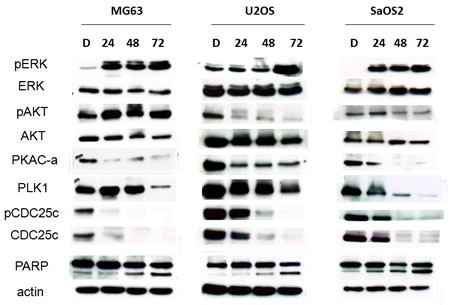
Figure 3: 15d-PGJ2 down-regulated both AKT and PKA-PLK1 pathways. Western blot analysis of MG63, SaOS2, and U2OS cells treated with DMSO or 15d-PGJ2 (20, 10, 10 µmol/L) for 72 h using antibodies against ERK, p-ERK, AKT, p-AKT, the PKA-PLK1-CDC25 pathway, and PARP.
Both AKT and PKA-PLK1 pathways are critical targets of 15d-PGJ2
To explore the functional significance of AKT and PKA-PLK1 pathway suppression in 15d-PGJ2–mediated cell death, U2OS cells were transiently transfected with a constitutively active AKT or PLK1 expression constructs as well as empty vector controls. Constitutively active AKT or PLK1 expression was confirmed by Western blot analysis (Figure 4A and 4C). Expression of constitutively active AKT or PLK1 partially protected cells from apoptosis induced by 15d-PGJ2 (Figure 4B and 4D). These findings indicate that down-regulation of both AKT and PLK1 plays a functional role in the death of OS cells resulting from treatment with 15d-PGJ2.
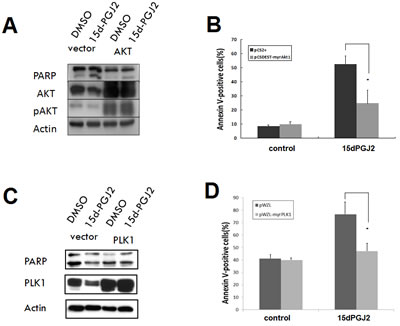
Figure 4: Overexpression of constitutively active AKT or PLK1 can partially reverse the cytotoxic effect of 15d-PGJ2. U2OS cells were transiently transfected with an empty vector or constitutively active AKT or PLK1 expression construct, with or without 15d-PGJ2 (20 µmol/L) for 48 h. (A, C) Immunoblot analysis using corresponding antibodies against (A) AKT or (C) PLK1. (B, D) Apoptosis assay. The percentage of apoptotic cells was determined using Annexin V-FITC/propidium iodide (PI) staining. All data represent the mean ± SD of three independent experiments. *P < 0.05.
15d-PGJ2-induced ROS generation in OS cell lines, and cytotoxic effects of 15d-PGJ2 on OS cell lines are ROS-dependent
ROS generation was considered the major cytotoxic mechanism of 15d-PGJ2 in tumor cell death [32,37]. Therefore, we measured ROS levels in U2OS cell lines exposed to 15d-PGJ2. 15d-PGJ2 induced production of ROS in U2OS cells after 2 h, peaking at 3-4 h (Figure 5A). To investigate a functional relationship between ROS generation and the cytotoxic effect of 15d-PGJ2, U2OS cells were exposed to 15d-PGJ2 in the absence or presence of N-Acetylcysteine (NAC), an antioxidant. As shown in Figure 5B, reduced suppression of the AKT and PKA-PLK1 pathways, as well as PARP degradation was observed in cells treated with 15d-PGJ2 and NAC. In addition, co-treatment of cells with NAC reduced 15d-PGJ2-induced ROS production (Figure 5C) and ameliorated the 15d-PGJ2-induced cell cycle arrest (Figure 5D) and apoptosis (Figure 5E). Thus, 15d-PGJ2 induced ROS generation in OS cell lines, and the cytotoxic effects of 15d-PGJ2 on OS cell lines were mediated by ROS-dependent down-regulation of the PKA-PLK1 and AKT pathways.
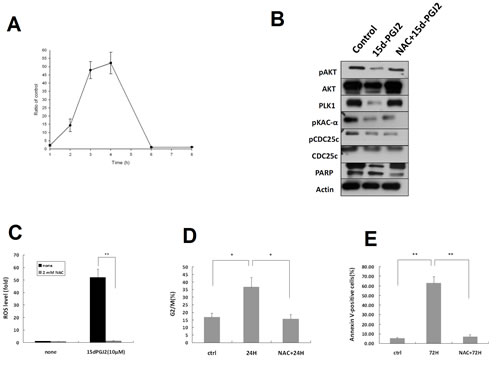
Figure 5: Cytotoxic effects of 15 d-PGJ2 on OS cell lines are ROS-dependent. (A) U2OS cells were incubated with 15d-PGJ2 (10 µmol/L) for the indicated time points, labeled with 8OHdG, and analyzed by flow cytometry. ROS level was expressed as an increased ratio in comparison with control. (B) Western blot analysis of USOS cells treated with DMSO or 15d-PGJ2 (20 µmol/L) for 72 h without or with NAC preteatment (2 mM) for 1 h using antibodies against AKT, p-AKT, the PKA-PLK1-CDC25 pathway, and PARP. (C) ROS level of U2OS cells at baseline or treated with 15d-PGJ2 (10 µmol/L) in the absence or presence of NAC (2 mM) for 8 h. (D) G2/M content was evaluated by flow cytometry, and (E) percentage of apoptotic cells was determined using Annexin V-FITC/propidium iodide (PI) staining of U2OS cells at baseline or treated with 15d-PGJ2 (10 µmol/L) with or without NAC (2 mM) for 72 h. All data represent the mean ± SD of three independent experiments. *P < 0.05; **P < 0.01.
15d-PGJ2 induced ROS-mediated c-Jun N-terminal kinases (JNK) activation contributes to apoptosis through down-regulation of the AKT and PKA-PLK1 pathways
Studies suggest that JNK plays an important role in ROS-induced apoptosis [30,37]. To investigate whether 15d-PGJ2–induced ROS leads to the activation of JNK in OS cells, we examined the phosphorylation state of JNK in OS cells treated with 15d-PGJ2. As shown in Figure 6A, 15d-PGJ2 treatment significantly increased the phosphorylation of JNK. Furthermore, pretreatment with JNK inhibitor, SP600125, for 1 h could prevent the phosphorylation of JNK caused by 15d-PGJ2, and block 15d-PGJ2-induced down-regulation of AKT as well as PKA-PLK1-CDC25 (Figure 6B). SP600125 also inhibited 15d-PGJ2–induced apoptosis (Figure 6C). These results indicate that ROS-mediated JNK activation may in part contribute to apoptosis through down-regulation of AKT and PLK1.
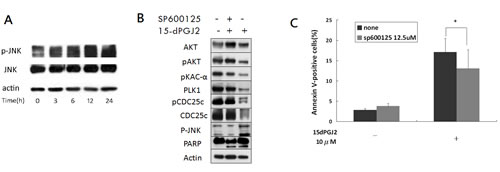
Figure 6: 15d-PGJ2-mediated cytotoxicity partially induced through JNK activation. (A) The phosphorylation state of JNK was analyzed in U2OS cells treated with 15d-PGJ2 (10 µmol/L) for various time points. (B) U2OS cells were either treated with DMSO, pretreated with SP600125 (12.5 µmol/L for 1 h) and then treated with 15d-PGJ2 (10 µmol/L for 72 h), or treated with 15d-PGJ2 alone. After treatment, the levels of protein expression were determined by Western blot analysis. (C) U2OS cells, with or without pretreatment of SP600125 (12.5 µmol/L for 1 h), were treated with 15d-PGJ2 (10 µmol/L) for 72 h. After treatment, the percentage of Annexin V–positive cells were determined by flow cytometry. All data represent the mean ± SD of three independent experiments. *P < 0.05.
DISCUSSION
In this study, 15d-PGJ2 inhibited OS cell proliferation by inducing apoptosis. This finding is in agreement with the results of previous studies of 15d-PGJ2 in other cell lines [28,37]. However, the molecular mechanisms underlying the cytotoxic mechanism of 15d-PGJ2–induced apoptosis remained unclear. The present study shows that 15d-PGJ2 induced generation of ROS and activated JNK in OS cells, resulting in the down-regulation of the AKT and PLK1 pathways with subsequent apoptosis.
Several pathways, such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [40-42], hypoxia-inducible factor-2α (HIF2α) [43], and AKT [33-37], have been identified as targets for 15d-PGJ2-induced cytotoxicity. AKT is a critical regulator of diverse cellular functions [44], and ROS may activate AKT through phosphatase and tensin homolog (PTEN) inactivation [45]. However, our study disclosed that 15d-PGJ2 down-regulated AKT in OS cells with resultant apoptosis, which is consistent with that of previous studies in other cancer types [34-37].
More importantly, we found that the PKA-PLK1-CDC25 pathway is a target of 15d-PGJ2 on OS cells. A previous study showed that 15d-PGJ2 inhibited PKA in renal proximal epithelial cells [40]. In addition, Zhu et al. [38] showed that exogenous PGD2 and 15d-PGJ2 diminished the viability of human chondrocytes through down-regulation of the PKA-PLK1 pathway [38]. These earlier studies prompted us to explore the possibility of targeting this pathway by 15d-PGJ2 in cancer. In this study, we demonstrated that 15d-PGJ2 down-regulated the PKA-PLK1-CDC25 pathway in OS cell lines. We also showed that constitutive activation of AKT or PLK1 partially reversed the apoptosis induced by 15d-PGJ2. These data confirmed the important role of both AKT and PLK1 in 15d-PGJ2–mediated cytotoxicity of cancer cells.
ROS has been implicated in several oncogenic pathways, such as promoting tumor growth [46], angiogenesis [45], mutagenesis [47,48], and drug resistance [49,50]. On the other hand, ROS-mediated cytotoxicity has also been identified as an important mechanism in some anti-cancer agents [51,52]. Several studies have linked ROS generation with 15d-PGJ2–induced apoptosis [31,32]. In this study, 15d-PGJ2 induced ROS generation in OS cell lines. Moreover, the anti-oxidant, NAC, could reverse the cytotoxic and cell cycle inhibition effects of 15d-PGJ2, confirming the critical role of ROS generation in 15d-PGJ2–induced apoptosis of cancer cell lines.
A previous study suggested that activation of JNK through ROS generation is important for 15d-PGJ2-induced apoptosis [30]. To investigate this hypothesis, we used immunoblot analysis to examine the expression of p-JNK after treatment with 15d-PGJ2. 15d-PGJ2 treatment activated JNK, and SP600125 pretreatment partially inhibited 15d-PGJ2–induced apoptosis. Taken together, these data suggest that activation of JNK through ROS may contribute to down-regulation of AKT and PLK1 to lethality.
In this study, we did not explore other possible mechanisms of 15d-PGJ2-induced cytotoxicity, such as the NF-κB pathway. Therefore, we cannot exclude the possibility that 15d-PGJ2 also inhibits NF-κB in OS. Also, we did not analyze the mechanism of 15d-PGJ2-induced down-regulation of PKA-PLK1-CDC25. Previous studies have shown that 15d-PGJ2 down-regulated AKT at the transcriptional level [35,37]. However, in a recent study analyzing 15d-PGJ2 target proteins, heat shock protein 90 (Hsp90) was found to be one of the targets [53]. Thus, it is also possible that 15d-PGJ2 may inhibit Hsp90 with subsequent degradation of PKA-PLK1-CDC25.
In summary, 15d-PGJ2 inhibited the growth and induced apoptosis of OS cells. 15d-PGJ2 generated ROS, activated JNK, and suppressed the AKT and PLK1 pathways. These results suggested that 15d-PGJ2 may be of therapeutic importance in the treatment of OS, and form the basis for further analysis of its role in animal studies and clinical applications.
Statement of translational relevance
Polo-like kinase 1 (PLK1) is a key component in cell cycle regulation, and has been identified as a potential target of osteosarcoma (OS). On the other hand, 15-deoxy-Δ12, 14-prostaglandin J2 (15d-PGJ2) has been found to mediate shear-induced chondrocyte apoptosis via protein kinase A (PKA)-dependent regulation of PLK1. In the current study, we explored the cytotoxic effect and mechanism of 15d-PGJ2 in an in vitro model of OS. In OS cell lines, 15d-PGJ2 induced a significant cytotoxic effect, including G2/M arrest and apoptosis. Moreover, the cytotoxicity of 15d-PGJ2 resulted from ROS-mediated, JNK-dependent down-regulation of both the AKT and PKA-PLK1 pathways in OS. The current study revealed a unique mechanism of 15d-PGJ2 against OS, and its efficacy in xenograft models and clinical usages deserved further exploration.
MATERIALS AND METHODS
Cell lines and reagents
Three OS cell lines, U2OS, MG63 and SaOS2, were chosen as our in vitro study model. They were kept in the DMEM or IMDM base media with 10% fetal bovine serum (FBS). 15d-PGJ2 was purchased from Calbiochem (San Diego, CA, USA). The following antibodies were used for immunoblotting: PKA C-α (Cell Signaling, Danvers, MA; #4782; 1:1000); PLK1 (Cell Signaling #4513; 1:1000); AKT (Cell Signaling #9272; 1:2000); p-AKT (Cell Signaling #9271; 1:1000); Cdc25c(5H9) (Cell Signaling #4688; 1:1000); P-cdc25c (Ser216) (Cell Signaling #4901; 1:1000); PARP (Cell Signaling #9542; 1:1000), and actin (Abs 24-100; 1:50000). The anti-8-hydroxy-2’-deoxyguanosine (8OHdG) antibody (Santa Cruz, #sc-66036; 1:200) and anti-mouse conjugated fluorescein isothiocyanate (FITC) antibody (Jackson ImmunoResearch West Grove, PA; 1:400) were used for 8OHdG detection.
Analysis of cell viability
Cells were seeded in triplicate 96-well plates in 100 μL complete media at a density of 2000-20000 cells per well. On the next day, drugs were added at different concentrations with variable times. Then, 10 μL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich) solution was added to the wells and the plates were incubated for an additional 4 h at 37°C. A detergent solution (200 μL/well) was next added and mixed thoroughly to dissolve the dark-blue crystals. Absorbance of the converted dye was measured spectrophotometrically using a microplate reader (Vmax, Molecular Devices, Sunnyvale, CA) at 570 nm (test) and 650 nm (reference). Cell survival was calculated as percentage of MTT inhibition as follows: % survival = (mean experimental absorbance/ mean control absorbance) × 100 [54].
Apoptosis assessment by annexin V staining
Drug-induced apoptosis was measured using annexin V-fluorescein isothiocyanate (Annexin V-FITC) and PI co-staining using an Annexin V-FITC apoptosis detection kit (BD Pharmingen, San Diego, CA). After 15d-PGJ2 or DMSO treatment, cells were washed and resuspended in 100 μL staining solution (containing Annexin V-FITC and PI in a HEPES buffer). After incubation in dark and at room temperature for 15 min, cells were diluted in 400 μL of 1x binding buffer, and the percentages of apoptotic cells were analyzed by flow cytometry using a FACS Calibur (Becton Dickinson & Co., Oxford, CA, USA) and CellQuest software (Becton Dickinson & Co.).
Cell cycle analysis
After 15d-PGJ2 or DMSO treatment, OS cells were trypsinized and fixed in 99% ethanol at -20°C for 2 h, washed and re-suspended in 420 μL PBS. Subsequently, samples were first incubated with RNase A (Sigma) (50 μL of a 10 mg/mL solution) at 37°C for 30 min, and then PI (20 μL of a 0.2 mg/mL solution) at room temperature for 10 min. DNA content was analyzed by flow cytometry using a FACS Calibur (Becton Dickinson & Co.) and CellQuest software (Becton Dickinson & Co.) [55].
Western blot analysis
Cell extracts were prepared with RIPA Lysis and Extraction Buffer (Thermo Scientific, Rockford, IL) containing a protease and phosphatase inhibitor cocktail (1:100 dilution; Thermo Scientific). Protein concentrations were determined using the BCA Protein Assay Kit (Thermo Scientific). Aliquots of protein lysates were electrophoretically separated on sodium dodecyl sulfate–polyacrylamide gels and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA), which were blocked with 5% blotting grade milk (Bio-Rad, Hercules, CA) in TBST (20mM Tris-HCl [pH 7.6], 137mM NaCl, 1% Tween 20). Membranes were then probed with the indicated primary antibodies, reacted with corresponding secondary antibodies, and were detected using an enhanced chemiluminescence system (Millipore) and X-ray films.
Transfection of the Myr-Akt or PLK1 vector
In order to explore the critical function of AKT or PLK1 in 15d-PGJ2-induced cytotoxicity, U2OS cells (7×105) were transfected with either 8 μg of myc-tagged myristoylated Akt expression vector (pCDEST-myrAkt) or pWZL NeoMyrFlag PLK1 expression vector or their corresponding empty vector (pCS2+ or pWZL-Neo-Myr-Flag-DEST) (all purchased from Addgene, Cambridge, MA, USA) using LipofectAMINE (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s procedure. After transfection, cells were cultured in 10% FBS–supplemented IMDM for 24 h and then subjected to 0.1% DMSO or 15d-PGJ2 treatment.
Measurement of ROS
The formation of the 8-hydroxy-2’-deoxyguanosine (8OHdG) base lesion, which is a biomarker for oxidative stress [56], was measured as previously described [57], with modification. Briefly, cells were trypsinized and harvested, washed twice with PBS and fixed by 1% paraformaldehyde at room temperature. The fixed cells were then washed with PBS, permeabilized by adding 0.1% Triton X100 for 15 min at room temperature, and washed with PBS again. Next, a blocking solution consisting of 1% BSA in PBS was added for 1 h at 37°C with gentle shaking, and cells were incubated with anti-8OHdG antibody for 1 h at 37°C after washing. Cells were washed again with PBS, and anti-mouse conjugated FITC antibody was then added and incubated for 1 h. Flow cytometry was used to detect the immunofluorescence.
ACKNOWLEDGMENTS
This work was supported by grants from the Yen Tjing Ling Medical Foundation (grant number CI-100-19), the National Science Council of Taiwan (grant number NSC 100-2314-B-075-081 and NSC 101-2314-B-075-029) and the Taipei Veterans General Hospital (grant number V102C-034) to Dr. CC Yen. This work was also partially supported by the Taiwan Clinical Oncology Research Foundation in a grant to CC Yen.
CONFLICT OF INTEREST STATEMENT
None declared.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012; 62 (1): 10-29.
2. Picci P, Mercuri M, Ferrari S, Alberghini M, Briccoli A, Ferrari C, Pignotti E, Bacci G. Survival in high-grade osteosarcoma: improvement over 21 years at a single institution. Ann Oncol. 2010; 21 (6): 1366-1373.
3. Janeway KA, Barkauskas DA, Krailo MD, Meyers PA, Schwartz CL, Ebb DH, Seibel NL, Grier HE, Gorlick R, Marina N. Outcome for adolescent and young adult patients with osteosarcoma: a report from the Children’s Oncology Group. Cancer. 2012; 118 (18): 4597-4605.
4. Anninga JK, Gelderblom H, Fiocco M, Kroep JR, Taminiau AH, Hogendoorn PC, Egeler RM. Chemotherapeutic adjuvant treatment for osteosarcoma: where do we stand? Eur J Cancer. 2011; 47 (16): 2431-2445.
5. Kempf-Bielack B, Bielack SS, Jurgens H, Branscheid D, Berdel WE, Exner GU, Gobel U, Helmke K, Jundt G, Kabisch H, Kevric M, Klingebiel T, Kotz R, Maas R, Schwarz R, Semik M et al. Osteosarcoma relapse after combined modality therapy: an analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). J Clin Oncol. 2005; 23 (3): 559-568.
6. Whelan JS, Jinks RC, McTiernan A, Sydes MR, Hook JM, Trani L, Uscinska B, Bramwell V, Lewis IJ, Nooij MA, van GM, Grimer RJ, Hogendoorn PC, Taminiau AH, Gelderblom H. Survival from high-grade localised extremity osteosarcoma: combined results and prognostic factors from three European Osteosarcoma Intergroup randomised controlled trials. Ann Oncol. 2012; 23 (6): 1607-1616.
7. Bielack SS, Kempf-Bielack B, Delling G, Exner GU, Flege S, Helmke K, Kotz R, Salzer-Kuntschik M, Werner M, Winkelmann W, Zoubek A, Jurgens H, Winkler K. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol. 2002; 20 (3): 776-790.
8. Toguchida J, Yamaguchi T, Ritchie B, Beauchamp RL, Dayton SH, Herrera GE, Yamamuro T, Kotoura Y, Sasaki MS, Little JB, . Mutation spectrum of the p53 gene in bone and soft tissue sarcomas. Cancer Res. 1992; 52 (22): 6194-6199.
9. Wadayama B, Toguchida J, Shimizu T, Ishizaki K, Sasaki MS, Kotoura Y, Yamamuro T. Mutation spectrum of the retinoblastoma gene in osteosarcomas. Cancer Res. 1994; 54 (11): 3042-3048.
10. Ladanyi M, Cha C, Lewis R, Jhanwar SC, Huvos AG, Healey JH. MDM2 gene amplification in metastatic osteosarcoma. Cancer Res. 1993; 53 (1): 16-18.
11. Pasic I, Shlien A, Durbin AD, Stavropoulos DJ, Baskin B, Ray PN, Novokmet A, Malkin D. Recurrent focal copy-number changes and loss of heterozygosity implicate two noncoding RNAs and one tumor suppressor gene at chromosome 3q13.31 in osteosarcoma. Cancer Res. 2010; 70 (1): 160-171.
12. Yen CC, Chen WM, Chen TH, Chen WY, Chen PC, Chiou HJ, Hung GY, Wu HT, Wei CJ, Shiau CY, Wu YC, Chao TC, Tzeng CH, Chen PM, Lin CH, Chen YJ et al. Identification of chromosomal aberrations associated with disease progression and a novel 3q13.31 deletion involving LSAMP gene in osteosarcoma. Int J Oncol. 2009; 35 (4): 775-788.
13. Kresse SH, Ohnstad HO, Paulsen EB, Bjerkehagen B, Szuhai K, Serra M, Schaefer KL, Myklebost O, Meza-Zepeda LA. LSAMP, a novel candidate tumor suppressor gene in human osteosarcomas, identified by array comparative genomic hybridization. Genes Chromosomes Cancer. 2009; 48 (8): 679-693.
14. Aue G, Muralidhar B, Schwartz HS, Butler MG. Telomerase activity in skeletal sarcomas. Ann Surg Oncol. 1998; 5 (7): 627-634.
15. Scheel C, Schaefer KL, Jauch A, Keller M, Wai D, Brinkschmidt C, van VF, Boecker W, Dockhorn-Dworniczak B, Poremba C. Alternative lengthening of telomeres is associated with chromosomal instability in osteosarcomas. Oncogene. 2001; 20 (29): 3835-3844.
16. Inaba H, Glibetic M, Buck S, Ravindranath Y, Kaplan J. Interferon-gamma sensitizes osteosarcoma cells to Fas-induced apoptosis by up-regulating Fas receptors and caspase-8. Pediatr Blood Cancer. 2004; 43 (7): 729-736.
17. Koshkina NV, Khanna C, Mendoza A, Guan H, DeLauter L, Kleinerman ES. Fas-negative osteosarcoma tumor cells are selected during metastasis to the lungs: the role of the Fas pathway in the metastatic process of osteosarcoma. Mol Cancer Res. 2007; 5 (10): 991-999.
18. Khanna C, Wan X, Bose S, Cassaday R, Olomu O, Mendoza A, Yeung C, Gorlick R, Hewitt SM, Helman LJ. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat Med. 2004; 10 (2): 182-186.
19. Wan X, Mendoza A, Khanna C, Helman LJ. Rapamycin inhibits ezrin-mediated metastatic behavior in a murine model of osteosarcoma. Cancer Res. 2005; 65 (6): 2406-2411.
20. Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006; 6 (4): 321-330.
21. Duan Z, Ji D, Weinstein EJ, Liu X, Susa M, Choy E, Yang C, Mankin H, Hornicek FJ. Lentiviral shRNA screen of human kinases identifies PLK1 as a potential therapeutic target for osteosarcoma. Cancer Lett. 2010; 293 (2): 220-229.
22. Yamaguchi U, Honda K, Satow R, Kobayashi E, Nakayama R, Ichikawa H, Shoji A, Shitashige M, Masuda M, Kawai A, Chuman H, Iwamoto Y, Hirohashi S, Yamada T. Functional genome screen for therapeutic targets of osteosarcoma. Cancer Sci. 2009; 100 (12): 2268-2274.
23. Hu K, Lee C, Qiu D, Fotovati A, Davies A, bu-Ali S, Wai D, Lawlor ER, Triche TJ, Pallen CJ, Dunn SE. Small interfering RNA library screen of human kinases and phosphatases identifies polo-like kinase 1 as a promising new target for the treatment of pediatric rhabdomyosarcomas. Mol Cancer Ther. 2009; 8 (11): 3024-3035.
24. Nappi TC, Salerno P, Zitzelsberger H, Carlomagno F, Salvatore G, Santoro M. Identification of Polo-like kinase 1 as a potential therapeutic target in anaplastic thyroid carcinoma. Cancer Res. 2009; 69 (5): 1916-1923.
25. Wang XQ, Zhu YQ, Lui KS, Cai Q, Lu P, Poon RT. Aberrant Polo-like kinase 1-Cdc25A pathway in metastatic hepatocellular carcinoma. Clin Cancer Res. 2008; 14 (21): 6813-6820.
26. Smith MR, Wilson ML, Hamanaka R, Chase D, Kung H, Longo DL, Ferris DK. Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochem Biophys Res Commun. 1997; 234 (2): 397-405.
27. Brockman JA, Gupta RA, Dubois RN. Activation of PPARgamma leads to inhibition of anchorage-independent growth of human colorectal cancer cells. Gastroenterology. 1998; 115 (5): 1049-1055.
28. Chen YX, Zhong XY, Qin YF, Bing W, He LZ. 15d-PGJ2 inhibits cell growth and induces apoptosis of MCG-803 human gastric cancer cell line. World J Gastroenterol. 2003; 9 (10): 2149-2153.
29. Clay CE, Atsumi GI, High KP, Chilton FH. Early de novo gene expression is required for 15-deoxy-Delta 12,14-prostaglandin J2-induced apoptosis in breast cancer cells. J Biol Chem. 2001; 276 (50): 47131-47135.
30. Lee SJ, Kim MS, Park JY, Woo JS, Kim YK. 15-Deoxy-delta 12,14-prostaglandin J2 induces apoptosis via JNK-mediated mitochondrial pathway in osteoblastic cells. Toxicology. 2008; 248 (2-3): 121-129.
31. Kitz K, Windischhofer W, Leis HJ, Huber E, Kollroser M, Malle E. 15-Deoxy-Delta12,14-prostaglandin J2 induces Cox-2 expression in human osteosarcoma cells through MAPK and EGFR activation involving reactive oxygen species. Free Radic Biol Med. 2011; 50 (7): 854-865.
32. Kondo M, Oya-Ito T, Kumagai T, Osawa T, Uchida K. Cyclopentenone prostaglandins as potential inducers of intracellular oxidative stress. J Biol Chem. 2001; 276 (15): 12076-12083.
33. Jung WK, Park IS, Park SJ, Yea SS, Choi YH, Oh S, Park SG, Choi IW. The 15-deoxy-Delta12,14-prostaglandin J2 inhibits LPS-stimulated AKT and NF-kappaB activation and suppresses interleukin-6 in osteoblast-like cells MC3T3E-1. Life Sci. 2009; 85 (1-2): 46-53.
34. Hasegawa H, Yamada Y, Komiyama K, Hayashi M, Ishibashi M, Sunazuka T, Izuhara T, Sugahara K, Tsuruda K, Masuda M, Takasu N, Tsukasaki K, Tomonaga M, Kamihira S. A novel natural compound, a cycloanthranilylproline derivative (Fuligocandin B), sensitizes leukemia cells to apoptosis induced by tumor necrosis factor related apoptosis-inducing ligand (TRAIL) through 15-deoxy-Delta 12, 14 prostaglandin J2 production. Blood. 2007; 110 (5): 1664-1674.
35. Han H, Shin SW, Seo CY, Kwon HC, Han JY, Kim IH, Kwak JY, Park JI. 15-Deoxy-delta 12,14-prostaglandin J2 (15d-PGJ 2) sensitizes human leukemic HL-60 cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis through Akt downregulation. Apoptosis. 2007; 12 (11): 2101-2114.
36. Fujita M, Tohji C, Honda Y, Yamamoto Y, Nakamura T, Yagami T, Yamamori M, Okamura N. Cytotoxicity of 15-deoxy-Delta(12,14)-prostaglandin J(2) through PPARgamma-independent pathway and the involvement of the JNK and Akt pathway in renal cell carcinoma. Int J Med Sci. 2012; 9 (7): 555-566.
37. Shin SW, Seo CY, Han H, Han JY, Jeong JS, Kwak JY, Park JI. 15d-PGJ2 induces apoptosis by reactive oxygen species-mediated inactivation of Akt in leukemia and colorectal cancer cells and shows in vivo antitumor activity. Clin Cancer Res. 2009; 15 (17): 5414-5425.
38. Zhu F, Wang P, Kontrogianni-Konstantopoulos A, Konstantopoulos K. Prostaglandin (PG)D(2) and 15-deoxy-Delta(12,14)-PGJ(2), but not PGE(2), mediate shear-induced chondrocyte apoptosis via protein kinase A-dependent regulation of polo-like kinases. Cell Death Differ. 2010; 17 (8): 1325-1334.
39. Chen ZY, Tseng CC. 15-deoxy-Delta12,14 prostaglandin J2 up-regulates Kruppel-like factor 4 expression independently of peroxisome proliferator-activated receptor gamma by activating the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signal transduction pathway in HT-29 colon cancer cells. Mol Pharmacol. 2005; 68 (5): 1203-1213.
40. Lee DR, Kwon CH, Park JY, Kim YK, Woo JS. 15-Deoxy-Delta(12,14)-prostaglandin J(2) induces mitochondrial-dependent apoptosis through inhibition of PKA/NF-kappaB in renal proximal epithelial cells. Toxicology. 2009; 258 (1): 17-24.
41. Kang DS, Kwon CH, Park JY, Kim JH, Woo JS, Jung JS, Kim YK. 15-deoxy-Delta12,14-prostaglandin J2 induces renal epithelial cell death through NF-kappaB-dependent and MAPK-independent mechanism. Toxicol Appl Pharmacol. 2006; 216 (3): 426-435.
42. Ciucci A, Gianferretti P, Piva R, Guyot T, Snape TJ, Roberts SM, Santoro MG. Induction of apoptosis in estrogen receptor-negative breast cancer cells by natural and synthetic cyclopentenones: role of the IkappaB kinase/nuclear factor-kappaB pathway. Mol Pharmacol. 2006; 70 (5): 1812-1821.
43. Zimmer M, Lamb J, Ebert BL, Lynch M, Neil C, Schmidt E, Golub TR, Iliopoulos O. The connectivity map links iron regulatory protein-1-mediated inhibition of hypoxia-inducible factor-2a translation to the anti-inflammatory 15-deoxy-delta12,14-prostaglandin J2. Cancer Res. 2010; 70 (8): 3071-3079.
44. Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle. 2009; 8 (16): 2502-2508.
45. Gao N, Ding M, Zheng JZ, Zhang Z, Leonard SS, Liu KJ, Shi X, Jiang BH. Vanadate-induced expression of hypoxia-inducible factor 1 alpha and vascular endothelial growth factor through phosphatidylinositol 3-kinase/Akt pathway and reactive oxygen species. J Biol Chem. 2002; 277 (35): 31963-31971.
46. Madureira PA, Hill R, Miller VA, Giacomantonio C, Lee PW, Waisman DM. Annexin A2 is a novel cellular redox regulatory protein involved in tumorigenesis. Oncotarget. 2011; 2 (12): 1075-1093.
47. Hei TK, Liu SX, Waldren C. Mutagenicity of arsenic in mammalian cells: role of reactive oxygen species. Proc Natl Acad Sci U S A. 1998; 95 (14): 8103-8107.
48. Warsch W, Grundschober E, Berger A, Gille L, Cerny-Reiterer S, Tigan AS, Hoelbl-Kovacic A, Valent P, Moriggl R, Sexl V. STAT5 triggers BCR-ABL1 mutation by mediating ROS production in chronic myeloid leukaemia. Oncotarget. 2012; 3 (12): 1669-1687.
49. Corazao-Rozas P, Guerreschi P, Jendoubi M, Andre F, Jonneaux A, Scalbert C, Garcon G, Malet-Martino M, Balayssac S, Rocchi S, Savina A, Formstecher P, Mortier L, Kluza J, Marchetti P. Mitochondrial oxidative stress is the Achille’s heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget. 2013; 4 (11): 1986-1998.
50. Posthumadeboer J, van Egmond PW, Helder MN, de Menezes RX, Cleton-Jansen AM, Belien JA, Verheul HM, van Royen BJ, Kaspers GJ, van B, V. Targeting JNK-interacting-protein-1 (JIP1) sensitises osteosarcoma to doxorubicin. Oncotarget. 2012; 3 (10): 1169-1181.
51. Kirshner JR, He S, Balasubramanyam V, Kepros J, Yang CY, Zhang M, Du Z, Barsoum J, Bertin J. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol Cancer Ther. 2008; 7 (8): 2319-2327.
52. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011; 475 (7354): 106-109.
53. Marcone S, Fitzgerald DJ. Proteomic identification of the candidate target proteins of 15-deoxy-delta12,14-prostaglandin J2. Proteomics. 2013; 13 (14): 2135-2139.
54. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983; 65 (1-2): 55-63.
55. Pozarowski P, Darzynkiewicz Z. Analysis of cell cycle by flow cytometry. Methods Mol Biol. 2004; 281 301-311.
56. Valavanidis A, Vlachogianni T, Fiotakis C. 8-hydroxy-2’ -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2009; 27 (2): 120-139.
57. De Iuliis GN, Thomson LK, Mitchell LA, Finnie JM, Koppers AJ, Hedges A, Nixon B, Aitken RJ. DNA damage in human spermatozoa is highly correlated with the efficiency of chromatin remodeling and the formation of 8-hydroxy-2’-deoxyguanosine, a marker of oxidative stress. Biol Reprod. 2009; 81 (3): 517-524.