
INTRODUCTION
Melanoma is the most aggressive form of skin cancer, and the incidence of melanoma continues to rise worldwide [1, 2]. Although surgical treatment of early melanoma leads to 90% cure rates, unresectable advanced melanoma is notorious for its intrinsic resistance to chemotherapy, aggressive clinical behavior and tendency to rapidly metastasize. Five-year survival rates for patients with distant metastatic disease remain below 20% [2]. Therefore, despite the variety of approaches used to treat melanoma, novel therapies and treatment strategies are needed. In the past decade, targeted inhibitors have been widely used for the treatment of melanoma. The effectiveness of these treatments depends on the presence of driver mutations in the tumor.
Melanoma patients frequently harbor somatic mutations in a number genes, including the BRAF gene that encodes the serine-threonine kinase [3]; the KIT gene that encodes the receptor tyrosine kinase [4]; the NRAS [5], GNA11 [6, 7] and GNAQ [7, 8] genes that encode the GTP-binding proteins; and the MAP2K1 [9] and MAP2K2 [10, 11] genes that encode the dual specificity mitogen-activated protein kinase kinases. The activating mutations in these genes lead to the constitutive activation of the RAS/RAF/MEK/ERK (MAPK) and the PI3K/PTEN/AKT (AKT) signaling pathways [7, 12–22]. Overall, mutations in these genes can be detected in approximately 70% of melanomas, depending on the site of the primary lesion [9, 23].
Tumor mutation status has been associated with the sensitivity of melanomas to specific targeted therapies. BRAF V600 mutations are linked with an increased sensitivity to BRAF (vemurafenib and dabrafenib) and MEK (trametinib and cobimetinib) inhibitors [24–32]. The type of BRAF mutation influences on sensitivity to targeted therapy. Thus, inhibition of BRAF with vemurafenib improves survival in patients with V600E and V600K mutations, but the response rate in patients with V600K was less than that in patients with BRAF V600E tumors [30]. The patients with rarer non-V600E BRAF mutations are showed objective responses to vemurafenib too; however, additional follow-up is required [33].
Tumors that harbor KIT mutations (W557R, V559D, K642E, L576P, and V559A) display a sensitivity to the KIT inhibitor, imatinib [4, 34–38].The KIT V559A and L576P mutations are sensitive to the multi-tyrosine kinase inhibitor, sunitinib [39, 40]. Tumors that harbor the KIT L576P mutation show a response to tyrosine kinase inhibitors, such as dasatinib [41] and nilotinib [34]. Melanoma patients with KIT D816H mutation, however, are not sensitive to imatinib [35]. Furthermore, KIT kinase mutations D816H, D816V and D816Y show drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients [35, 42–45]. Preclinical data suggest that MEK inhibition may be effective for uveal melanomas carrying GNAQ or GNA11 mutations [23]. MAP2K1/2 mutations confer resistance to MEK and BRAF inhibition [10, 11, 46, 47]. In most cases, mutations in the BRAF, NRAS, KIT, GNAQ and GNA11 genes are mutually exclusive [8, 23, 35, 48] In contrast, MAP2K1 and MAP2K2 mutations often occur together with BRAF mutations [9, 46, 49].
Several techniques are currently used to detect somatic mutations in cancer cells. Among the most common techniques are Sanger sequencing, pyrosequencing, allele-specific real-time PCR and next generation sequencing (NGS). These methods have some advantages and disadvantages. The Sanger sequencing and pyrosequencing are inexpensive methods but have low analytical sensitivity and require setting the parallel reactions for each of the analyzed loci. The allele-specific real-time PCR is highly sensitive for the detection of any known mutation, but it is expensive and has the limitation of multiplex analyses. NGS has a high analytical sensitivity and allows the simultaneous detection of a large number of somatic mutations but is very expensive and time-consuming. Some of the approaches are fully automated [50–52], which makes them more attractive to clinicians but significantly increases the cost of analysis.
DNA microarray technology is successfully applied for the multiple testing of genetic markers in tumor cells. The high-density microarrays, for example, the OncoScan assay (Affymetrix, a Thermo Fisher Scientific company, Waltham, USA), allow simultaneous analysis of numerous genes, including structural, copy number and single nucleotide variations [53, 54]. The technology is very powerful and reliable but the high cost limits routine clinical use. Besides, the analysis is multistage and requires sophisticated bioinformatics. Low-density microarrays [55–60] allow to detect much less targets, however, they offer an inexpensive, fast and easy way to analyze somatic mutations, and so are more suitable for routine applications. Some of low-density platforms are commercially available [55–57], but none of them is specific focused to determine most clinically relevant mutations in melanoma patients.
Thus, there is a need to develop fast, inexpensive and highly sensitive method, based on low-density microarrays technology, which provides the detection of significant somatic mutations in melanomas to meet the demands for modern melanoma treatment. BRAF, NRAS, KIT, MAP2K1/2, GNAQ and GNA11 mutations may be clinically useful for selecting patients for different targeted therapies. In the present study, we proposed a reliable biochip-based approach designed to simultaneously detect 39 recurrent mutations in melanoma genes. The high sensitivity has been reached by combining, in one assay, the LNA PCR clamp technique and the mutation-specific hybridization on a hydrogel biochip, which allows the identification of somatic mutations in a large excess of wild type (WT) DNA. Previously, this approach has been developed and applied for the analysis of somatic mutations in EGFR, KRAS, BRAF, PI3K genes in lung cancer cells [61].
This method was validated and used to analyze the frequency of mutations in 253 melanoma patients, using tumor-derived DNA from fresh-frozen and formalin-fixed paraffin-embedded (FFPE) tissues. The results obtained with this new approach were compared to the results obtained by traditional Sanger sequencing, NGS and ARMS/Scorpion real-time PCR.
RESULTS
Biochip assay for BRAF, NRAS, KIT, MAP2K1/2, GNAQ and GNA11 mutation detection
A scheme of the biochip is presented in Figure 1. The examples of hybridization patterns for samples with the V600K mutation of BRAF and a Q61K NRAS mutation are shown in Figure 2. The mutated sequences were predominantly amplified because the LNA PCR clamp was used. Insignificant wild-type sequence amplification occurred during the second round of PCR. Further, the amplified DNA fragments carrying the fluorescent label were bound to the oligonucleotide probes on the biochip; therefore, the fluorescent label was accumulated in the gel. The biochip assay was able to detect 39 different mutations in the BRAF, NRAS, KIT, MAP2K1/2, GNAQ and GNA11 genes simultaneously.
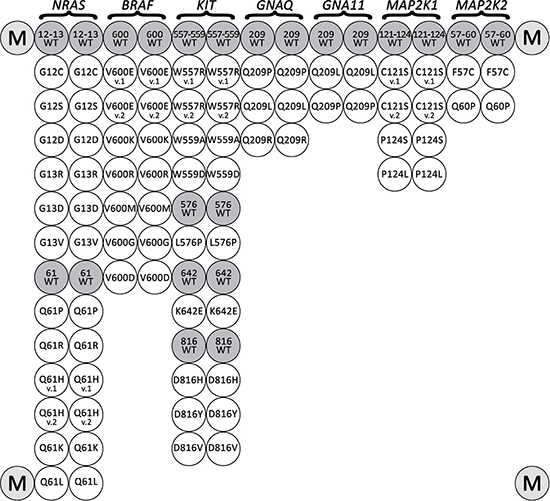
Figure 1: Biochip spotting scheme. The biochip includes paired probes for the detection of each somatic mutations and corresponding wild-type sequences. Marker spots with Cy5 are located in the corners.
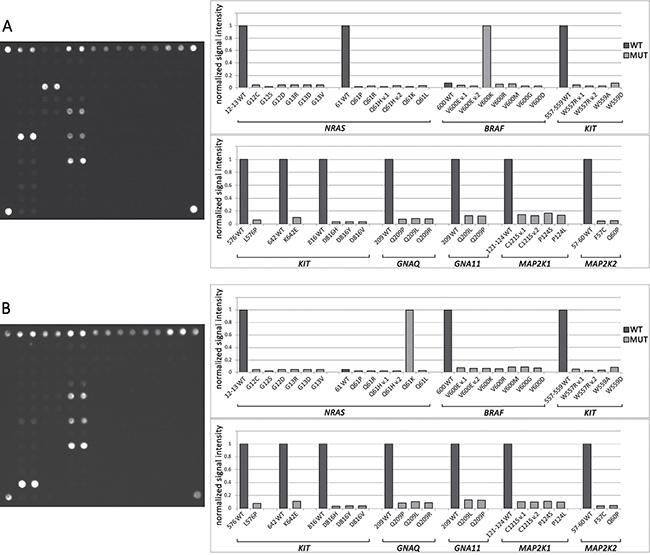
Figure 2: Mutational analysis by biochip. (A) Biochip image and histogram of normalized signal intensity obtained for a sample with a V600K BRAF mutation. The bright spots in the hybridization image correspond to the outstanding bars on the histogram. All gel spots correspond to one analyzed site with common primers, and LNA oligonucleotides are combined into one group. Fluorescent signals from the paired probes are averaged. The fluorescent signal is normalized to the maximum signal in a group of gel spots. The sample contains a V600K BRAF mutation because J(V600K)>J(600WT). (B) Biochip images and histograms of normalized signal intensity obtained for a sample with a Q61K NRAS mutation.
Before using the biochip assay for clinical analysis, it was tested by an analysis of 40 control samples with known genotypes, which were characterized by direct sequencing as follows: WT sample and samples harbored all variations of the mutations localized on the biochip. All genotypes were identified by the biochips correctly.
Sensitivity of the biochip assay
The biochip assay sensitivity was determined by analyzing serially diluted mutant DNA (BRAF V600E) in a background of wild-type DNA in different ratios (0%, 0.25%, 0.5%, 1%, 5%, 10%, 50% and 100%). The samples were analyzed in triplicate. Our results showed that the biochip assay was able to detect 0.5% mutated DNA in wild-type DNA (Figure 3).
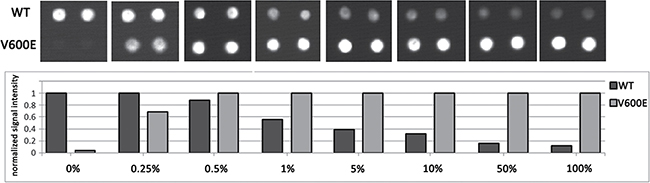
Figure 3: Biochip assay sensitivity. The assay sensitivity for BRAF V600E mutation detection was determined by an analysis of serially diluted mutant DNA in the background of WT DNA: 0%, 0.25%, 0.5%, 1%, 5%, 10%, 50% and 100%. Fragment of a biochip images (in the upper part) and the normalized signal intensity (in the lower part) only for the spots corresponding to BRAF 600WT and V600E are present for each dilution.
Analysis of clinical samples by biochip assay
The clinical characteristics of the patients are reported in Table 1. In 177 of 253 (70.0%) melanoma patients, somatic mutations in the analyzed genes were detected by the biochip assay (Table 2): 129 (51.0%) BRAF, 45 (17.8%) NRAS, 6 (2.4%) KIT, 4 (1.6%) GNAQ, 2 (0.8%) GNA11, 2 (0.8%) MAP2K1 and none of MAP2K2. The biochip allows the detection of only the most common somatic mutations, which are listed in Table 2.
Table 1: Patient clinical characteristics
Characteristic |
No. of patients |
|---|---|
Age (median ± SD) |
53.7 ± 16.4 |
Sex |
|
Male |
97 |
Female |
140 |
NA (NA - not available) |
16 |
Location |
|
Trunk |
82 |
Upper limb |
23 |
Lower limb |
66 |
Head and Neck |
30 |
NA |
52 |
Tumor subtype |
|
superficial spreading melanoma |
24 |
acral-lentiginuous |
1 |
lentigo maligna |
10 |
nodular |
43 |
NA |
175 |
Stage |
|
0 |
1 |
I A + B |
14 |
II A + B |
44 |
III A + B |
17 |
IV |
5 |
NA |
172 |
Breslow thickness |
|
≤ 1 mm |
26 |
1.01–2.0 mm |
19 |
2.01–4.0 mm |
28 |
> 4 mm |
44 |
NA |
136 |
Clark level |
|
I |
3 |
II |
13 |
III |
65 |
IV |
18 |
V |
20 |
NA |
133 |
Ulceration |
|
Yes |
68 |
No |
50 |
NA |
135 |
Table 2: Analysis of the 253 melanoma patients using the biochip assay
Mutations |
No. of patients (%) |
|---|---|
BRAF |
129 (51.0%) |
V600E (c.1799T>A) |
111 |
V600E (c.1799_1800delTGinsAA) |
0 |
V600K (c.1798_1799delGTinsAA) |
14 |
V600R (c.1798_1799delGTinsAG) |
3 |
V600M (c.1798G>A) |
1 |
V600G (c.1799T>G) |
0 |
V600D (c.1799_1800delTGinsAT) |
0 |
NRAS |
45 (17.8%) |
G12C (c.34G>T) |
0 |
G12S (c.34G>A) |
0 |
G12D (c.35G>A) |
0 |
G13R (c.37G>C) |
1 |
G13D (c.38G>A) |
1 |
G13V (c.38G>T) |
0 |
Q61P (c.182A>C) |
0 |
Q61R (c.182A>G) |
17 |
Q61H (c.183A>C) |
2 |
Q61H (c.183A>T) |
1 |
Q61K (c.181C>A) |
20 |
Q61L (c.182A>T) |
3 |
KIT |
6 (2.4%) |
W557R (c.1669T>A) |
0 |
W557R (c.1669T>C) |
0 |
V559A (c.1676T>C) |
0 |
V559D (c.1676T>A) |
0 |
L576P (c.1727T>C) |
5 |
K642E (c.1924A>G) |
1 |
D816H (c.2446G>C) |
0 |
D816Y (c.2446G>T) |
0 |
D816V (c.2447A>T) |
0 |
GNAQ |
4 (1.6%) |
Q209P (c.626A>C) |
2 |
Q209L (c.626A>T) |
2 |
Q209R (c.626A>G) |
0 |
GNA11 |
2 (0.8%) |
Q209L (c.626A>T) |
2 |
Q209P (c.626A>C) |
0 |
MAP2K1 |
2 (0.8%) |
C121S (c.361T>A) |
0 |
C121S (c.362G>C) |
0 |
P124S (c.370C>T) |
2 |
P124L (c.371C>T) |
0 |
MAP2K2 |
0 (0%) |
F57C (c.170T>G) |
0 |
Q60P (c.179A>C) |
0 |
WT |
76 (30.0%) |
Comparison with ARMS/Scorpion real-time PCR
The 98 melanoma samples were screened for BRAF V600E/K/R/D mutations by ARMS/Scorpion real-time PCR (BRAF RGQ PCR Kit, Qiagen, Germany). In 58 of 98 (59.2%) samples, somatic mutations in the BRAF gene were detected. Discordance with the biochip data was shown in 10 samples (Table 3). The specimens with discordant results were subjected to Sanger sequencing with and without the enrichment of mutant DNA by LNA PCR clamp. In 8/10 cases, the sequencing method (with or without the enrichment of mutant DNA) confirmed the biochip data. In 2 cases, the samples harbored the rare mutations V600V (c.1800G>A; COSM249890) and T599_V600insT (c.1797_1798insACA; COSM144982). The specific hybridization probes were absent on the biochip; therefore, the mutations could not be detected.
Table 3: Identification of BRAF mutations using the ARMS/Scorpion real-time PCR, biochip assay, sequencing and LNA PCR clamp with sequencing (samples that yielded discordant results are included only)
Patient No |
Biochip |
ARMS/Scorpion real-time PCR |
Sanger sequencing |
LNA PCR clamp + Sanger sequencing |
|---|---|---|---|---|
163 |
V600E (c.1799T>A) |
WT |
V600E (c.1799T>A) |
V600E (c.1799T>A) |
191 |
V600K (c.1798_1799delGTinsAA) |
WT |
V600K (c.1798_1799delGTinsAA) |
V600K (c.1798_1799delGTinsAA) |
219 |
WT |
V600R (c.1798_1799delGTinsAG) |
WT |
V600V (c.1800G>A) |
223 |
WT |
V600E (c.1799T>A) |
WT |
WT |
226 |
WT |
V600K (c.1798_1799delGTinsAA) |
WT |
WT |
235 |
V600M (c.1798G>A) |
WT |
WT |
V600M (c.1798G>A) |
238 |
WT |
V600E (c.1799T>A) |
T599_V600insT (c.1797_1798insACA) |
T599_V600insT (c.1797_1798insACA) |
239 |
V600E (c.1799T>A) |
V600K (c.1798_1799delGTinsAA) |
WT |
V600E (c.1799T>A) |
241 |
WT |
V600K (c.1798_1799delGTinsAA) |
WT |
WT |
242 |
WT |
V600K (c.1798_1799delGTinsAA) |
WT |
WT |
Comparison with Next generation sequencing
A total of 25 melanoma samples and 6 melanoma cell lines were tested by NGS (GS Junior, 454 Life Sciences, Branford, USA) (Table 4). In 4 samples, somatic mutations in the BRAF or NRAS genes were detected only by the biochip assay. In all cases, the results of the biochip assay were confirmed by sequencing with the enrichment of the mutant DNA by LNA PCR clamp. The NGS failed to detect these mutations, probably due to very low percentage of tumor cells in a sample, because the fresh-frozen tissue samples did not subjected to histological control before the analysis. In cell line Mel Cher a low percentage of cells carrying NRAS mutation may be explained by clonal heterogeneity or cross-contamination between different cell lines. In over 5 controversial cases, the genetic alterations were detected only by NGS. These cases represent germinal mutations and SNPs in the KIT and BRAF genes. The specific hybridization probes were absent on the biochip; therefore, the mutations could not be detected.
Table 4: Comparison NGS with biochip assay
Patient No |
Biochip |
NGS |
Sanger sequencing |
LNA PCR clamp + Sanger sequencing |
|---|---|---|---|---|
2 |
BRAF V600E (c.1799T>A) |
BRAF V600E (c.1799T>A) |
- |
- |
31 |
WT |
WT |
- |
- |
52 |
BRAF V600E (c.1799T>A) |
BRAF V600E (c.1799T>A) |
- |
- |
91 |
WT |
WT |
- |
- |
92 |
BRAF V600E (c.1799T>A) |
BRAF V600E (c.1799T>A) |
- |
- |
93 |
NRAS Q61K (c.181C>A) |
WT |
WT |
NRAS Q61K (c.181C>A) |
94 |
BRAF V600E (c.1799T>A);MAP2K1 P124S (c.370C>T) |
BRAF V600E (c.1799T>A);MAP2K1 P124S (c.370C>T) |
- |
- |
95 |
BRAF V600E (c.1799T>A) |
BRAF V600E (c.1799T>A) |
- |
- |
96 |
BRAF V600E (c.1799T>A) |
WT |
- |
- |
97 |
BRAF V600E (c.1799T>A) |
BRAF V600E (c.1799T>A) |
- |
- |
98 |
WT |
WT |
- |
- |
99 |
BRAF V600K (c.1798_1799delGTinsAA) |
BRAF V600K (c.1798_1799delGTinsAA) |
- |
- |
100 |
BRAF V600E (c.1799T>A) |
BRAF V600E (c.1799T>A) |
- |
- |
101 |
WT |
KIT M541L (c.1621A>C) |
- |
- |
102 |
BRAF V600E (c.1799T>A) |
BRAF V600E (c.1799T>A) |
- |
- |
103 |
NRAS Q61H (c.182A>C) |
NRAS Q61H (c.182A>C) |
- |
- |
104 |
WT |
WT |
- |
- |
105 |
BRAF V600E (c.1799T>A) |
BRAF V600E (c.1799T>A) |
- |
- |
106 |
BRAF V600K (c.1798_1799delGTinsAA) |
KIT V50L (c.148G>T) |
WT |
BRAF V600K (c.1798_1799delGTinsAA) |
107 |
BRAF V600E (c.1799T>A) |
BRAF V600E (c.1799T>A) |
- |
- |
108 |
NRAS G13R (c.37G>C) |
NRAS G13R (c.37G>C) |
- |
- |
109 |
NRAS Q61K (c.181C>A) |
NRAS Q61K (c.181C>A) |
- |
- |
110 |
BRAF V600E (c.1799T>A) |
BRAF V600E (c.1799T>A) |
- |
- |
111 |
BRAF V600E (c.1799T>A) |
KIT M541L (c.1621A>C);BRAF V600E (c.1799T>A) |
- |
- |
112 |
NRAS Q61K (c.181C>A) |
WT |
WT |
NRAS Q61K (c.181C>A) |
cell line SK-MEL2 |
NRAS Q61R (c.182A>G) |
NRAS Q61R (c.182A>G); KITM541L (c.1621A>C), G245S (c.733G>A) |
- |
- |
cell line Mel Il |
BRAF V600K (c.1798_1799delGTinsAA) |
BRAF V600K (c.1798_1799delGTinsAA);BRAF R389C (c.1165C>T) |
- |
- |
cell line Mel Ibr |
BRAF V600E (c.1799T>A) |
BRAF V600E (c.1799T>A) |
- |
- |
cell line Mel Rac |
NRAS Q61R (c.182A>G) |
NRAS Q61R (c.182A>G) |
- |
- |
cell line Mel Cher |
NRAS Q61R (c.182A>G) |
WT |
WT |
NRAS Q61R (c.182A>G) |
cell line Mel Z |
BRAF V600E (c.1799T>A) |
BRAF V600E (c.1799T>A) |
- |
- |
Sequencing and LNA PCR clamp with sequencing were used us control methods
Comparison with sanger sequencing
In total, 119 of the 253 samples were screened for BRAF, NRAS, KIT, GNAQ, GNA11 and MAP2K1/2 mutations by Sanger sequencing. In 87/119 samples, the genotype coincided with the data obtained by the biochip. In 2 (1.7%) cases, the rare mutations in the BRAF gene were identified by Sanger sequencing only: V600D (c.1799_1800TG>AC, COSM308550) and A598_T599insV (c.1794_1795insGTT, COSM26625). The 30 specimens with discordant results were subjected to LNA PCR clamp followed by sequencing. In 29/30 samples, the results were identical to the data obtained with the biochip assay. In 1 sample, a rare mutation, G60G (c.180A>T), was detected in the NRAS gene.
Genetic alterations in melanoma patients
Summarizing the results obtained by all genotyping methods, 185/253 (73.1%) melanoma patients harbored somatic mutations in the analyzed genes.
-BRAF mutations
In total, 134/253 (53.0%) melanoma patients carried BRAF mutations. In most cases, the frequent mutations were identified using the biochip (111/134 [82.8%] V600E mutation, 14/134 [10.4%] V600K, 3/134 [2.2%] V600R, and 1/134 [0.7%] V600M). In five cases (5/253, 2.0%), we detected rare mutations only by Sanger sequencing (1/134 [0.7%] V600D (c.1799_1800TG>AC, COSM308550), 1/134 [0.7%] V600V (c.1800G>A, COSM249890), 1/134 [0.7%] A598V (c.1793C>T, COSM21549), 1/134 [0.7%] A598_T599insV (c.1794_1795insGTT, COSM26625), 1/134 [0.7%] p.T599_V600insT (c.1797_1798insACA, COSM144982) (Figure 4). Thus, 17.2% of BRAF-mutated patients showed a rare mutation (non-V600E). The frequency of rare BRAF mutations increased with age (P = 0.05). Only 4.3% of patients ≤ 41 years old harbored non-V600E mutation, while 14.4% of patients ≥ 61 years old were non-V600E (Figure 5).
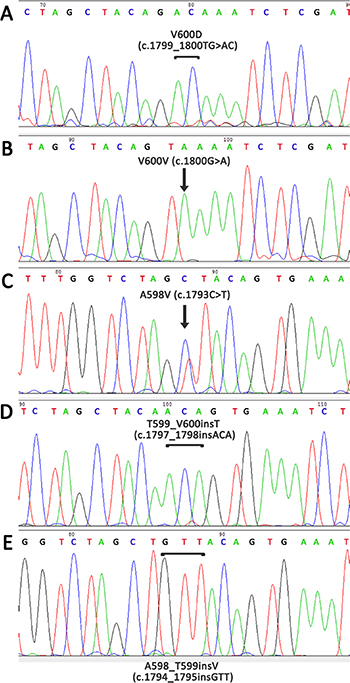
Figure 4: Detection of rare mutations in the BRAF gene using Sanger sequencing with preliminary enrichment with mutant allele. (A) V600D, c.1799_1800TG>AC, COSM308550; (B) V600V, c.1800G>A, COSM249890; (C) A598V, c.1793C>T, COSM21549; (D) T599_V600insT, c.1797_1798insACA, COSM144982; (E) A598_T599insV, c.1794_1795insGTT, COSM26625.
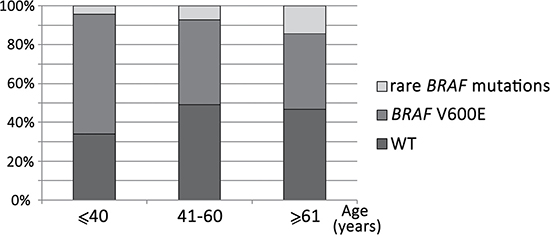
Figure 5: Frequency of rare BRAF mutations in different age groups of patients. Rare mutations are more common in older patients (P = 0.05).
Women showed a higher frequency of BRAF mutations compared to men (85/140 [60.7%] vs. 46/97 [47.4%], P = 0.047). The BRAF mutated patients, compared with the WT BRAF patients, showed a slightly lower median age as follows: 52.7±14.2 years (range 17-83 years) vs. 55.1±18.8 years (range 0.1–88 years) (P = 0.108). There were no significant associations among the BRAF mutation status, tumor localization, subtype, stage, Breslow thickness, Clark level or ulceration.
-NRAS mutations
NRAS mutations were found in 45/253 (17.8%) patients. In one case, the patient harbored two mutations, Q61K and G60G (c.180A>T), in the NRAS gene simultaneously. In total, 43/46 (93.4%) of these mutations were observed in codon 61 as follows: 20/46 (43.5%) Q61K, 17/46 (37.0%) Q61R, 3/46 (6.5%) Q61L, 2/46 (4.3%) Q61H (c.183A>C) and 1/46 (2.2%) Q61H (c.183A>T) mutations. Mutations in codon 13 were detected only in 2 patients as follows: 1/46 (2.2%) G13R and 1/46 (2.2%) G13D. The rare mutation G60G (c.180A>T) was detected only by Sanger sequencing.
The NRAS mutations occurred more frequently in men than in women; however, the difference was not statistically significant (22/97 [22.7%] vs. 19/140 [13.6%], P = 0.0813). NRAS mutations were more common in nodular melanoma than in superficial spreading melanoma (7/43 [16.3%] vs. 0/24 [0%], P=0.0367). There were no associations between tumor NRAS mutation status, location, age, stage, Breslow thickness, Clark level or ulceration.
-KIT mutations
Six (2.4%) patients had KIT mutations. Five of these patients harbored the mutation in codon 576 (83.3%), and one (16.7%) harbored the mutation in codon 624 (K642E). The KIT mutation status did not correlate with gender, age or ulceration.
-GNAQ mutations
The GNAQ mutations in codon 209 were detected in 2.0% (5/253) of our melanoma cases. The Q209P (c.626A>C) and Q209L (c.626A>T) mutations were identified in 2 cases, and the Q209L (c.625_626CA>TT) mutation was identified in 1 case. Q209L (c.625_626CA>TT) is a rare mutation detected only by Sanger sequencing. Mutations in the GNAQ gene were not associated with age, gender or ulceration.
-GNA11 mutations
The Q209L GNA11 gene mutation was detected in 2 (0.8%) of 253 patients. In both cases, the mutations were identified in females. The GNA11 mutations were mutually exclusive, with mutations in the BRAF, NRAS, KIT, GNAQ and MAP2K1/2 genes. Mutations in the GNA11 gene were not associated with age or gender. The other correlations were invalid due to the small number of analyzed groups.
-MAP2K1 mutations
The P124S MAP2K1 gene mutation was found in 2/253 (0.8%) patients. In both cases, the MAP2K1 mutation was identified in tumors simultaneously harboring the BRAF V600E mutation. MAP2K1 mutation status did not correlate with age, gender or ulceration.
-MAP2K2 mutations
The F57C and Q60P mutations in the MAP2K2 gene were not found in the 253 melanoma patients.
DISCUSSION
The mutation status of a tumor is critical for considering targeted therapy for patients with melanoma. In this study, we proposed a biochip-based assay for the detection of somatic mutations in melanoma. Although several methodologies are currently used to test somatic mutations (Sanger sequencing, NGS, PCR-related technologies and others), many of them have flaws in comparison to the biochip assay. Sanger sequencing has a relatively low sensitivity with a detection limit of approximately 15–30% mutant alleles [62, 63]. NGS is becoming more widely adopted as a valuable method for somatic mutation analysis in cancer. NGS offers high sensitivity and accurate data quality for identifying even rare mutations successfully. These advantages are driving increased adoption of NGS in clinical cancer research, but this method is time-consuming and, thus far, rather expensive.
The real-time PCR based commercial kits, such as the COBAS 4800 BRAF V600 Mutation Test (Roche Molecular Diagnostics, Pleasanton, USA) and BRAF RGQ PCR Kit (TheraScreen, Qiagen, Hilden, Germany), are used worldwide and have sensitivity levels of approximately 5% but are expensive, allowing for the identification of a limited number of mutations.
Microarrays remain useful and accurate tools for parallel analysis of actionable mutations in cancer. The Affymetrix OncoScan array has been optimized for whole genome copy number, loss of heterozygosity and somatic mutation detection from highly degraded FFPE samples. Based on molecular inversion probe technology, the assay currently detects 74 somatic mutations commonly found in 9 cancer genes (BRAF, KRAS, EGFR, IDH1, IDH2, PTEN, PIK3CA, NRAS and TP53). This high-density platform is reliable and powerful, but is rather labor-consuming and requires expensive equipment [53, 54]. The claimed sensitivity of the assay in relation to variant allele frequency is about 5%, which may be not enough while working with the bulk of the tumor tissue [64]. Although the low-density microarrays allow to analyze much less genes, they are more suitable for routine use due to simple laboratory procedure, reliability and low cost of analysis [55–60].
We proposed the biochip-based assay, which combined multiplex LNA PCR clamp followed by hybridization with an array of allele-specific immobilized probes, allowing for the identification of 39 mutations in the BRAF, NRAS, KIT, MAP2K1/2, GNAQ and GNA11 genes. Our data showed that this approach could detect at least 0.5% of mutated sequences in a background of WT DNA. The biochips are inexpensive (approximately $15 per chip) and may be useful as a routine laboratory test. The workflow included isolation of DNA from fresh frozen or FFPE tumor, LNA PCR clamp, hybridization on the biochip and image analysis and requires no more than 20–24 h. Twenty-four clinical samples could be tested in parallel. To reach the highest accuracy of the mutation analysis, verification of the hybridization results may be recommended using the same primer set in LNA PCR clamp, followed by Sanger sequencing.
In our study, 253 melanoma patients were analyzed by developing a biochip-based approach. Mutations in the BRAF, NRAS, KIT, GNAQ, GNA11 and MAP2K1 genes were found in 51.0%, 17.8%, 2.4%, 1.6%, 0.8% and 0.8% of the cases, respectively. Mutations in the MAP2K2 gene were not detected.
The biochip-based assay was compared with the following three widely used methods: NGS, ARMS/Scorpion real-time PCR and Sanger sequencing.
In this study, we analyzed 25 melanoma patients and 6 melanoma cell by NGS and biochip assay in parallel. Despite a 100-fold coverage in NGS, in 4 samples somatic mutations in BRAF or NRAS genes were detected only by the biochip assay. The results were confirmed by Sanger sequencing with the preliminary enrichment of mutant DNA. In 5 cases, the genetic alterations were detected only by NGS and represented allelic variants in the KIT and BRAF genes, which were not considered valuable for choice of therapy. Thereby, the biochip assay was not inferior to the NGS approach in the detection of clinically relevant driver mutations in melanoma samples.
The 98 melanoma samples were screened for BRAF mutations by ARMS/Scorpion real-time PCR and hybridization with biochips. The mutation rate of ARMS/Scorpion real-time PCR in these samples was 59.2% compared with 56,1% detected by biochips. Discordance in the results was obtained in 10 samples. In 1/10 case, the two methods showed different types of mutations. In 3/10 cases, we detected mutations only by biochip assay. In 6/10 cases, we detected mutations only by the BRAF RGQ PCR Kit. The specimens with discordant results were subjected to Sanger sequencing (with and without the enrichment of mutant DNA), and, in 8/10 cases, sequencing confirmed the biochip data. In 2/10 cases, samples harbored rare BRAF mutations, V600V and T599_V600insT, which were not included in the biochip and, therefore, could not be detected.
The biochip assay was compared with Sanger sequencing. The biochip allowed detecting somatic mutations in 70% of samples, while the Sanger sequencing without a preliminary enrichment by mutant DNA revealed mutations only in 53% of cases. The sequencing approach with preliminary enrichment by mutant allele using LNA-clamp PCR made possible the identification of mutations in a high percentage of cases similar to biochip assay.
Based on these results, we can conclude that the biochip-based assay is a reliable, accurate, reproducible and highly sensitive method for the detection of frequent mutations in melanoma.
In total, somatic mutations in the BRAF, NRAS, KIT, GNAQ, GNA11, MAP2K1 and MAP2K2 genes were found in 53.0%, 17.8%, 2.4%, 2.0%, 0.8%, 0.8% and 0% of the 253 cases, respectively. These frequencies are very close to those previously reported in melanoma patients [23, 35, 65–67]. In general, BRAF, NRAS, GNAQ and GNA11 mutations were not found in one tumor sample. Moreover, we found that mutations in BRAF with NRAS (P < 0.0001) and BRAF with GNAQ (P = 0.022) are mutually exclusive. In contrast, MAP2K1 mutations were present together with mutations in the BRAF gene.
In addition, in this study, we defined a relationship between the mutation status and the clinical characteristics of the patients. In our study, the age of BRAF mutated patients was found to be slightly lower than BRAF WT patients (P = 0.108). However, in many studies, this difference was statistically significant [68–70]. The lack of significant difference in the two groups of patients might be explained by the inclusion of children with melanoma in our study. By analyzing only the adult patients (over 21 years), we obtained a statistically significant difference between the MUT and WT BRAF groups (53.21 ± 13.59 vs. 58.51 ± 13.99, P = 0.0148).
The higher proportion of non-V600E genotypes in BRAF-mutated melanomas was observed in older patients (P = 0.05). This trend is in accordance with the publication of Menzies et al. [71].
Women showed a higher frequency of BRAF mutations compared to men (85/140 [60.7%] vs. 46/97 [47.4%], P = 0.047), but other studies did not demonstrate statistically significant associations between BRAF mutation status and gender [70, 72, 73].
The NRAS mutations occurred more frequently in men than in women; however, the difference was not statistically significant (22/97 [22.7%] vs. 19/140 [13.6%], P = 0.0813) in our study and other studies [74, 75]. NRAS mutations were more common in nodular melanoma than in superficial spreading melanoma (7/43 [16.3%] vs. 0/24 [0%], P = 0.0367). Similar data were obtained in other studies [76–78], but several groups found no difference in NRAS mutation rates among nodular and superficial spreading melanoma [79, 80].
In general, the association between the tumor genotype of patients and their clinical characteristics also corresponds to previously published data.
In this study, we proposed a highly sensitive, reproducible and fast method for BRAF, NRAS, KIT, GNAQ, GNA11 and MAP2K1/2 mutation detection based on a hydrogel biochip platform. This approach is easy-to-use, inexpensive and does not require expensive equipment. The mutation detection rate using this approach is as great as 70% in melanoma patients. The method is suitable for rapid and effective mutation screening in large research centers and small distant laboratories.
We have compared this method with NGS, ARMS/Scorpion real-time PCR and Sanger sequencing and concluded that the biochip-based approach has a great potential for routine clinical application and can be used in a rapid screening for the presence of somatic mutations in melanoma patients prior to targeted therapy appointment.
MATERIALS AND METHODS
Tumor samples and cell lines
The tumor samples were obtained from 253 melanoma patients who were treated at the Blokhin Cancer Research Center, Ministry of Health of the Russian Federation between 1984 and 2016 and the P. Hertsen Moscow Oncology Research Institute between 2003 and 2016. The mean age of the melanoma patients was 53.7 ± 16.4 years. All patients provided written informed consent. Ethical approval for the study was provided by the Ethical Committees of the Blokhin Cancer Research Center and the P. Hertsen Moscow Oncology Research Institute.
The tumor tissues were collected by surgical resection. In total, 63/253 tissue samples were frozen directly after surgery and were stored at −20°C until DNA extraction, 190/253 tissue samples were fixed in formalin and embedded in paraffin.
In addition, genomic DNA was derived from the following 6 melanoma cell lines: Mel Rac [81], Mel Z [82], Mel Cher [83], Mel Il [84], Mel Ibr [85] and SK-MEL-2 (ATCC® HTB-68™, Guernsey, Ireland, United Kingdom).
Biochip assay
The procedure of the biochip approach analysis consisted of the following successive steps: (1) DNA isolation from the tumor tissue; (2) LNA clamped multiplex PCR to predominant amplification of the mutant DNA in the presence of large excess of wild-type DNA; (3) asymmetric multiplex nested PCR to yield fluorescently labeled single-stranded DNA fragments; (4) hybridization of labeled PCR-products on a biochip; and (5) hybridization image analysis.
DNA isolation
Genomic DNA from the tumor tissues was isolated using a QIAamp DNA Micro Kit, QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) or blackPREP FFPE DNA Kit (Analytik Jena, Jena, Germany) according to the manufacturer’s instructions. The quantity of nucleic acids was controlled by the Qubit fluorometer (Invitrogen, Life Technologies Corporation, Carlsbad, USA).
Oligonucleotide probes and biochip fabrication
Oligonucleotide probes for immobilization on the biochip (Supplementary Table 1) were synthesized on a 394 DNA/RNA synthesizer (Applied Biosystems, Carlsbad, USA) using standard phosphoramidite chemistry. All oligonucleotides carry an amino group at the 5′-terminus for immobilization in the polyacrylamide gel. The amino group was introduced during synthesis using a 3′-amino-modifier C7 CPG 500 (Glen Research, Sterling, USA). Some oligonucleotides included the LNA residues (Exiqon, Vedbaek, Denmark) in addition to the DNA residues to increase the sensitivity and specificity of the hybridization. Microarrays of polyacrylamide gel drops were prepared using a copolymerization method [86]. Each gel drop is duplicated to improve the reliability of the analysis.
Primers for amplification (Supplementary Table 2) were synthesized commercially (Evrogen, Moscow, Russian Federation).
We used LNA-containing clamping oligomers, which consisted of LNA and DNA residues or LNA residues only (Supplementary Table 3). The 3′-terminus was phosphorylated to prevent extension by Taq-polymerase. LNA-oligomers were synthesized on a 394 DNA/RNA synthesizer.
Preparation of DNA targets for hybridization
Target DNA samples were prepared through four parallel nested (two-round) multiplex PCR reactions. The first reaction was used to amplify KIT (codons 576 and 816) fragments; the second reaction was used to amplify BRAF and NRAS fragments; the third reaction was used to amplify fragments of KIT (codons 557 and 559, 642); and the fourth reaction was used to amplify the GNAQ, GNA11, MAP2K1 and MAP2K2 loci. The SNPdetect polymerase was used for the amplification because it does not have 5′ to 3′ exonuclease activity (Evrogene, Moscow, Russian Federation). The multiplex PCR was performed in a total volume of 12 μl, containing 1× SNPdetect buffer, 2.5 units SNPdetect polymerase, 0.2 mM dNTPs, 0.2 μM primers, 0.02–0.2 μM LNA-oligomers (the optimal concentration for each LNA-oligonucleotide was selected individually) and 5–12 ng genomic DNA. The cycling conditions were as follows: for 3 min and 30 s at 94°C, followed by 35 cycles of 30 s at 94°C, 20 s at 60°C, 10 s at 72°C and a final elongation at 72°C for 3 min. The PCR was performed on a T100 thermal cycler (Bio-Rad, Hercules, USA).
Each multiplex PCR (12 μl) in the second round contained 1× SNPdetect buffer, 2.5 units SNPdetect polymerase, 0.2 mM dNTPs, 0.2 μM forward primers and 2 μM reverse primers, 0.2 nM Cy5-dTTP and 1 μl of the first-round PCR product. The same program was used for the amplification. Thus, single-stranded fluorescently labeled PCR products were obtained.
Hybridization and image analysis
The hybridization and image analysis were performed as described before [61]. Briefly, hybridization was performed in 40 μl of 25% formamide (Serva, Oklahoma City, USA), 6× saline-sodium phosphate-EDTA (Promega, Fitchburg, USA) and 20 μl lebeled PCR products. The hybridization mixture was denatured at 95°C (5 min), briefly cooled on ice (2 min) and then applied on a biochip under a hybridization chamber and left overnight at 37°C. The chamber was disassembled, and the biochip was washed for 10 min with 50 ml of 1× saline-sodium phosphate-EDTA at room temperature and then dried. Hybridization signals were monitored by a portable chip analyzer (BIOCHIP-IMB, Moscow, Russian Federation). Data processing and image analysis were performed using dedicated software ImaGeWare version 3.5 (BIOCHIP-IMB, Moscow, Russian Federation). The fluorescence signals produced by the biochip’s gel spots were used as the input data as follows: Jm = (Im − I0)/(Bm − I0), where Im is the signal intensity per unit area in the internal region of a gel pad, Bm is the counterpart background intensity, I0 is the dark current in the charge-coupled device and m is the gel pad number. Because the gel spots were duplicated, the signal intensity was averaged. The assignment of mutation in a sample can be done if J(mut) >J(wt) and J(mut) / Bm ≥ 2.
Methods for validation
Sanger Sequencing
Sanger sequencing was used as a routine reference method for detecting mutations in the BRAF, NRAS, KIT, GNAQ, GNA11, MAP2K1 and MAP2K2 genes. For the identification of mutations in samples with low percentages of mutant DNA, we used LNA PCR clamp amplification followed by direct Sanger sequencing. The primers and LNA-oligomers for the sequencing analysis are shown in Supplementary Tables 4 and 3, respectively. For the amplification of most loci, we used the same primers that were used in the first-round of PCR for the biochip analysis.
PCR products were purified by ethanol precipitation of DNA with ammonium acetate and sequenced with the BigDyeTM Terminator 3.1 Cycle Sequencing Kit (Life Technologies Corporation, Carlsbad, USA) on an Applied Biosystems 3730 DNA Analyzer (Life Technologies Corporation, Carlsbad, USA) according to the manufacturer’s instructions. The sequencing results were interpreted using Chromas Lite software V.2.1 (Technelysium Pty, Helensvale, Australia).
Next generation sequencing
NGS (GS Junior, 454 Life Sciences, Branford, USA) was used to analyze mutations in 25 melanoma samples and 6 melanoma cell lines. NimbleGen technology (Roche, Basel, Switzerland) was used to target regions enrichment, including BRAF, NRAS, KIT, MAP2K1 and MAP2K2 genes.
A bioinformatic analysis of the results was performed using the GS Reference Mapper. Sequence reads were aligned to the human reference genome (HG19). Sequence coverage was, on average, 100-fold for target regions. Variants obtaining a frequency of detection ≥ 10% were considered for the analysis.
ARMS/Scorpion real-time PCR
A BRAF RGQ PCR Kit (Qiagen, Hilden, Germany) was used according to the manufacturer’s instructions. This assay is designed to detect a wild-type control and the four most common BRAF mutations V600E/K/R/D. Real-time PCR was performed on a Rotor-Gene Q Real-time PCR Platform (Qiagen, Hilden, Germany). The cycling conditions for quality control runs and mutation assays were as follows: 15 min at 95°C, followed by 40 cycles at 95°C for 30 s and 60°C for 1 min. Fluorescence was measured at 60°C. Data on each mutation were interpreted according to the kit manual after a curve analysis and calculation of ΔCt values.
Statistical analyses
The χ2 with Yates correction and two-sided Fisher exact tests were used to determine correlations between the qualitative clinicopathologic characteristics and mutation status of the patients. The Mann-Whitney U test was used to analyze the relationship between age and mutation status. All statistical tests were two-sided. A P-value of 0.05 was the statistical significance threshold. The GraphPad InStat, version 3.05 (GraphPad Software Inc., San Diego, USA) was used for all analyses.
Abbreviations
LNA, locked nucleic acid; NGS, next generation sequencing; WT, wild type; FFPE, formalin-fixed paraffin-embedded; PCR, polymerase chain reaction.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by the Russian Science Foundation (grant # 14-35-00107).
REFERENCES
1. MacKie RM, Hauschild A, Eggermont AM. Epidemiology of invasive cutaneous melanoma. Ann Oncol. 2009; 20:vi1–7.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7–30.
3. Woodman SE, Lazar AJ, Aldape KD, Davies MA. New strategies in melanoma: molecular testing in advanced disease. Clin Cancer Res. 2012; 18:1195–200.
4. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006; 24:4340–46.
5. Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Bröcker EB, LeBoit PE, Pinkel D, Bastian BC. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005; 353:2135–47.
6. Fisher DE, Barnhill R, Hodi FS, Herlyn M, Merlino G, Medrano E, Bastian B, Landi MT, Sosman J. Melanoma from bench to bedside: meeting report from the 6th international melanoma congress. Pigment Cell Melanoma Res. 2010; 23:14–26.
7. Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, Simpson EM, Barsh GS, Bastian BC. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009; 457:599–602.
8. Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, Sozen MM, Baimukanova G, Roy R, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010; 363:2191–99.
9. Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, Harshman K, Guipponi M, Bukach O, Zoete V, Michielin O, Muehlethaler K, Speiser D, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet. 2011; 44:133–39.
10. Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, Howle J, Saw R, Thompson JF, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014; 20:1965–77.
11. Trunzer K, Pavlick AC, Schuchter L, Gonzalez R, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, Kim KB, Weber JS, Hersey P, Long GV, Lawrence D, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013; 31:1767–74.
12. Wang H, Daouti S, Li WH, Wen Y, Rizzo C, Higgins B, Packman K, Rosen N, Boylan JF, Heimbrook D, Niu H. Identification of the MEK1(F129L) activating mutation as a potential mechanism of acquired resistance to MEK inhibition in human cancers carrying the B-RafV600E mutation. Cancer Res. 2011; 71:5535–45.
13. Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R, Cancer Genome Project. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004; 116:855–67.
14. Growney JD, Clark JJ, Adelsperger J, Stone R, Fabbro D, Griffin JD, Gilliland DG. Activation mutations of human c-KIT resistant to imatinib mesylate are sensitive to the tyrosine kinase inhibitor PKC412. Blood. 2005; 106:721–24.
15. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998; 279:577–80.
16. Hirota S, Nishida T, Isozaki K, Taniguchi M, Nakamura J, Okazaki T, Kitamura Y. Gain-of-function mutation at the extracellular domain of KIT in gastrointestinal stromal tumours. J Pathol. 2001; 193:505–10.
17. Kitayama H, Kanakura Y, Furitsu T, Tsujimura T, Oritani K, Ikeda H, Sugahara H, Mitsui H, Kanayama Y, Kitamura Y, Matsuzawa Y. Constitutively activating mutations of c-kit receptor tyrosine kinase confer factor-independent growth and tumorigenicity of factor-dependent hematopoietic cell lines. Blood. 1995; 85:790–98.
18. Taparowsky E, Shimizu K, Goldfarb M, Wigler M. Structure and activation of the human N-ras gene. Cell. 1983; 34:581–86.
19. Kalinec G, Nazarali AJ, Hermouet S, Xu N, Gutkind JS. Mutated alpha subunit of the Gq protein induces malignant transformation in NIH 3T3 cells. Mol Cell Biol. 1992; 12:4687–93.
20. Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989; 340:692–96.
21. Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, McCormick F, Rauen KA. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006; 311:1287–90.
22. Rodriguez-Viciana P, Rauen KA. Biochemical characterization of novel germline BRAF and MEK mutations in cardio-facio-cutaneous syndrome. Methods Enzymol. 2008; 438:277–89.
23. Lovly CM, Dahlman KB, Fohn LE, Su Z, Dias-Santagata D, Hicks DJ, Hucks D, Berry E, Terry C, Duke M, Su Y, Sobolik-Delmaire T, Richmond A, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS One. 2012; 7:e35309.
24. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011; 364:2507–16.
25. Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC, O’Day SJ, Blackman SC, Curtis CM, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012; 379:1893–901.
26. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363:809–19.
27. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA 3rd, Falchook G, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012; 367:1694–703.
28. Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, Dummer R, Trefzer U, Larkin JM, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012; 367:107–14.
29. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr, Kaempgen E, Martín-Algarra S, Karaszewska B, Mauch C, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012; 380:358–65.
30. Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, Hersey P, Kefford R, Lawrence D, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012; 366:707–14.
31. Eroglu Z, Ribas A. Combination therapy with BRAF and MEK inhibitors for melanoma: latest evidence and place in therapy. Ther Adv Med Oncol. 2016; 8:48–56.
32. Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014; 371:1867–76.
33. Hallmeyer S, Hamid O, Sorof TA, Mun Y, Liu S, Abhyankar S, Gibney GT, Puzanov I. Phase II study of vemurafenib in patients with locally advanced, unresectable stage IIIc or metastatic melanoma and activating exon 15 BRAF mutations other than V600E. J Clin Oncol. 2014; 32:5.
34. Antonescu CR, Busam KJ, Francone TD, Wong GC, Guo T, Agaram NP, Besmer P, Jungbluth A, Gimbel M, Chen CT, Veach D, Clarkson BD, Paty PB, Weiser MR. L576P KIT mutation in anal melanomas correlates with KIT protein expression and is sensitive to specific kinase inhibition. Int J Cancer. 2007; 121:257–64.
35. Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, Town A, Harlow A, Cruz F 3rd, Azar S, Rubin BP, Muller S, West R, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008; 14:6821–28.
36. Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, Panageas KS, Busam KJ, Chmielowski B, Lutzky J, Pavlick AC, Fusco A, Cane L, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011; 305:2327–34.
37. Lutzky J, Bauer J, Bastian BC. Dose-dependent, complete response to imatinib of a metastatic mucosal melanoma with a K642E KIT mutation. Pigment Cell Melanoma Res. 2008; 21:492–93.
38. Terheyden P, Houben R, Pajouh P, Thorns C, Zillikens D, Becker JC. Response to imatinib mesylate depends on the presence of the V559A-mutated KIT oncogene. J Invest Dermatol. 2010; 130:314–16.
39. Zhu Y, Si L, Kong Y, Chi Z, Yuan X, Cui C, Sheng X, Guo J, Shen L. Response to sunitinib in Chinese KIT-mutated metastatic mucosal melanoma. J Clin Oncol. 2009; 27:e20017.
40. Minor DR, Kashani-Sabet M, Garrido M, O’Day SJ, Hamid O, Bastian BC. Sunitinib therapy for melanoma patients with KIT mutations. Clin Cancer Res. 2012; 18:1457–63.
41. Woodman SE, Trent JC, Stemke-Hale K, Lazar AJ, Pricl S, Pavan GM, Fermeglia M, Gopal YN, Yang D, Podoloff DA, Ivan D, Kim KB, Papadopoulos N, et al. Activity of dasatinib against L576P KIT mutant melanoma: molecular, cellular, and clinical correlates. Mol Cancer Ther. 2009; 8:2079–85.
42. Gajiwala KS, Wu JC, Christensen J, Deshmukh GD, Diehl W, DiNitto JP, English JM, Greig MJ, He YA, Jacques SL, Lunney EA, McTigue M, Molina D, et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc Natl Acad Sci USA. 2009; 106:1542–47.
43. Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, Town A, McKinley A, Ou WB, Fletcher JA, Fletcher CD, Huang X, Cohen DP, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008; 26:5352–59.
44. Zhao J, Quan H, Xu Y, Kong X, Jin L, Lou L. Flumatinib, a selective inhibitor of BCR-ABL/PDGFR/KIT, effectively overcomes drug resistance of certain KIT mutants. Cancer Sci. 2014; 105:117–25.
45. Guo T, Hajdu M, Agaram NP, Shinoda H, Veach D, Clarkson BD, Maki RG, Singer S, Dematteo RP, Besmer P, Antonescu CR. Mechanisms of sunitinib resistance in gastrointestinal stromal tumors harboring KITAY502-3ins mutation: an in vitro mutagenesis screen for drug resistance. Clin Cancer Res. 2009; 15:6862–70.
46. Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB, MacConaill LE, Zhang J, Gray NS, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci USA. 2009; 106:20411–16.
47. Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen HY, Li B, Swoboda RK, Wilson M, Vultur A, Fukunaba-Kalabis M, Wubbenhorst B, Chen TY, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Reports. 2013; 4:1090–99.
48. Griewank KG, Yu X, Khalili J, Sozen MM, Stempke-Hale K, Bernatchez C, Wardell S, Bastian BC, Woodman SE. Genetic and molecular characterization of uveal melanoma cell lines. Pigment Cell Melanoma Res. 2012; 25:182–87.
49. Shi H, Moriceau G, Kong X, Koya RC, Nazarian R, Pupo GM, Bacchiocchi A, Dahlman KB, Chmielowski B, Sosman JA, Halaban R, Kefford RF, Long GV, et al. Preexisting MEK1 exon 3 mutations in V600E/KBRAF melanomas do not confer resistance to BRAF inhibitors. Cancer Discov. 2012; 2:414–24.
50. Harlé A, Salleron J, Franczak C, Dubois C, Filhine-Tressarieu P, Leroux A, Merlin JL. Detection of BRAF Mutations Using a Fully Automated Platform and Comparison with High Resolution Melting, Real-Time Allele Specific Amplification, Immunohistochemistry and Next Generation Sequencing Assays, for Patients with Metastatic Melanoma. PLoS One. 2016; 11:e0153576.
51. Skorokhod A, Helmbold P, Brors B, Schirmacher P, Enk A, Penzel R. Automated universal BRAF state detection within the activation segment in skin metastases by pyrosequencing-based assay U-BRAF(V600). PLoS One. 2013; 8:e59221.
52. Therascreen BRAF Pyro Kit Handbook. Qiagen. 2015. Available from: https://www.qiagen.com/kr/resources/resourcedetail?id=689cc7e9-cf08-4cc7-98a2-750a8009fc43&lang=en.
53. Foster JM, Oumie A, Togneri FS, Vasques FR, Hau D, Taylor M, Tinkler-Hundal E, Southward K, Medlow P, McGreeghan-Crosby K, Halfpenny I, McMullan DJ, Quirke P, et al. Cross-laboratory validation of the OncoScan® FFPE Assay, a multiplex tool for whole genome tumour profiling. BMC Med Genomics. 2015; 8:5.
54. Jung HS, Lefferts JA, Tsongalis GJ. Utilization of the oncoscan microarray assay in cancer diagnostics. Applied Cancer Research. 2017; 31:1.
55. INFINITI ® KRAS-BRAF Assay. AutoGenomics. 2013. Available from: http://www.autogenomics.com/pdf/EnglishPackageInsertforKRAS-BRAF.pdf.
56. GENOMICA. MULTIPLEX. MICROARRAY MDX. GENOMICA. 2014; Version 3. Available from: http://genomica.es/en/documents/GENOMICA_2014_English.pdf.
57. KRAS/BRAF/PIK3CA Array. Randox Molecular. Available from: http://www.randox.com/kras-colorectal-molecular-test/.
58. Kim IJ, Kang HC, Jang SG, Ahn SA, Yoon HJ, Park JG. Development and applications of a BRAF oligonucleotide microarray. J Mol Diagn. 2007; 9:55–63.
59. Galbiati S, Damin F, Pinzani P, Mancini I, Vinci S, Chiari M, Orlando C, Cremonesi L, Ferrari M. A new microarray substrate for ultra-sensitive genotyping of KRAS and BRAF gene variants in colorectal cancer. PLoS One. 2013; 8:e59939.
60. Weyant GW, Wisotzkey JD, Benko FA, Donaldson KJ. BRAF mutation testing in solid tumors: a methodological comparison. J Mol Diagn. 2014; 16:481–85.
61. Emelyanova M, Arkhipova K, Mazurenko N, Chudinov A, Demidova I, Zborovskaya I, Lyubchenko L, Zasedatelev A, Nasedkina T. Sensitive genotyping of somatic mutations in the EGFR, KRAS, PIK3CA, BRAF genes from NSCLC patients using hydrogel biochips. Appl Immunohistochem Mol Morphol. 2015; 23:255–65.
62. Tsiatis AC, Norris-Kirby A, Rich RG, Hafez MJ, Gocke CD, Eshleman JR, Murphy KM. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. J Mol Diagn. 2010; 12:425–32.
63. Angulo B, García-García E, Martínez R, Suárez-Gauthier A, Conde E, Hidalgo M, López-Ríos F. A commercial real-time PCR kit provides greater sensitivity than direct sequencing to detect KRAS mutations: a morphology-based approach in colorectal carcinoma. J Mol Diagn. 2010; 12:292–99.
64. Wood HM, Foster JM, Taylor M, Tinkler-Hundal E, Togneri FS, Wojtowicz P, Oumie A, Spink KG, Brew F, Quirke P. Comparing mutation calls in fixed tumour samples between the affymetrix OncoScan® array and PCR based next-generation sequencing. BMC Med Genomics. 2017; 10:17.
65. Colombino M, Capone M, Lissia A, Cossu A, Rubino C, De Giorgi V, Massi D, Fonsatti E, Staibano S, Nappi O, Pagani E, Casula M, Manca A, et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J Clin Oncol. 2012; 30:2522–29.
66. Yilmaz I, Gamsizkan M, Kucukodaci Z, Berber U, Demirel D, Haholu A, Narli G. BRAF, KIT, NRAS, GNAQ and GNA11 mutation analysis in cutaneous melanomas in Turkish population. Indian J Pathol Microbiol. 2015; 58:279–84.
67. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, et al. A landscape of driver mutations in melanoma. Cell. 2012; 150:251–63.
68. Ekedahl H, Cirenajwis H, Harbst K, Carneiro A, Nielsen K, Olsson H, Lundgren L, Ingvar C, Jönsson G. The clinical significance of BRAF and NRAS mutations in a clinic-based metastatic melanoma cohort. Br J Dermatol. 2013; 169:1049–55.
69. Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, Hughes TM, Thompson JF, Scolyer RA, Kefford RF. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011; 29:1239–46.
70. Jung JE, Falk TM, Bresch M, Matias JE, Böer A. BRAF mutations in cutaneous melanoma: no correlation with histological prognostic factors or overall survival. J Bras Patol Med Lab. 2010; 46:487–93.
71. Menzies AM, Haydu LE, Visintin L, Carlino MS, Howle JR, Thompson JF, Kefford RF, Scolyer RA, Long GV. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin Cancer Res. 2012; 18:3242–49.
72. Lee HW, Song KH, Hong JW, Jeon SY, Ko DY, Kim KH, Kwon HC, Lee S, Kim SH, Kim DC. Frequency of BRAF Mutation and Clinical Relevance for Primary Melanomas. Korean J Pathol. 2012; 46:246–52.
73. Chang DZ, Panageas KS, Osman I, Polsky D, Busam K, Chapman PB. Clinical significance of BRAF mutations in metastatic melanoma. J Transl Med. 2004; 2:46.
74. Johnson DB, Lovly CM, Flavin M, Panageas KS, Ayers GD, Zhao Z, Iams WT, Colgan M, DeNoble S, Terry CR, Berry EG, Iafrate AJ, Sullivan RJ, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res. 2015; 3:288–95.
75. Sheen YS, Liao YH, Liau JY, Lin MH, Hsieh YC, Jee SH, Chu CY. Prevalence of BRAF and NRAS mutations in cutaneous melanoma patients in Taiwan. J Formos Med Assoc. 2016; 115:121–27.
76. Edlundh-Rose E, Egyházi S, Omholt K, Månsson-Brahme E, Platz A, Hansson J, Lundeberg J. NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: a study based on mutation screening by pyrosequencing. Melanoma Res. 2006; 16:471–78.
77. Lee JH, Choi JW, Kim YS. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: a meta-analysis. Br J Dermatol. 2011; 164:776–84.
78. Jafari M, Papp T, Kirchner S, Diener U, Henschler D, Burg G, Schiffmann D. Analysis of ras mutations in human melanocytic lesions: activation of the ras gene seems to be associated with the nodular type of human malignant melanoma. J Cancer Res Clin Oncol. 1995; 121:23–30.
79. Omholt K, Platz A, Kanter L, Ringborg U, Hansson J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003; 9:6483–88.
80. Reifenberger J, Knobbe CB, Sterzinger AA, Blaschke B, Schulte KW, Ruzicka T, Reifenberger G. Frequent alterations of Ras signaling pathway genes in sporadic malignant melanomas. Int J Cancer. 2004; 109:377–84.
81. Mikhaylova I, Baryshnikov A, Demidov L, Kiselev S, Burova O, Morozova L. Cell line of human melanoma mel rac, applied for obtaining antitumour vaccine. Russia; Patent RU 2402602, 2010.
82. Mikhaylova I, Baryshnikov A, Demidov L, Kiselev S, Burova O, Morozova L. Cell line of human melanoma mel z, applied for obtaining antitumour vaccine. Russia; Patent RU 2390556, 2010.
83. Mikhaylova I, Baryshnikov A, Demidov L, Kiselev S, Burova O, Morozova L, Vorojtsov G. Cell line of human melanoma mel cher, applied for obtaining antitumour vaccine. Russia; Patent RU 2364624, 2009.
84. Mikhaylova I, Baryshnikov A, Morozova L, Burova O, Demidov L, Lukashina M, Larin S, Georgiev G, Vorojtsov G, Gnuchev N. Cell line of human melanoma mel il, applied for obtaining antitumour vaccine. Russia; Patent RU 2287577, 2006.
85. Mikhaylova I, Baryshnikov A, Morozova L, Berejnoy A, Kozlova A, Larin S, Georgiev G, Vorojtsov G, Gnuchev N. Cell line of human melanoma mel ibr, applied for obtaining antitumour vaccine. Russia; Patent RU 2287576, 2006.
86. Rubina AY, Pan’kov SV, Dementieva EI, Pen’kov DN, Butygin AV, Vasiliskov VA, Chudinov AV, Mikheikin AL, Mikhailovich VM, Mirzabekov AD. Hydrogel drop microchips with immobilized DNA: properties and methods for large-scale production. Anal Biochem. 2004; 325:92–106.