
INTRODUCTION
CD20 is a glycosylated phosphoprotein expressed on most mature B-cells and a clinically validated target for non-Hodgkin’s lymphoma (NHL) and autoimmune diseases. Rituximab was the first anti-CD20 monoclonal antibody approved by the US Food and Drug Administration (FDA) for the treatment of patients with B-cell lymphomas [1]. Typically, anticancer monoclonal antibodies (mAbs) mediate antitumor effects via a variety of mechanisms, including cell cycle arrest signals, direct induction of apoptosis, and sensitization to cytotoxic drugs [2]. Antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-mediated cytotoxicity (CDC) are additional important mechanisms in tumor therapy [2]. All clinically approved antitumor antibodies have the IgG isotype because of their long half-life, and technologies have been established for large-scale production and purification.
IgA, the most abundant antibody class found on mucosal surfaces, plays an important role in the immune system [2]. FcαRI (CD89) is the most important IgA receptor expressed on polymorphonuclear cells (PMNs), monocytes, macrophages, and Kupffer cells [3]. FcαRI plays an important role in triggering various immunological effector functions, including induction of an oxidative burst, phagocytosis, and ADCC [2].
IgA antitumor antibodies have been shown to mediate efficient tumor lysis both in vitro [2] and in vivo using human FcαRI transgenic (Tg) mice [2]. However, daily injections of IgA antibodies are needed to achieve ideal antitumor effects in vivo due to the short half-life (only 15 h) of IgA in mice. Moreover, a tandem molecule containing IgG1 and IgA2, which has a half-life similar to that of IgG, has been shown to exhibit more potent antitumor activity, regulating natural killer (NK) cell- or PMN-mediated ADCC and macrophage-mediated antibody-dependent cell-mediated phagocytosis (ADCP), as described by Borrok et al [2]. However, the antitumor activity of IgG1/IgA2 antibodies has not been evaluated in vivo.
The innate mononuclear phagocyte network is the predominant effector cell population utilized by rituximab (anti-CD20) in vivo [2]. In histological sections of tumors, tumor-associated macrophages constitute the major proportion of the leukocyte tumor infiltrate, particularly for solid tumors [2]. In vitro studies have also found that both tumor-killing M1 and tumor-helper M2 macrophages are able to kill tumor cells in the presence of rituximab [2]. Additionally, IgA anti-epidermal growth factor receptor (EGFR) antibodies induce potent antitumor activity via M0, M1, and M2 macrophages [2].
In this study, we investigated the in vivo antitumor effects of an anti- CD20-IgG/IgA molecule using transgenic mice expressing CD89 on monocytes and macrophages. Our results establish a unique model to study the in vivo interactions of both IgG and IgA Fc with mononuclear phagocytes and have implications in the improved treatment of solid tumors.
RESULTS
Anti-CD20 antibodies mediated ADCC by mouse effector cells in vitro
To assess the antitumor activity of anti-CD20 antibodies, we previously generated CD20-IgGA by fusing the IgA2-CH2-CH3 region to rituximab (CD20-IgG) with the (G4S)3 linker. For CD20-IgA, the variable region of rituximab was fused to the IgA2m (1) constant region (Supplementary Figure 1). Rituximab was used as a CD20-IgG control. Consistent with Her2-IgGA, which was constructed in another study [2], CD20-IgGA and CD20-IgA were confirmed to have similar target affinity (Supplementary Table 1) and were as potent as rituximab in eliciting Fab-mediated antitumor effects because both constructs shared the same variable region. The antibodies were produced by transient transfection of HEK293F cells with similar levels of expression level exhibited by both CD20-IgG and CD20- IgGA constructs (Supplementary Figure 1). In accordance with previous findings, CD20-IgGA could induce ADCC mediated by either peripheral blood mononuclear cells (PBMCs) or PMNs [2]. Tumor lysis by PBMCs, which include NK cells, was only efficient with CD20-IgG. Notably, PMNs are the only blood-resident effector cells that mediate CD20-IgA activity [2]. CD20-IgGA exhibited stronger ADCC activity than CD20-IgG and CD20-IgA using human whole blood (PBMCs and PMNs) as effectors (Supplementary Figure 2).
To investigate whether anti-CD20 antibodies mediate antitumor activity by mouse blood effector cells, we generated a human FcαRI (CD89) Tg mouse strain using an authentic murine CD14 promoter with CD89 expression restricted to blood and tissue monocytes/macrophages [4]. A previous study showed that monocytes/macrophages mediate ADCC in an overnight assay [2]. Therefore, we used isolated CD14-positive monocytes from FcαRI Tg mice as effector cells in a 20-h assay. CD20-IgGA and CD20-IgA mediated potent tumor lysis, which exceeded tumor lysis mediated by CD20-IgG (Figure 1A). Wild-type (WT) monocytes were not able to lyse target cells using CD20-IgA, while tumor lysis by CD20-IgGA was similar to that mediated by CD20-IgG (Figure 1B), indicating that the cytotoxic activity of IgA Fc ex vivo was fully dependent on the presence of FcαRI. Monocyte-depleted PBMCs were able to efficiently lyse target cell utilizing both CD20-IgG and CD20-IgGA, through mechanisms likely mediated by NK cells, indicating that NK-mediated ADCC activity could be induced by CD20-IgGA (Figure 1A). As expected, CD20-IgGA exhibited greater cytotoxicity than CD20-IgG and CD20-IgA using PBMCs from Tg mice (which include monocytes and NK cells) as effector cells against Raji cells in an overnight assay. Because mouse PMNs do not express FcγRIa or FcγRIIIb [2], mouse PMN assays were not carried out.
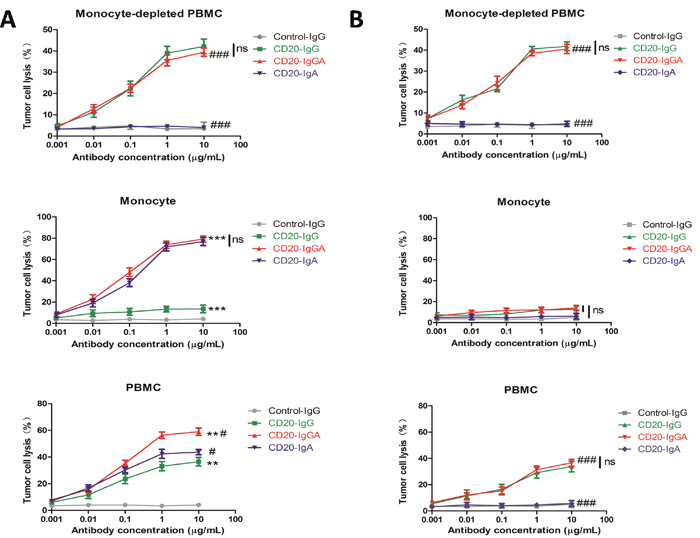
Figure 1: ADCC with different mouse effector cells ex vivo. ADCC results are shown using Raji cells as targets and the mouse monocyte-depleted PBMC fraction, isolated CD14+monocytes, or PBMCs (E:T = 40:1) from FcaRI Tg mice (A) or wild-type C57BL/6 mice (B) in the presence of anti-CD20 antibodies. For monocytes and PBMCs, a 20-h ADCC assay was used. For monocyte-depleted PBMCs, a 4-h ADCC assay was used. All data are presented as the mean ± SEM (n = 3) from one of three representative experiments. *CD20-IgGA group versus CD20-IgG group; #CD20-IgGA group versus CD20-IgA group. **P < 0.01; ***P < 0.001; #P < 0.05; ###P < 0.001; ns, not statistically significant by two-way ANOVA.
Taken together, these results showed that CD20-IgGA was more efficient than CD20-IgG or CD20-IgA in tumor cell killing by both human myeloid effector cells and Tg mouse PBMCs.
Macrophage-mediated ADCP induced by anti-CD20 antibodies in vitro
Next, we investigated whether macrophages could be recruited by anti-CD20 antibodies to mediate ADCP against Raji cells. Macrophages express high levels of FcαRI/FcγRs and can eliminate pathogenic cells primarily via phagocytosis [2]. Bone marrow-derived macrophages (BMDMs) of FcαRI Tg and WT C57BL/6 mice were used to evaluate the ability of anti-CD20 antibodies to facilitate ADCP. After a 4-h incubation in the presence of anti-CD20 antibodies, phagocytosis of carboxyfluorescein succinimidyl ester (CFSE)-Raji cells by macrophages was observed by confocal microscopy (Figure 2A). Phagocytosis was also evaluated by fluorescence-activated cell sorting (FACS); macrophages were labeled with APC-conjugated anti-mouse F4/80 antibodies and incubated with target cells labeled with CFSE in the presence of anti-CD20 antibodies. A PE anti-CD19 antibody was also used as the control to discriminate between target cell adhesion and phagocytosis. Phagocytosis was reported as the fraction of triple-positive cells relative to the total number of tumor cells in the sample (Figure 2B). CD20-IgGA and CD20-IgA were able to effectively clear up to 60% of the targeted tumor cells, a much higher degree of phagocytosis than observed with CD20-IgG. The results shown in Figure 2A mirrored those of ADCP assays analyzed by FACS. Only the IgA2-Fc-containing formats, CD20-IgA and CD20-IgGA, exhibited obvious phagocytosis compared with the CD20-IgG and control IgG. These observations were consistent with those of previous studies [2].
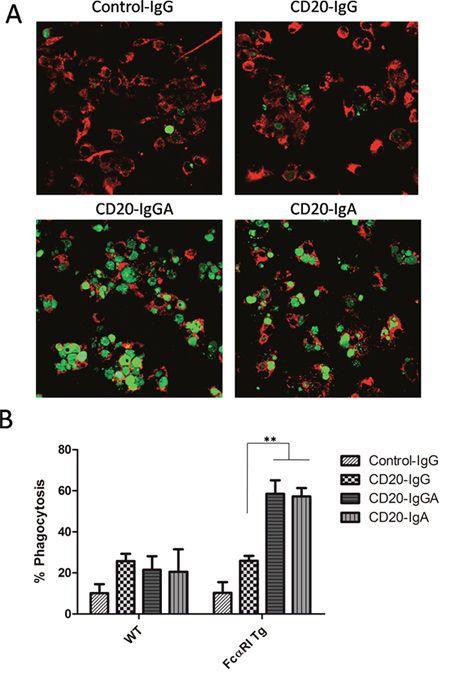
Figure 2: ADCP assays with mouse BMDMs. In vitro assay to determine elimination of Raji cells by macrophages. (A) BMDMs from WT and FcαRI Tg C57BL/6 mice were incubated with CFSE-labeled Raji cells at an E:T ratio of 5:1 in the presence of 10 μg/mL anti-CD20 antibodies. After a 4-h incubation, cells were transferred to a new tube and visualized via confocal microscopy. Representative images of ADCP mediated by anti-CD20 antibodies are shown. (B) BMDMs were labeled with APC-F4/80 antibodies and cocultured with CFSE-labeled Raji cells in the presence of the indicated antibodies. Phagocytosis of Raji cells was analyzed by FACS and quantified as the percentage of double-positive cells relative to total CFSE-positive cells and F4/80+cells. **P < 0.01 using unpaired two-tailed t tests.
Pharmacokinetics (PK) analysis of anti-CD20 antibodies in C57BL/6 mice
Human IgA antibodies do not bind to FcRn; therefore, the half-life of mouse or human IgA in mice is much shorter than that of human IgG, estimated to be in the range of 12–48 h [2]. Because CD20-IgGA contains both IgG Fc and IgA Fc, the IgGA antibody is expected to be recycled efficiently by FcRn, resulting in IgG1-like serum persistence (Supplementary Table 1). To determine the serum half-life of anti-CD20 antibodies in C57BL/6 mice, we injected 100 μg CD20-IgGA, CD20-IgG, or CD20-IgA intravenously (i.v.) and measured the serum antibody concentration at the indicated time points (Figure 3). As expected, CD20-IgGA exhibited a clearance rate similar to that of the parental IgG1 molecule, with half-lives of 4.2 and 4.4 days (Table 1), respectively, suggesting that FcRn recycling was not influenced by the IgA Fc region. In contrast, CD20-IgA exhibited rapid clearance, showing a substantially shorter serum half-life in mice than IgGA and IgG (half-life of 10.3 h; Table 1). These findings were consistent with a previously published study [5].
Table 1: Parameter of pharmacokinetics
Parameter |
CD20-IgG |
CD20-IgGA |
CD20-IgA |
|---|---|---|---|
t1/2 (h) |
106.54 |
100.57 |
10.33 |
CL(L/h) |
14.96 |
17.9 |
36.88 |
MRT (h) |
147.92 |
136.43 |
24.05 |
t1/2: Elimination Half-life
CL: clearance
MRT: mean residence time.
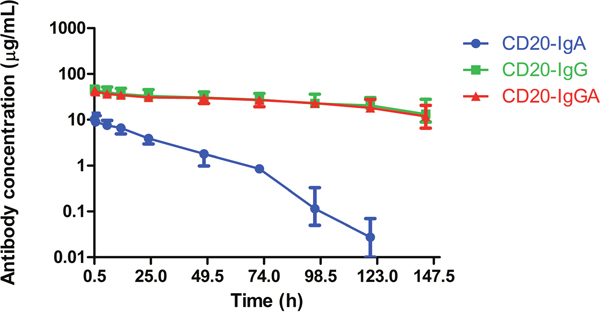
Figure 3: Serum half-life of anti-CD20 antibodies in C57BL/6 mice. Serum levels for CD20-IgG (green), CD20-IgGA (red), and CD20-IgA (blue) in C57BL/6 mice (n = 6/group) were quantified by ELISA for detection of human IgG and IgA. Mice were intravenously (i.v.) injected with 100 μg anti-CD20 antibodies, and blood samples were collected at the indicated time points.
Comparison of the in vivo antitumor activities of three types of anti-CD20 antibodies
Because CD20-IgGA and CD20-IgA exhibited stronger ADCC and ADCP than CD20-IgG in vitro with both human and mouse effector cells, we next performed in vivo studies to assess the antitumor activities of the panel of anti-CD20 antibodies. To study the ADCC and ADCP effects mediated by IgG- and IgA-Fc domains in the C57BL/6 background, LLC-CD20 mice were generated using the mouse LLC cell line, which was transduced with a human CD20 gene to facilitate tumor growth in C57BL/6 mice. Because CD20-transfected LLC cells lacked signaling pathways related to cell growth and cell apoptosis, Fab-dependent effector mechanisms did not contribute to the antitumor effects in this model, which allowed us to focus on Fc-dependent killing.
Mice were inoculated with LLC-CD20 cells and treated with CD20-IgGA, CD20-IgG, or CD20-IgA. Treatment of tumor-bearing mice with CD20-IgGA induced significant and persistent tumor regression in Tg mice; this antitumor activity was better than that of CD20-IgG and CD20-IgA (Figure 4A). Thus, the IgA Fc exerted greater antitumor activities than IgG Fc in the Tg model. In contrast, no differences were observed between CD20-IgGA and IgG treatment, and CD20-IgA treatment had almost no effect in WT mice (Figure 4B), indicating that in vivo antitumor activity was strictly dependent on the presence of FcαRI.
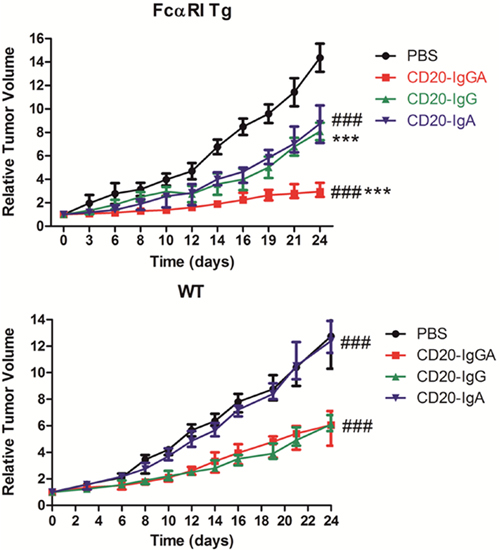
Figure 4: Comparison of the efficacy of anti-CD20 antibodies in the LLC-CD20 xenograft model. FcαRI Tg (A) or WT (B) C57BL/6 mice were injected s.c. with 1 × 106 LLC-CD20 cells. Mice bearing xenografts (tumor size ~100–150 mm3) were administered 5 mg/kg CD20-IgG and CD20-IgGA (n = 6–8/group) via i.v. injection on days 0, 4, 8, 12, 16, and 20 or 5 mg/kg CD20-IgA daily from day 0 to day 23. *CD20-IgGA-treated group versus CD20-IgG-treated group; #CD20-IgGA-treated group versus CD20-IgA-treated group. ***P < 0.001 and ###P < 0.001 using two-way ANOVA.
Macrophage-mediated anti-CD20 antibody activity in an intraperitoneal model
Next, we used a peritoneal model to analyze effector-target interactions and antitumor mechanisms of antibodies (Figure 5A), as previously reported [2]. Our results showed that CD20-IgG treatment did not induce significant killing of LLC cells (Figure 5C). However, CD20-IgGA and CD20-IgA still showed significant cytotoxicity in this intraperitoneal model in the FcαRI Tg background. In WT mice, IgGA and IgA were not effective, indicating that the Fab region did not contribute to the antitumor activity under these conditions (Figure 5D). Effector cells in the peritoneum include macrophages, resident monocytes, and PMNs. Thus, it is likely that peritoneal macrophages and monocytes were the main effector cells responsible for the cytotoxicity induced by IgA Fc in this model because PMNs do not express mouse FcγRs and human FcαRI.
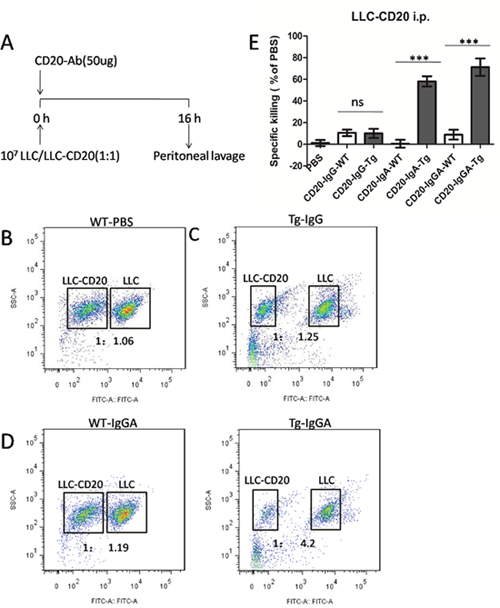
Figure 5: Macrophages mediated in vivo anti-CD20 antibody activity in a peritoneal model using LLC-CD20 cells. (A) The LLC-CD20 i.p. model was generated by injection of a 1:1 mixture of LLC and LLC-CD20 cells (1 × 107 cells) differentially labeled with high and low concentrations of CFSE into the peritoneum of FcαRI Tg or WT BALB/c mice (n = 6/group). Immediately after injection of the cells, the mice received 50 μg anti-CD20 antibodies. After 16 h, the peritoneum was washed, and the ratios of CSFE+LLC to CSFE++LLC-CD20 cells were analyzed to calculate specific cytotoxicity. (B–D) Representative dot plots showing cells retrieved from the peritoneum. LLC and LLC-CD20 cells could be identified based on CFSE labeling. (E) Specific cytotoxicity was calculated from the ratio of LLC to LLC-CD20 cells in each treatment group. ***P < 0.001; ns, not statistically significant using unpaired two-tailed t tests.
Taken together, these findings suggested that IgA Fc mediated efficient in vivo cytotoxicity of LLC-CD20 cells in a short-term peritoneal model and that these effects were fully dependent on the IgA Fc interaction with FcαRI on macrophages.
DISCUSSION
Currently, all of the therapeutic antibodies approved by the FDA are IgG antibodies. IgG can induce NK cell-mediated ADCC, CDC, and apoptosis in vivo. Although IgG can efficiently activate FcγRs (FcγRIIIa and FcγRIIa) and induce ADCC, IgG may interact with inhibitory FcγRIIb and FcγRIIIb on several types of effector cells, thereby weakening the cytotoxicity of effector cells [2]. IgA is the most abundantly expressed immunoglobulin on mucosal surfaces and exhibits antimicrobial effects [2]. Additionally, IgA can activate effector cells to stimulate a respiratory burst, phagocytosis, antigen presentation, and ADCC by binding to CD89 [6]. However, IgA antibody therapy faces development changes due problems with aggregation and a shorter half-life versus IgG antibodies [2]. Therefore, IgG/IgA “cross-isotype” molecules may be benefitted by providing advantages of both IgG and IgA [2]. Moreover, some studies have shown that FcγR pathways and functions do not overlap with those of FcαRI on the same effector cells [2]. In an in vitro study, monocytes, which co-express FcγRs and FcαRI, were found to display increased cytotoxicity with a combination of IgG and IgA antibodies in an overnight killing assay [7].
In this study, we constructed hybrid CD20-IgG/IgA molecules having the structure of CD20 IgG-linker-IgACH2CH3. The IgGA construct exhibited an enhanced ability to induce ADCC by human blood cells by engaging cytotoxic PMNs and NK cells. Importantly, CD20-IgGA had a half-life similar to that of IgG (about 5 days), whereas CD20-IgA had a half-life of only 16.8 h, consistent with published studies showing a half-life of 12–48 h [7]. IgGA Fc also retained C1q binding (Supplementary Table 1) and exhibited CDC activity against CD20-expressing Raji cells (data not shown). These results showed that the hybrid IgG/IgA molecules possessed the advantages of both IgG and IgA antibody types.
The hybrid IgG/IgA construct exhibited antitumor activity in a unique Tg mouse model expressing CD89 controlled by the murine CD14 promotor, restricting expression to monocytes, macrophages, and Kupffer cells. In addition to PMNs and NK cells, monocytes and macrophages have been shown to function as potent effector cells in ADCC and phagocytosis [2]. Although monocytes and macrophages express both IgG and IgA FcR, CD20-IgG/IgA and CD20-IgA showed better ADCC and ADCP effects than IgG in vitro, and CD20-IgG/IgA showed significant antitumor effects in vivo using LLC-CD20 cells in the Tg-C57BL/6 model. Thus, the mononuclear phagocyte network was the predominant effector cell population in vivo.
Despite the absence of PMN-mediated cytotoxicity, IgGA could facilitate ADCC and ADCP from both NK cells and monocytes/macrophages, consistent with a previous study in which direct activation of macrophages by IgA was demonstrated in vivo [2]. Boross et al provided evidence that in CD89 Tg mice, IgA antibodies could induce phagocytosis of tumor cells by macrophages in a short-term peritoneal model [2]. Similarly, CD20-IgGA and IgA also showed significant cytotoxicity in our intraperitoneal model in the FcαRI Tg background.
Although our model may not mimic the situation in humans because of the restriction of CD89 expression to monocytes and macrophages, our findings of the activation of monocytes and macrophages are important, particularly considering the abundant infiltration of tumor-associated macrophages in solid tumors. Future studies are needed to explore the possibility of using CD89 as a potent activator of PMNs, monocytes, and macrophages to augment antibody-mediated antitumor therapy.
MATERIALS AND METHODS
Mice
Human FcaRI Tg mice were generated with the murine CD14 promoter controlling the expression of CD89, as previously described [4]. A knock-in targeting vector was designed that contained a cDNA encoding the human CD89 cDNA, the 2A self-processing peptide, and 5.2 kb of a sequence homologous to the murine CD14 gene. Homologous recombinant embryonic stem (ES) cells were micro-injected into C57BL/6 blastocysts. The resulting chimeric mice were crossed with C57BL/6 mice, and a stable line was established in the C57BL/6 background. Littermates not expressing the human CD89 gene were used as controls.
All animal experiments were approved by the Animal Ethics Committee of Tongji University and were in accordance with the Association for Research in Vision and Ophthalmology (ARVO) statement for the use of Animals in Ophthalmic and Vision Research. C57BL/6 mice were housed in a pathogen-free animal facility at the experimental animal center of Tongji University. Mice were fed standard chow and provided with distilled water ad libitum. Cages were refreshed weekly. Mice were euthanized using sodium pentobarbital, and appropriate efforts were made to minimize animal suffering.
Cell lines
Raji and Lewis lung cancer (LLC) cell lines (ATCC) were cultured in RPMI1640 or Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS). LLC cells were transfected with the human CD20 gene (Addgene), and CD20-expressing clones were selected using cytofluorimetry.
Antibody expression and purification
Rituximab (accession number: DB00073) was used as the CD20-IgG control in this paper. The CD20-IgGA used in this study was generated by fusing human IgA2-CH2-CH3 to the C-terminus of rituximab with a (G4S)3 linker. The variable region of the HC and LC for the generation of CD20-IgA was derived from rituximab. All antibodies were transiently expressed in HEK293F cells in Freestyle medium (Life Technologies). At 5–6 days after transfection, the cell suspension was centrifuged for purification. IgG and IgGA antibodies were purified by standard protein A affinity chromatography (Life Technologies). IgA2 was purified via Peptide M agarose resin (Invivogen). Samples were buffer-exchanged into 1× phosphate-buffered saline (PBS) using Amicon Ultra-4 (Millipore) spin columns with a 30-kDa cutoff, and the purity of purified samples was assessed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using 10% gradient gels.
ADCC assay
CytoTox96 nonradioactive assays (Promega) were used to evaluate the capacity of mouse blood cells to trigger lysis of tumor cells. PBMCs were isolated from freshly drawn peripheral blood of CD89 Tg and WT mice using mouse Percoll (Sigma) following the manufacturer’s directions. Monocytes were isolated from the PBMC fraction using CD14-positive microbeads (Miltenyi Biotech). PBMCs without monocytes were used as effector cells. Effector cells were plated (8 × 104cells/well) in 96-well plates. Raji cells (2 × 103 cells/well) were also transferred to the same plates, and cultures were incubated with recombinant anti-CD20 antibodies at different concentrations (0.001, 0.01, 0.1, 1.0, or 10.0 μg/mL) at 37°C for 4 h for monocyte-depleted PBMC assays or 20 h for monocytes and PBMCs. An IgG isotype (Abcam) was used as a negative control. The lactate dehydrogenase (LDH) released into the medium following challenge with anti-CD20 antibodies was quantified by measuring the absorbance at 490 nm. The LDH activity in supernatants from cocultures of effector cells and Raji cells, effector cells cultured in the absence of the cell lysis target, and Raji cells cultured in the absence of effector cells was used to calculate the corrected experimental, effector, and target values, respectively. These values were then used to calculate cytotoxicity expressed as a percentage of the target cells according to routine procedures.
ADCP analysis
BMDMs of mice were isolated and cultured as previously described [8]. Macrophages were seeded at 5 × 104 cells/well in 96-well plates, and CSFE-Raji cells (1 × 104cells/well; 10 mM; Life Technologies) were also transferred to the same plates and were incubated with 5.0 μg/mL anti-CD20 antibodies at 37°C. After 4 h, adherent cells were detached and stained with APC-conjugated anti-mouse F4/80 antibody (F4/80-APC; BD Pharmingen). The PE-conjugated anti-human CD19 antibody (CD19-PE; BD Pharmingen) was used as a control to discriminate between target cell adhesion and phagocytosis. Phagocytosis was evaluated by FACS on a FACSVerse (BD Bioscience) and reported as the fraction of triple-positive cells over the total number of tumor cells in the sample.
For microscopy experiments, macrophages and CFSE-Raji cells were incubated on cover slips in 24-well plates at the appropriate effector tumor cell ratio (5:1) along with 5.0 μg/mL anti-CD20 antibodies at 37°C for 4 h. Subsequently, cells were fixed, followed by permeabilization, staining of macrophages using a rabbit anti-mouse F4/80 antibody and a goat anti-rabbit IgG-Cy3 antibody (Boster Biotech, Wuhan, China), nuclear staining by 4',6-diamidino-2-phenylindole (DAPI; Boster Biotech), and confocal microscopy according to routine procedures.
PK study
IgG1 CD20, Ig1/IgA2 CD20, or IgA2 CD20 (100 μg/mouse) was injected intravenously into C57BL/6 mice (four mice/group). Blood was collected via the tail vein from alternating mice at the indicated time points. The human IgA antibody concentrations in the sera were determined by enzyme-linked immunosorbent assays (ELISAs) using human IgA ELISA kits (Bethyl Laboratories), and human IgG and IgGA antibody concentrations in the sera were determined using human IgG ELISA Kits (Bethyl Laboratories) according to the manufacturer’s instructions. The data were analyzed by PK solver software, as previously described [2].
LLC-CD20 models
LLC cells were subjected to lentivirus-mediated transduction of human CD20. A clone was established that reproducibly grew in WT C57BL/6 mice. LLC-CD20 cells (1 × 106) were subcutaneously (s.c.) injected into WT and FcαRI Tg C57BL/6 mice. Animals were randomly assigned into treatment groups (6–8 per group), with the mean tumor volume for each group being 100–150 mm3. Tumor size was monitored twice a week, and tumor volumes were determined according to the formula: tumor volume (mm3) = longer diameter × (shorter diameter)2 × 0.5. PBS was administered as the vehicle control for all groups. IgG-CD20 and IgGA-CD20 (5 mg/kg) were administered intravenously (i.v.) twice a week for 3 weeks (days 0, 4, 8, 12, 16, and 20). CD20- IgA (5 mg/kg) was administered i.v. daily beginning from day 0 to day 23.
LLC peritoneal model
The LLC peritoneal model was established as previously described [9]. LLC and LLC-CD20 cells were labeled with 0.1 μM and 1 μM CFSE (Life Technologies) and mixed at a 1:1 ratio. Next, 107 cells were intraperitoneally injected into WT and FcαRI Tg C57BL/6 mice (six per group). Anti-CD20 antibodies (50 μg/mouse) were injected intraperitoneally into mice directly after injection of tumor cells. Sixteen hours later, the mice were euthanized, and the peritoneum was washed. The ratio of LLC cells (CSFE+) to LLC-CD20 cells (CSFE++) was determined by cytofluorimetry.
Statistical analysis
Data were graphed and analyzed using GraphPad Prism 6.0 (Graph Pad Software). Data are expressed as the median and range or mean ± standard error of the mean (SEM). Quantitative data between groups were compared using Student’s t test with unpaired two-tailed t tests or two-way analysis of variance (ANOVA). Differences with P values of less than 0.05 were considered statistically significant.
Abbreviations
NHL, non-Hodgkin’s lymphoma; FDA, US Food and Drug Administration; ADCC, antibody-dependent cell-mediated cytotoxicity; CDC, complement-mediated cytotoxicity; PMN, polymorphonuclear leukocyte; Tg, transgenic; NK, natural killer; ADCP, antibody-dependent cell-mediated phagocytosis; EGFR, epidermal growth factor receptor; ES, embryonic stem; LLC, Lewis lung cancer; FBS, fetal bovine serum; DMEM, Dulbecco’s modified Eagle’s medium; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; PBS, phosphate-buffered saline; PBMCs, peripheral blood mononuclear cells; WT, wild-type; LDH, lactate dehydrogenase; BMDM, bone marrow-derived macrophage; PK, pharmacokinetics; CFSE, carboxyfluorescein succinimidyl ester.
ACKNOWLEDGMENTS
We thank all members of our laboratory for their helpful suggestions and support. This study was supported by grants from the National Natural Science Foundation of China (NSFC31470896, and NSFC81402399), National Basic Research Program of China (973 Program) 2015CB553706, and Suzhou Applicational Basic Project, China (SYG201509).
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
1. Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene. 2003; 22:7359-7368.
2. Weiner GJ. Rituximab: mechanism of action. Seminars in hematology. 2010; 47:115-123.
3. Cerny T, Borisch B, Introna M, Johnson P, Rose AL. Mechanism of action of rituximab. Anti-cancer drugs. 2002; 13:S3-10.
4. Mathas S, Rickers A, Bommert K, Dorken B, Mapara MY. Anti-CD20- and B-cell receptor-mediated apoptosis: evidence for shared intracellular signaling pathways. Cancer research. 2000; 60:7170-7176.
5. Hatjiharissi E, Xu L, Santos DD, Hunter ZR, Ciccarelli BT, Verselis S, Modica M, Cao Y, Manning RJ, Leleu X, Dimmock EA, Kortsaris A, Mitsiades C, et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood. 2007; 110:2561-2564.
6. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nature reviews Cancer. 2012; 12:278-287.
7. Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, Hengartner H, Zinkernagel RM. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science. 2000; 288:2222-2226.
8. Morton HC, Brandtzaeg P. CD89: the human myeloid IgA Fc receptor. Archivum immunologiae et therapiae experimentalis. 2001; 49:217-229.
9. Guettinger Y, Barbin K, Peipp M, Bruenke J, Dechant M, Horner H, Thierschmidt D, Valerius T, Repp R, Fey GH, Stockmeyer B. A recombinant bispecific single-chain fragment variable specific for HLA class II and Fc alpha RI (CD89) recruits polymorphonuclear neutrophils for efficient lysis of malignant B lymphoid cells. Journal of immunology. 2010; 184:1210-1217.
10. Dechant M, Beyer T, Schneider-Merck T, Weisner W, Peipp M, van de Winkel JG, Valerius T. Effector mechanisms of recombinant IgA antibodies against epidermal growth factor receptor. Journal of immunology. 2007; 179:2936-2943.
11. Lohse S, Brunke C, Derer S, Peipp M, Boross P, Kellner C, Beyer T, Dechant M, van der Winkel JG, Leusen JH, Valerius T. Characterization of a mutated IgA2 antibody of the m(1) allotype against the epidermal growth factor receptor for the recruitment of monocytes and macrophages. The Journal of biological chemistry. 2012; 287:25139-25150.
12. Boross P, Lohse S, Nederend M, Jansen JH, van Tetering G, Dechant M, Peipp M, Royle L, Liew LP, Boon L, van Rooijen N, Bleeker WK, Parren PW, et al. IgA EGFR antibodies mediate tumour killing in vivo. EMBO molecular medicine. 2013; 5:1213-1226.
13. Borrok MJ, Luheshi NM, Beyaz N, Davies GC, Legg JW, Wu H, Dall'Acqua WF, Tsui P. Enhancement of antibody-dependent cell-mediated cytotoxicity by endowing IgG with FcalphaRI (CD89) binding. mAbs. 2015; 7:743-751.
14. Uchida J, Hamaguchi Y, Oliver JA, Ravetch JV, Poe JC, Haas KM, Tedder TF. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. The Journal of experimental medicine. 2004; 199:1659-1669.
15. Algars A, Irjala H, Vaittinen S, Huhtinen H, Sundstrom J, Salmi M, Ristamaki R, Jalkanen S. Type and location of tumor-infiltrating macrophages and lymphatic vessels predict survival of colorectal cancer patients. International journal of cancer. 2012; 131:864-873.
16. Leidi M, Gotti E, Bologna L, Miranda E, Rimoldi M, Sica A, Roncalli M, Palumbo GA, Introna M, Golay J. M2 macrophages phagocytose rituximab-opsonized leukemic targets more efficiently than m1 cells in vitro. Journal of immunology. 2009; 182:4415-4422.
17. Kelton W, Mehta N, Charab W, Lee J, Lee CH, Kojima T, Kang TH, Georgiou G. IgGA: a “cross-isotype” engineered human Fc antibody domain that displays both IgG-like and IgA-like effector functions. Chemistry & biology. 2014; 21:1603-1609.
18. Peipp M, Lammerts van Bueren JJ, Schneider-Merck T, Bleeker WW, Dechant M, Beyer T, Repp R, van Berkel PH, Vink T, van de Winkel JG, Parren PW, Valerius T. Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood. 2008; 112:2390-2399.
19. Xu L, Li B, Huang M, Xie K, Li D, Li Y, Gu H, Fang J. Critical Role of Kupffer Cell CD89 Expression in Experimental IgA Nephropathy. PloS one. 2016; 11:e0159426.
20. Biburger M, Aschermann S, Schwab I, Lux A, Albert H, Danzer H, Woigk M, Dudziak D, Nimmerjahn F. Monocyte subsets responsible for immunoglobulin G-dependent effector functions in vivo. Immunity. 2011; 35:932-944.
21. van Egmond M, Hanneke van Vuuren AJ, van de Winkel JG. The human Fc receptor for IgA (Fc alpha RI, CD89) on transgenic peritoneal macrophages triggers phagocytosis and tumor cell lysis. Immunology letters. 1999; 68:83-87.
22. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, Peggs KS, Ravetch JV, Allison JP, Quezada SA. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. The Journal of experimental medicine. 2013; 210:1695-1710.
23. Keler T, Wallace PK, Vitale LA, Russoniello C, Sundarapandiyan K, Graziano RF, Deo YM. Differential effect of cytokine treatment on Fc alpha receptor I- and Fc gamma receptor I-mediated tumor cytotoxicity by monocyte-derived macrophages. Journal of immunology. 2000; 164:5746-5752.
24. Monteiro RC, Van De Winkel JG. IgA Fc receptors. Annual review of immunology. 2003; 21:177-204.
25. Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93:5512-5516.
26. Sancho J, Gonzalez E, Rivera F, Escanero JF, Egido J. Hepatic and kidney uptake of soluble monomeric and polymeric IgA aggregates. Immunology. 1984; 52:161-167.
27. Boross P, Jansen JH, de Haij S, Beurskens FJ, van der Poel CE, Bevaart L, Nederend M, Golay J, van de Winkel JG, Parren PW, Leusen JH. The in vivo mechanism of action of CD20 monoclonal antibodies depends on local tumor burden. Haematologica. 2011; 96:1822-1830.
28. de Haij S, Jansen JH, Boross P, Beurskens FJ, Bakema JE, Bos DL, Martens A, Verbeek JS, Parren PW, van de Winkel JG, Leusen JH. In vivo cytotoxicity of type I CD20 antibodies critically depends on Fc receptor ITAM signaling. Cancer research. 2010; 70:3209-3217.
29. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nature medicine. 2000; 6:443-446.
30. Hamaguchi Y, Xiu Y, Komura K, Nimmerjahn F, Tedder TF. Antibody isotype-specific engagement of Fcgamma receptors regulates B lymphocyte depletion during CD20 immunotherapy. The Journal of experimental medicine. 2006; 203:743-753.
31. Chintalacharuvu KR, Vuong LU, Loi LA, Larrick JW, Morrison SL. Hybrid IgA2/IgG1 antibodies with tailor-made effector functions. Clinical immunology. 2001; 101:21-31.
32. Rifai A, Mannik M. Clearance kinetics and fate of mouse IgA immune complexes prepared with monomeric or dimeric IgA. Journal of immunology. 1983; 130:1826-1832.
33. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O'Dwyer PJ, Vonderheide RH. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011; 331:1612-1616.
34. Weischenfeldt J, Porse B. Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH protocols. 2008; 2008:pdb.prot5080.
35. Li H, Sun Y, Chen D, Zhao H, Zhao M, Zhu X, Ke C, Zhang G, Jiang C, Zhang L, Zhang F, Wei H, Li W. Synergistic anti-tumor therapy by a comb-like multifunctional antibody nanoarray with exceptionally potent activity. Scientific reports. 2015; 5:15712.
36. Meyer S, Nederend M, Jansen JH, Reiding KR, Jacobino SR, Meeldijk J, Bovenschen N, Wuhrer M, Valerius T, Ubink R, Boross P, Rouwendal G, Leusen JH. Improved in vivo anti-tumor effects of IgA-Her2 antibodies through half-life extension and serum exposure enhancement by FcRn targeting. mAbs. 2016; 8:87-98.