
INTRODUCTION
Ischemic neuron death is presumably initiated by neural excitotoxicity, in which glutamate elevation [1–7] and GABAergic neuron impairment [8–12] have been found to be involved. The elevation of the synaptic glutamate during ischemia may be due to its increased release and impaired reuptake. For instance, the expression of glutamate-transporters changes in ischemia [13–17] and their functions are deficit under pathological conditions [9, 18], such that the impaired glutamate reuptake occurs in ischemia. Moreover, GABAergic neurons are vulnerable to various pathogenic factors and functionally impaired in the early stage of ischemia [9, 12, 19–24]. Their ischemic impairment shifts the balance between excitation and inhibition toward neuronal over-excitation [25], leading to the elevated glutamate release and subsequently neuronal excitotoxicity for ischemic neuronal death. The enhancement of GABAergic synapse transmission reduces loss of hippocampal CA1 pyramidal neurons [26]. In this regard, the elucidation of mechanisms underlying the functional impairment of cerebral GABAergic neurons as well as their protection will provide the clues for developing therapeutic strategies of ischemic neuron death.
Cerebral ischemia triggers a complex series of biochemical and molecular mechanisms that impairs neuronal functions mediated by excitotoxic glutamatergic signaling, ionic balance and free-radical reaction. These intricate processes activate the signaling mechanisms, such as calcium/calmodulin-dependent kinases (CaMK), protein kinase C (PKC) and mitogen-activated protein kinases, which in turn lead to the neuronal stroke [27–31]. In addition to the excitatory neurons, protein kinases, such as PKC and CaMK-II, are located in GABAergic neurons and regulate their functions [32–37]. Based on these studies, we hypothesized that the activated PKC and CaMK-II may be involved in ischemic impairment of GABAergic neurons, and the inhibition of these protein kinases might be able to secure the ischemia-induced deficits of GABAergic neurons.
With questions and hypothesis above, we aim to investigate whether the inhibitions of CaMK-II and PKC are able to prevent the ischemia-induced deficits of GABAergic neurons. In terms of strategies for this study, the functions of cortical GABAergic neurons were measured by whole-cell recording in cortical slices in terms of their abilities of sequential spikes (input-output) and reception (excitatory postsynaptic currents, EPSC). A mimic of cortical ischemia was conducted by reducing perfusion rate to brain slices. The inhibitions of PKC and CaMK-II were fulfilled by applying chelerythrine chloride (PKC inhibitor) and 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazine (CaMK-II inhibitor).
RESULTS
Ischemia attenuates excitatory synaptic transmission and excitability of cortical GABAergic neurons
The functions of GABAergic neurons in cortical slices were measured by whole-cell recording under the conditions of control and subsequent ischemia. Spontaneous excitatory postsynaptic currents (sEPSC) were recorded under voltage-clamp to analyze their receptions to excitatory inputs. Sequential spikes were induced by injecting depolarization pulses in various intensities into these neurons under current-clamp to assess their ability to convert excitatory input into spikes.
Figure 1 illustrates the ischemia-induced change of sEPSCs on cortical GABAergic neurons. sEPSC amplitudes and frequencies appear to be substantially increased by ischemia (Figure 1A). Figure 1B shows cumulative probability versus sEPSC amplitude in the control (red symbols) and subsequent ischemia (blue, n=13 neurons from 3 mice). Insert figure shows that sEPSC amplitudes at 50% cumulative probability are 8.54±0.21 pA under the control (red bar) and 23.11±0.10 pA after ischemia for 5 min (blue; two asterisks, p<0.01). Figure 1C shows cumulative probability versus inter-sEPSC intervals in the control (red symbols) and subsequent ischemia (blue). Insert figure shows that sEPSC intervals at 50% cumulative probability are 215.27±4.00 ms under the control (red bar) and 78.74±2.13 ms after ischemia for 5 min (blue; two asterisks, p<0.01). This result indicates that ischemia leads to excitatory synaptic transmission at cortical GABAergic neurons to be upregulated.
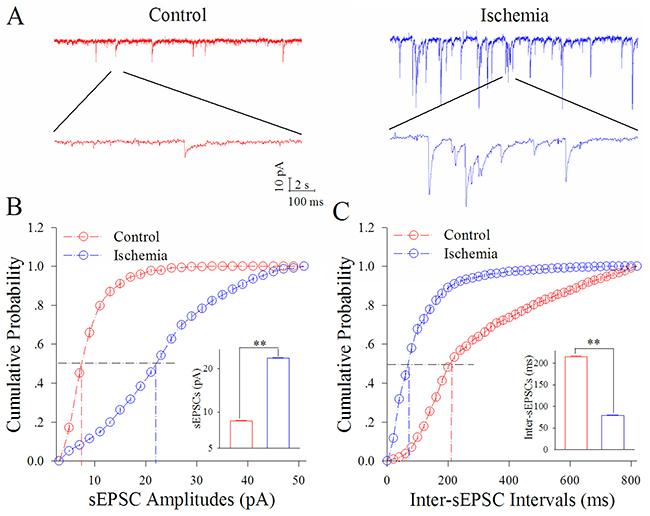
Figure 1: Ischemia upregulates excitatory synaptic transmission at cortical GABAergic neurons. sEPSCs were recorded by whole-cell voltage-clamp in cortical GABAergic neurons and analyzed in terms of their amplitudes and frequencies. (A) shows sEPSCs recorded in GABAergic neurons during control (red trace) and subsequent ischemia (blue). Calibration bars are 10 pA, 2 seconds (top traces) and 100 ms (bottom trace). (B) shows cumulative probability versus sEPSC amplitudes in the control (red symbols) and ischemia (blue). The insert shows sEPSC amplitudes at 50% cumulative probability under the control (red bar) and after ischemia (blue bar; two asterisks, p<0.01; n=13). (C) shows cumulative probability versus inter-sEPSC intervals in the control (red symbols) and ischemia (blue). The insert shows that sEPSC intervals at 50% cumulative probability under the control (red bar) and after ischemia (blue bar; two asterisks, p<0.01).
Figure 2 shows sequential spike capacity at GABAergic neurons under the conditions of control and ischemia. Sequential spikes in Figure 2A were recorded under the conditions of control (top panel) and ischemia (bottom) at a GABAergic neuron in response to the same stimulus intensity of depolarization pulse. Figure 2B shows that spike frequencies versus normalized stimuli at GABAergic neurons (n=13 cells from 3 mice) under the control (red symbols) and subsequent ischemia (blue; two asterisks, p<0.01). This result indicates that spiking ability of cortical GABAergic neurons is substantially impaired after ischemia.
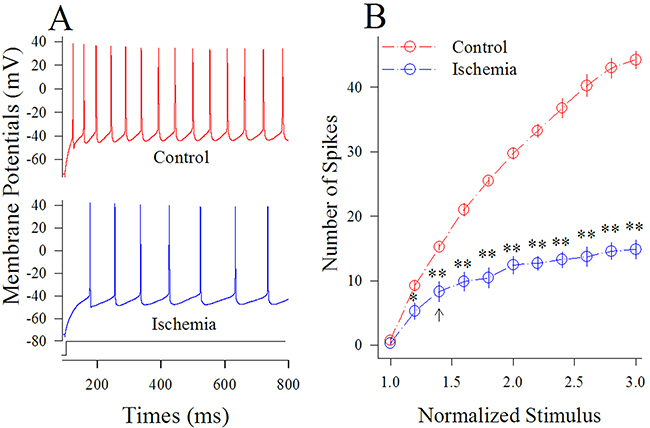
Figure 2: Ischemia impairs spiking ability at cortical GABAergic neurons. Sequential spikes were recorded by whole-cell current-clamp in cortical GABAergic neurons and analyzed in terms of spikes per second. (A) shows sequential spikes of GABAergic neurons in response to depolarization pulse with the same intensity under the conditions of control (red trace on top panel) and control (blue trace on bottom) at a GABAergic neuron. (B) shows that spike frequencies versus normalized stimuli at GABAergic neurons (n=13) under the control (red symbols) and subsequent ischemia (blue; two asterisks, p<0.01).
Therefore, ischemia induces the upregulation of excitatory synapses on cortical GABAergic neurons and the downregulation of their spiking ability, indicating ischemic excitotoxicity. If protein kinases, such as CaMK-II and PKC, are involved in this ischemic neuronal impairment, their inhibitors should block the upregulated excitatory synaptic transmission and the downregulated spiking ability, which we have studied.
The inhibition of CaMK-II partially blocks ischemic over-excitation in cortical GABAergic neurons
The selective inhibitor of CaMK-II, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4- phenylpiperazine (KN-62) [38–40], was used to examine the role of CaMK-II in the ischemic impairment of cortical GABAergic neurons. The experiments were conducted under the conditions of KN-62 presence and subsequent KN-62 plus ischemia.
Figure 3 shows the influence of KN-62 on excitatory synaptic transmission at cortical GABAergic neurons in ischemia. KN-62 at 0.9 μM appears to reduce ischemic over-excitation at GABAergic neurons (Figure 3A). Figure 3B illustrates cumulative probability versus sEPSC amplitudes under the conditions of KN-62 application (red symbols) and KN-62 plus ischemia (green; n=13 neurons from 5 mice) as well as ischemia (blue). Insert figure show that sEPSC amplitudes at 50% cumulative probability are 8.97±0.32 pA in KN-62 application (red bar), 17.02±0.12 pA in KN-62 plus ischemia (green) and 23.11±0.10 pA during ischemia only (blue; two asterisks, p<0.01). Figure 3C shows cumulative probability versus inter-sEPSC intervals under the conditions of KN-62 application (red symbols), KN-62 plus ischemia (green) and ischemia (blue). Insert figure shows that sEPSC intervals at 50% cumulative probability are 222.52±2.35 ms in KN-62 application (red bar), 117.21±1.31 ms in KN-62 plus ischemia (green) and 78.74±2.13 ms during ischemia only (blue; two asterisks, p<0.01). Therefore, the inhibition of CaMK-II partially blocks the ischemic upregulation of excitatory synaptic transmission at cortical GABAergic neurons.
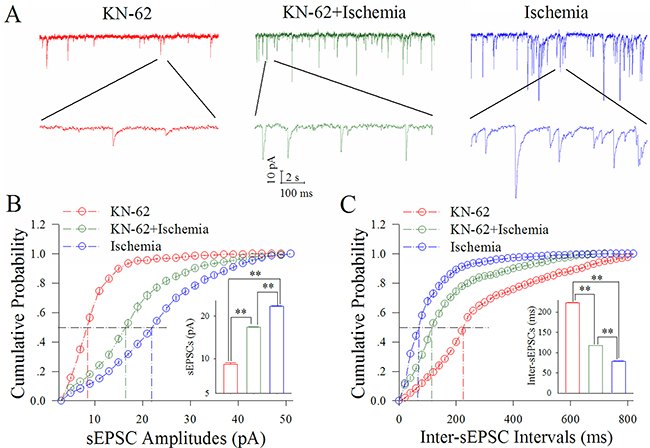
Figure 3: KN-62 partially blocks the ischemic upregulation of excitatory synaptic transmission at cortical GABAergic neurons. KN-62 was added into the ACSF with a final concentration at 0.9 μM. (A) illustrates sEPSC recorded on GABAergic neurons under the conditions of KN-62 application (red traces), KN-62 plus ischemia (green) and ischemia (blue). (B) illustrates cumulative probability versus sEPSC amplitudes under the conditions of KN-62 application (red symbols), KN-62 plus ischemia (green) as well as ischemia (blue). Insert figure shows sEPSC amplitudes at 50% cumulative probability in KN-62 application (red bar), KN-62 plus ischemia (green) and during ischemia only (blue; two asterisks, p<0.01; n=13). (C) illustrates cumulative probability versus inter-sEPSC intervals under the conditions of KN-62 application (red symbols), KN-62 plus ischemia (green) and ischemia (blue). Insert figure demonstrates sEPSC intervals at 50% cumulative probability in KN-62 application (red bar), KN-62 plus ischemia (green) and during ischemia only (blue; two asterisks, p<0.01; n=13).
Figure 4 shows the influence of KN-62 on spiking ability at cortical GABAergic neurons in ischemia. Sequential spikes in Figure 4A-4C were recorded under the conditions of KN-62 application (A), KN-62 plus ischemia (B) and ischemia only (C) at a GABAergic neuron in response to the same stimulus intensity of depolarization pulse. Figure 4D shows that spike frequencies versus normalized stimuli at GABAergic neurons under the conditions of KN-62 application (red symbols) and KN-62 plus ischemia (green; n=13 neurons from 3 mice) as well as ischemia (blue; two asterisks, p<0.01). Thus, the inhibition of CaMK-II partially blocks the ischemic impairment of spiking ability at cortical GABAergic neurons.
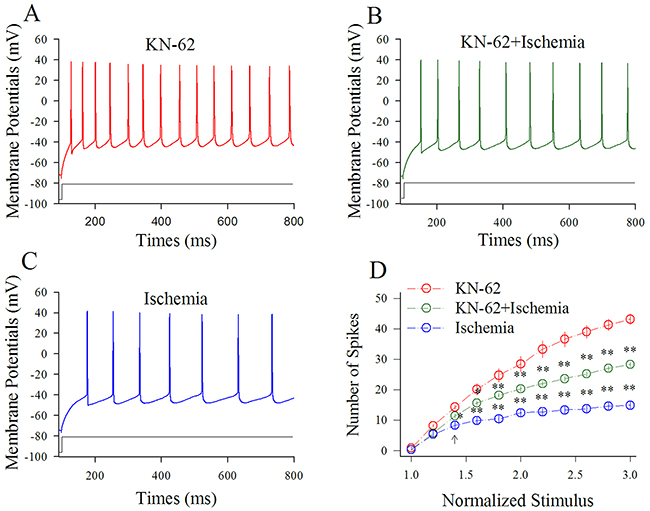
Figure 4: KN-62 prevents the ischemic impairment of spiking capability at cortical GABAergic neurons. KN-62 was added into the ACSF with a final concentration at 0.9 μM. (A) shows sequential spikes in presence of KN-62 application. (B) shows sequential spikes under the condition of KN-62 plus ischemia. (C) shows sequential spikes during ischemia. Spikes at the GABAergic neuron in A~C respond to the same stimulus intensity of depolarization pulse. (D) shows that spike frequencies versus normalized stimuli at GABAergic neurons (n=13) under the conditions of KN-62 application (red symbols), KN-62 plus ischemia (green) and ischemia (blue; two asterisks, p<0.01).
These results indicate that CaMK-II plays a role in ischemic over-excitation at cortical GABAergic neurons. We subsequently examine the effect of PKC on the ischemic impairment of cortical GABAergic neurons.
The inhibition of PKC partially blocks ischemic over-excitation in cortical GABAergic neurons
The selective and potent inhibitor of PKC, chelerythrine chloride (CHE) [41, 42], was used to examine the role of PKC in the ischemic impairment of cortical GABAergic neurons. The experiments were conducted under the conditions of CHE presence and subsequent CHE plus ischemia.
Figure 5 shows the influence of CHE on excitatory synaptic transmission at cortical GABAergic neurons in ischemia. CHE at 0.6 μM appears to reduce ischemic over-excitation at GABAergic neurons (Figure 5A). Figure 5B illustrates cumulative probability versus sEPSC amplitudes under the conditions of CHE application (red symbols) and CHE plus ischemia (green; n=13 neurons from 4 mice) as well as ischemia (blue). Insert figure shows that sEPSC amplitudes at 50% cumulative probability are 8.69±0.30 pA in CHE application (red bar), 16.99±0.24 pA in CHE plus ischemia (green) and 23.11±0.10 pA during ischemia only (blue; two asterisks, p<0.01). Figure 5C shows cumulative probability versus inter-sEPSC intervals under the conditions of CHE application (red symbols), CHE plus ischemia (green) and ischemia (blue). Insert figure shows that sEPSC intervals at 50% cumulative probability are 216.21±2.37 ms in CHE application (red bar), 118.68±0.95 ms in CHE plus ischemia (green) and 78.74±2.13 ms during ischemia only (blue; two asterisks, p<0.01). Thus, the inhibition of PKC partially blocks the ischemic upregulation of excitatory synaptic transmission at cortical GABAergic neurons.
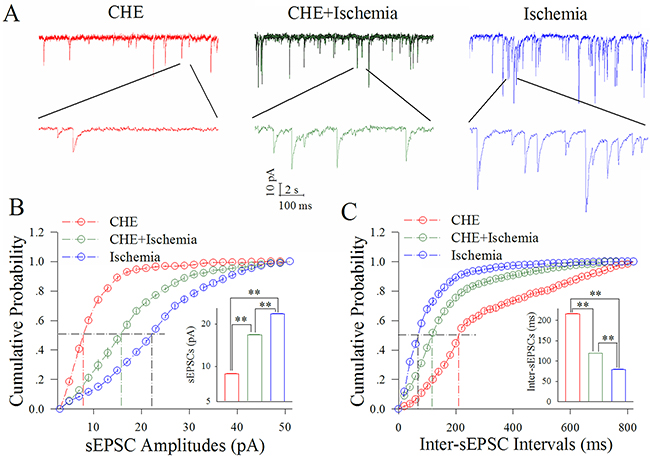
Figure 5: CHE partially blocks the ischemic upregulation of excitatory synaptic transmission at cortical GABAergic neurons. CHE was added into the ACSF with a final concentration at 0.6 μM. (A) illustrates sEPSC recorded on GABAergic neurons under the conditions of CHE application (red traces), CHE plus ischemia (green) and ischemia (blue). (B) illustrates cumulative probability versus sEPSC amplitudes under the conditions of CHE application (red symbols), CHE plus ischemia (green) as well as ischemia (blue). Insert figure shows sEPSC amplitudes at 50% cumulative probability in CHE application (red bar), CHE plus ischemia (green) and during ischemia only (blue; two asterisks, p<0.01; n=13). (C) shows cumulative probability versus inter-sEPSC intervals under the conditions of CHE application (red symbols), CHE plus ischemia (green) and ischemia (blue). Insert figure demonstrates sEPSC intervals at 50% cumulative probability in CHE application (red bar), CHE plus ischemia (green) and during ischemia only (blue; two asterisks, p<0.01; n=13).
Figure 6 shows the influence of CHE on spiking ability at cortical GABAergic neurons in ischemia. Sequential spikes in Figure 6A-6C were recorded under the conditions of CHE application (A), CHE plus ischemia (B) and ischemia only (C) at a GABAergic neuron in response to the same stimulus intensity of depolarization pulse. Figure 6D shows that spike frequencies versus normalized stimuli at GABAergic neurons under the conditions of CHE application (red symbols) and CHE plus ischemia (green; n=13 cells from 4 mice) as well as ischemia (blue; two asterisks, p<0.01). Therefore, the inhibition of PKC partially blocks the ischemic impairment of spiking ability at cortical GABAergic neurons.
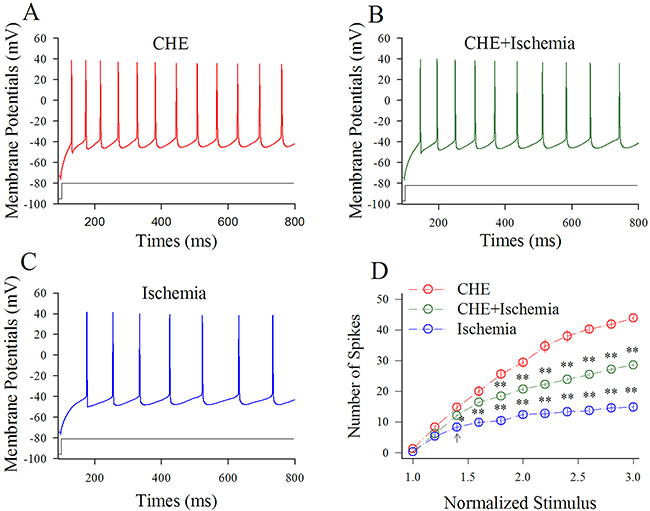
Figure 6: CHE prevents the ischemic impairment of spiking capability at cortical GABAergic neurons. CHE was added into the ACSF with a final concentration at 0.6 μM. (A) shows sequential spikes in presence of CHE application. (B) shows sequential spikes under the condition of CHE plus ischemia. (C) shows sequential spikes during ischemia. Spikes at the GABAergic neuron in A~C respond to the same stimulus intensity of depolarization pulse. (D) shows that spike frequencies versus normalized stimuli at GABAergic neurons (n=13) under the conditions of CHE application (red symbols), CHE plus ischemia (green) and ischemia (blue; two asterisks, p<0.01).
The results indicate that PKC plays a role in ischemic over-excitation at cortical GABAergic neurons. We subsequently test whether PKC and CaMK-II synergistically block the ischemic impairment of cortical GABAergic neurons.
The inhibitions of PKC and CaMK-II synergistically block ischemic over-excitation in GABAergic cells
The joint application of CaMK-II and PKC inhibitors was used to examine the role of CaMK-II and PKC in the ischemic impairment of cortical GABAergic neurons. The experiments were conducted under the conditions of KN-62/CHE presence and subsequent KN-62/CHE plus ischemia.
Figure 7 illustrates the influence of KN-62/CHE on excitatory synaptic transmission at the cortical GABAergic neurons during ischemia. The joint application of KN-62 (0.9 μM) and CHE (0.6 μM) appears to synergistically reduce ischemic over-excitation at GABAergic neurons (Figure 7A). Figure 7B illustrates cumulative probability versus sEPSC amplitudes under the conditions of KN-62/CHE applications (red symbols) and KN-62/CHE plus ischemia (green; n=13 neurons from 5 mice) as well as ischemia (blue). Insert figure shows that sEPSC amplitudes at 50% cumulative probability are 8.46±0.26 pA in KN-62/CHE applications (red bar), 10.99±0.27 pA in KN-62/CHE plus ischemia (green) and 23.11±0.10 pA during ischemia (blue; two asterisks, p<0.01). Figure 7C shows cumulative probability versus inter-sEPSC interval under the conditions of KN-62/CHE applications (red symbols), KN/62/CHE plus ischemia (green) and ischemia (blue). Insert figure shows that sEPSC intervals at 50% cumulative probability are 220.10±11.84 ms in KN-62/CHE applications (red bar), 156.43±3.14 ms in KN-62/CHE plus ischemia (green) and 78.74±2.13 ms during ischemia only (blue; two asterisks, p<0.01). Therefore, the inhibitions of CaMK-II and PKC synergistically blocks the ischemic upregulation of excitatory synaptic transmission at cortical GABAergic neurons.
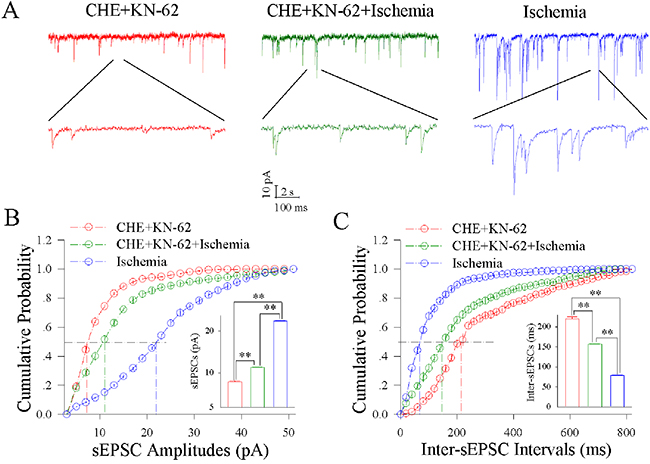
Figure 7: CHE and KN-62 synergistically block ischemic upregulation of excitatory synaptic transmission at cortical GABAergic neurons. Final concentrations of CHE and KN-62 in the ACSF are 0.6 μM and 0.9 μM, respectively. (A) shows sEPSC recorded on GABAergic neurons under the conditions of CHE/KN-62 application (red traces), CHE/KN-62 plus ischemia (green) and ischemia (blue). (B) illustrates cumulative probability versus sEPSC amplitudes under the conditions of CHE/KN-62 application (red symbols), CHE/KN-62 plus ischemia (green) and ischemia (blue). Insert figure illustrates sEPSC amplitudes at 50% cumulative probability in CHE/KN-62 application (red bar), CHE/KN-62 plus ischemia (green) and during ischemia (blue; two asterisks, p<0.01; n=13). (C) shows cumulative probability versus inter-sEPSC intervals under the conditions of CHE/KN-62 application (red symbols), CHE/KN-62 plus ischemia (green) and ischemia (blue). Insert figure shows sEPSC intervals at 50% cumulative probability in CHE/KN-62 application (red bar), CHE/KN-62 plus ischemia (green) and during ischemia (blue; two asterisks, p<0.01; n=13).
Figure 8 shows the influence of KN-62/CHE on spiking ability at cortical GABAergic neurons in ischemia. Sequential spikes in Figure 8A-8C were recorded under the conditions of KN-62/CHE application (A), KN-62/CHE plus ischemia (B) and ischemia only (C) at a GABAergic neuron in response to the same stimulus intensity of depolarization pulse. Figure 8D shows that spike frequency versus normalized stimuli at GABAergic neurons under the conditions of KN-62/CHE application (red symbols) and KN-62/CHE plus ischemia (green; n=13 cells from 5 mice) as well as ischemia (blue; two asterisks, p<0.01). Therefore, the inhibitions of CaMK-II and PKC synergistically blocks the ischemic impairment of spiking capability at the cortical GABAergic neurons.
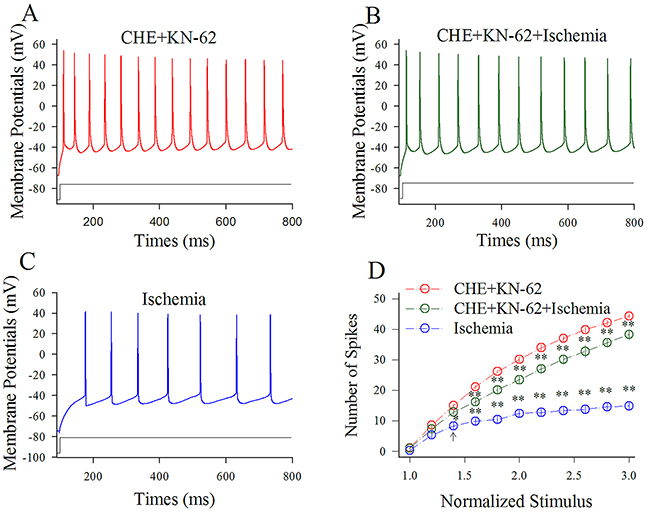
Figure 8: CHE and KN-62 synergistically prevent the ischemic impairment of spiking capability at cortical GABAergic neurons. Final concentrations of CHE and KN-62 in the ACSF are 0.6 μM and 0.9 μM, respectively. (A) illustrates sequential spikes in presence of CHE/KN-62 application. (B) illustrates sequential spikes under the condition of CHE/KN-62 plus ischemia. (C) illustrates sequential spikes during ischemia. Spikes at the GABAergic neuron in A~C respond to the same stimulus intensity of depolarization pulse. (D) shows that spike frequencies versus normalized stimuli at GABAergic neurons (n=13) under the conditions of CHE/KN-62 application (red symbols), CHE/KN-62 plus ischemia (green) and ischemia only (blue; two asterisks, p<0.01).
DISCUSSION
Our study demonstrates that the ischemia induces the overexcited impairment of cortical GABAergic neurons (Figures 1-2), consistent to previous studies [8–10, 12], which may lead to the excitotoxicity of GABAergic neurons in the early stage of ischemia and subsequently ischemic death of all neurons for cerebral stroke. In terms of molecular mechanisms underlying their impairment, our study indicates that the inhibition of PKC or CaMK-II blocks the ischemic impairment of cortical GABAergic neurons partially (Figures 3-6). The joint application of PKC and CaMK-II inhibitors blocks these pathological changes further. Thus, PKC and CaMK-II prevent the functional impairment of cortical GABAergic neurons synergistically, and the joint application of PKC and CaMK-II inhibitors may be a potential therapeutic strategy to protect cortical neurons from ischemic death.
In our study, ischemia upregulates sEPSC amplitudes and frequencies as well as downregulates spike capability. As we known, sEPSC amplitudes are influenced by the density and conductance of postsynaptic ionotropic glutamate receptors, and sEPSC frequencies are affected by the number and release probability of presynaptic terminals [43, 44]. Moreover, spike ability is influenced by voltage-gated sodium channels [45–48]. In this regard, a sequence of chain reactions for the ischemic dysfunction of cortical GABAergic neurons may be that ischemia leads to intracellular Ca2+ increase [8] as well as PKC and CaMK-II activations [49, 50], which further upregulate the glutamate receptor-channels and glutamate release as well as downregulate voltage-gated sodium channels [36, 51–53], such that these GABAergic neurons are dysfunction. In addition to these signaling pathways for the ischemic deficit of cortical GABAergic neurons, other mechanisms remain to be studied.
Our previous study indicates that an impairment of cortical GABAergic neurons induced by ischemia and acidosis is also due to the ischemic dysfunction of glutamate transporters on astrocytes [9]. As the neuron-glia interaction is critical to fulfill brain functions [54–61], the astrocytes may play an important role in the ischemia-induced impairment of cortical GABAergic neurons. Together these data, we suggest that both intracellular signaling pathways and extracellular glia cell supports are critical for protecting GABAergic neurons from ischemic neuron death. In other words, multi-target therapy can also be used in the protection of ischemia neuron injury [62].
In terms of treatment of ischemic stroke, therapeutic strategies include anticoagulation, thrombolysis and neuroprotection [63–73]. Based on the preclinical studies about ischemia-related molecules and biochemical reactions in brain cells, many approaches to interrupt injurious cellular and molecular processes were applied to the clinical trials [74–76]. These efforts have not shown to fully improve stroke patients [1, 4, 74, 75]. In addition to anticoagulation and thrombolysis, the protection of ischemic neuron death by improving multiple targets is needed, since numerous signaling pathways are activated during ischemia. However, the functions of the GABAergic neurons and astrocytes are impaired immediately after ischemia [9, 62]. The protection of neuronal functions in the early stage of ischemia should be focused on the astrocytes and GABAergic neurons in the cerebral cortex. Based on our studies, the joint applications of PKC and CaMK-II inhibitors as well as glutamate transporter activator in a low dosage may be a potential way to pursue.
MATERIALS AND METHODS
Cortical slices and GABAergic neurons
The experimental procedures were approved by Institutional Animal Care Use Committee in Bengbu Anhui China. The cortical slices (400 μm) were prepared from C57 GAD-GFP mice (Jackson Lab, USA) whose cortical GABAergic neurons are genetically labeled by green fluorescent protein [77]. The mice in postnatal days 19~21 were anesthetized by inhaling isoflurane and decapitated by a guillotine. Cortical slices were cut with a Vibratome in the oxygenated (95% O2 and 5% CO2) artificial cerebrospinal fluid (ACSF) in the concentrations (mM) of 124 NaCl, 3 KCl, 1.2 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 4 MgSO4, 10 dextrose and 5 HEPES (pH 7.35) at 4°C. The brain slices were held in the oxygenized ACSF (124 NaCl, 3 KCl, 1.2 NaH2PO4, 26 NaHCO3, 2.4 CaCl2, 1.3 MgSO4, 10 dextrose and 5 HEPES, pH 7.35) at 25°C for 1 hour. A slice was then transferred to the submersion chamber (Warner RC-26G) that was perfused with the oxygenated ACSF at 31°C for whole-cell recording [36, 78–82]. Chemical reagents were purchased from Sigma.
GFP-labeled GABAergic neurons were recorded by whole-cell clamp in the sensory cortices under DIC-fluorescent microscope (Nikon FN-E600, Tokyo, Japan). A wavelength at 488 nm was used to excite the fluorescence of GFP-labeled neurons. GABAergic neurons expressed fast spikes with less adaptation in their amplitude and frequency, the typical properties for the interneurons [83–86].
Whole-cell recording and neuronal functions
The neurons were recorded by a MultiClamp-700B amplifier under voltage-clamp for their synaptic activity and the current-clamp for their intrinsic properties. Electrical signals were inputted to pClamp-10 (Axon Instrument Inc.) for data acquisition and analysis. An output bandwidth of this amplifier was set at 3 kHz. The pipette solution of recording spikes and excitatory synaptic events included (mM) 150 K-gluconate, 5 NaCl, 5 HEPES, 0.4 EGTA, 4 Mg-ATP, 0.5 Tris-GTP, and 5 phosphocreatine (pH 7.35; [79, 87–89]. These pipette solutions were freshly made and filtered (0.1 μm). The osmolarity was 295~305 mOsmol and pipette resistance was 5~6 MΩ [20, 77].
The functions of GABAergic neurons were assessed by recording their active intrinsic properties (spiking ability) and excitatory synaptic events (reception to excitatory input) [12, 22, 81]. Their excitatory inputs were analyzed by recording spontaneous excitatory postsynaptic currents (sEPSC) on GABAergic neurons in the presence of 10 μM bicuculline in the ACSF to block GABAAR [12, 81]. 10 μM CNQX and 40 μM D-AP5 were added in the ACSF at the end of experiments to examine whether synaptic responses were mediated by glutamate receptor, which blocked sEPSCs in our studies.
The recording of spontaneous postsynaptic currents, instead of evoked synaptic currents, is based on the following reasons. sEPSC and sIPSC amplitudes represent the responsiveness and the densities of postsynaptic receptors. The frequencies imply the probability of transmitter release from an axon terminal and the number of presynaptic axons innervated on the recorded neurons [43, 44]. These parameters can be used to analyze presynaptic and postsynaptic mechanisms. Evoked postsynaptic currents cannot separate these mechanisms. We did not apply TTX into the ACSF to record miniature postsynaptic currents since we had to record neuronal excitability. The synaptic events in our recording are presumably miniature postsynaptic currents. This point is supported by a single peak of the postsynaptic currents in our study.
Action potentials at GABAergic neurons were induced by injecting depolarization pulse under the current-clamp. Their excitability was assessed by input-outputs (spikes versus normalized stimuli) when various stimuli were given [24, 90, 91]. We did not measure a rheobase to show cellular excitability, since this strength-duration relationship was used to assess the ability to fire single spike. We measured the ability of firing sequential spikes [80, 92].
Data were analyzed if the recorded neurons had resting membrane potentials negatively more than -60 mV and action potential amplitudes more than 90 mV. Criteria for the acceptance of each experiment also included less than 5% changes in resting membrane potential, spike magnitude, and input resistance throughout each recording. Series and input resistances in all of the neurons were monitored by injecting hyperpolarization pulses (5 mV/50 ms), as well as calculated by voltage pulses versus instantaneous and steady-state currents. The values in the amplitudes and inter-event intervals of sEPSCs were read at 50% of cumulative probability (P0.5) for their statistical comparisons [93]. It is noteworthy that sEPSC frequencies were applied to merit presynaptic transmitter release and sEPSC amplitudes were used to merit postsynaptic receptor functions [24].
In vitro ischemia
To simulate the artery occlusion and intracranial anastomotic circulation during in vivo ischemic stroke, we reduced the perfusion rate to cortical slices from 2 ml/min to 0.2 ml/min for 6 min [8, 9, 12]. We measured the functions of GABAergic neurons before and during reducing perfusion rate. Subsequently, the perfusion rate was reinstalled to the normal rate before an obvious decrease of resting membrane potentials. In the experiments to examine the influences of protein kinase C (PKC) and Ca2+/CaM-dependent protein kinase II (CaMK-II) on neuronal functions, the procedures were the perfusion of the oxygenized ACSF at 2 ml/min for 5 min, the perfusion of the mixture of the oxygenized ACSF plus the inhibitors of PKC and/or CaMK-II at 2 ml/min, and the perfusion of this mixture solution at 0.2 ml/min.
The effects of PKC on sEPSC and spiking ability in GABAergic neurons and their ischemia-induced deficit were examined by using its selective and potent inhibitor, chelerythrine chloride (CHE IC50=0.6 μM; Sigma, USA) [41, 42], which lowered PKC activity [94–97]. CHE was dissolved in Dimethyl Sulphoxide with a concentration at 0.6 μM. The influences of CaMK-II on sEPSC and spiking ability in cortical GABAergic neurons and their ischemia-induced deficit were examined by applying its selective inhibitor, 1-[N,O-bis (5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazine (KN-62; IC50=0.9 μM; Sigma, USA) [38–40]. KN-62 was dissolved in Dimethyl Sulphoxide with concentration at 0.9 μM. As the concentrations of CHE and KN-62 being used in our study were 0.6 and 0.9 μM, respectively, i.e., IC50, such low concentrations were thought to be specific. Moreover, these concentrations of reagents do not affect basal synaptic transmission and neuronal spiking ability (Supplementary Figure 1).
Statistical analyses
The data of electrophysiological recordings are presented as mean±SEM. The paired t-test was used in the comparisons of experimental data before and after the ischemia or kinase inhibitor application in each of the mice. One-way ANOVA was used to make statistical comparisons in neuronal activity among control, PKC inhibitor, CaMK-II inhibitor and their mixtures.
Authors' contributions
LH, CW, SZ, RG and SG contribute to experiments and data analyses. JHW contributes to experimental design and paper writing.
ACKNOWLEDGMENTS
This study is supported by the National Basic Research Program (2013CB531304 and 2016YFC1307100) and Natural Science Foundation China (81671071 and 81471123) to Jin-Hui Wang. Anhui Natural Science Foundation (1308085QH147) to Li Huang and (1408085MH185) to Shidi Zhao, as well as Natural Science Foundation of Bengbu Medical College (BYKY201622ZD) to Li Huang and (BYKY201635ZD) to Chun Wang.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
COMPETING INTERESTS
All authors declare no competing interest. All authors have read and approved the final version of the manuscript.
REFERENCES
1. Candelario-Jalil E. Injury and repair mechanisms in ischemic stroke: considerations for the development of novel neurotherapeutics. Curr Opin Investig Drugs. 2009; 10: 644-54.
2. Metha SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Research Review. 2007; 54: 34-66.
3. Schwartz-Bloom RD, Sah R. r-aminobutyric acid A neurotransmission and cerebral ischemia. Journal of neurochemistry. 2001; 77: 353-71.
4. Taoufik E, Probert L. Ischemic neuronal damage. Current Pharm Des. 2008; 14: 3565-73.
5. Welsh JP, Yuen G, Placantonkis DG, Yu TQ, Haiss F, O’Heaen E, Molliver ME, Aicher SA. Why do Purkinje cells die so easily after global brain ischemia? Aldolase C, EAAT4, and the cerebellar contribution to posthypoxic myoclonus. Advanced Neurology. 2002; 89: 331-59.
6. White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW, Grossman LI, Rafols JA, Krause GS. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. Journal of the Neurological Sciences. 2000; 179: 1-33.
7. Won SJ, Kim DY, Gwag BJ. Cellular and molecular pathways of ischemic neuronal death. Journal of Biochemical and Molecular Biology. 2002; 35: 67-86.
8. Huang L, Chen N, Ge M, Zhu Y, Guan S, Wang JH. Ca2+ and acidosis synergistically lead to the dysfunction of cortical GABAergic neurons during ischemia. Biochemical and Biophysical Research Communications. 2010; 394: 709-14.
9. Huang L, Zhao S, Lu W, Guan S, Zhu Y, Wang JH. Acidosis-Induced Dysfunction of Cortical GABAergic Neurons through Astrocyte-Related Excitotoxicity. PLoS One. 2015; 10: e0140324. doi: 10.1371/journal.pone.0140324.
10. Johansen FF, Tonder N, Zimmer J, Baimbridge KG, Diemer NH. Short-term changes of parvalbumin and calbindin immunoreactivity in the rat hippocampus following cerebral ischemia. Neurosci Lett. 1990; 120: 171-4. doi: 0304-3940(90)90030-D.
11. Li H, Siegel RE, Schwartz RD. Rapid decline of GABAA receptor subunit mRNA expression in hippocampus following transient cerebral ischemia in the gerbil. Hippocampus. 1993; 3: 527-37. doi: 10.1002/hipo.450030412.
12. Wang JH. Short-term cerebral ischemia causes the dysfunction of interneurons and more excitation of pyramidal neurons. Brain Research Bulletin. 2003; 60: 53-8.
13. Bruhn T, Levy LM, Nielsen M, Christensen T, Johansen FF, Diemer NH. Ischemia induced changes in expression of the astrocyte glutamate transporter GLT1 in hippocampus of the rat. Neurochem Int. 2000; 37: 277-85. doi: S0197-0186(00)00029-2.
14. Camacho A, Massieu L. Role of glutamate transporters in the clearance and release of glutamate during ischemia and its relation to neuronal death. Arch Med Res. 2006; 37: 11-8.
15. Inage YW, Itoh M, Wada K, Takashima S. Expression of two glutamate transporters, GLAST and EAAT4, in the human cerebellum: their correlation in development and neonatal hypoxia-ischemic damage. Journal Neuropathology and Experimental Neurology. 1998; 57: 554-62.
16. Swanson RA, Ying W, Kauppinen TM. Astrocyte influences on ischemic neuronal death. Curr Mol Med. 2004; 4: 193-205.
17. Yamashita A, Makita K, Kuroiwa T, Tanaka K. Glutamate transporters GLAST and EAAT4 regulate postischemic Purkinje cell death: an in vivo study using a cardiac arrest model in mice lacking GLAST or EAAT4. Neuroscience Research. 2006; 55: 264-70.
18. Dallas M, Boycott HE, Atkinson L, Miller A, Boyle JP, Pearson HA, Peers C. Hypoxia suppresses glutamate transport in astrocytes. J Neurosci. 2007; 27: 3946-55. doi: 27/15/3946.
19. Akaike N. Time-dependent rundown of GABA response in mammalian cns neuron during experimental anoxia. Obes Res. 1995; 3: 769S-77S.
20. Ma K, Xu A, Cui S, Sun M, Xue Y, Wang JH. Impaired GABA synthesis, uptake and release are associated with depression-like behaviors induced by chronic mild stress. Translational Psychiatry. 2016; 6: 1-10. doi: 10.1038/tp.2016.181.
21. Luscher B, Shen Q, Sahir N. The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry. 2011; 16: 383-406. doi: 10.1038/mp.2010.120.
22. Wang GY, Zhu ZM, Cui S, Wang JH. Glucocorticoid Induces Incoordination between Glutamatergic and GABAergic Neurons in the Amygdala. PLoS One. 2016; 11: e0166535. doi: 10.1371/journal.pone.0166535.
23. Wang JH, Lu W, Wen B. Neuron-specific mechanisms for epilepsy self-termination. Molecular & Cellular Epilepsy. 2015; 2: e716. doi: 10.14800/mce.716.
24. Xu A, Cui S, Wang J. Incoordination among subcellular compartments is associated to depression-like behavior induced by chronic mild stress. International Journal of Neuropsychopharmacology. 2015; 19. doi: 10.1093/ijnp/pyv122.
25. Luhmann HJ. Ischemia and lesion induced imbalances in cortical function. Prog Neurobiol. 1996; 48: 131-66. doi: 0301-0082(95)00042-9.
26. Johansen FF, Diemer NH. Enhancement of GABA neurotransmission after cerebral ischemia in the rat reduces loss of hippocampal CA1 pyramidal cells. Acta Neurol Scand. 1991; 84: 1-6.
27. Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. 2007; 54: 34-66. doi: S0165-0173(06)00117-2.
28. Mengesdorf T, Althausen S, Mies G, Olah L, Paschen W. Phosphorylation state, solubility, and activity of calcium/calmodulin-dependent protein kinase II alpha in transient focal ischemia in mouse brain. Neurochem Res. 2002; 27: 477-84.
29. Morioka M, Fukunaga K, Yasugawa S, Nagahiro S, Ushio Y, Miyamoto E. Regional and temporal alterations in Ca2+/calmodulin-dependent protein kinase II and calcineurin in the hippocampus of rat brain after transient forebrain ischemia. J Neurochem. 1992; 58: 1798-809.
30. Onodera H, Yamasaki Y, Kogure K, Miyamoto E. Calcium/calmodulin-dependent protein kinase II and protein phosphatase 2B (calcineurin) immunoreactivity in the rat hippocampus long after ischemia. Brain Res. 1995; 684: 95-8. doi: 0006-8993(95)00382-Z.
31. Sieber FE, Traystman RJ, Brown PR, Martin LJ. Protein kinase C expression and activity after global incomplete cerebral ischemia in dog. Stroke. 1998; 29: 1445-53.
32. Kawaguchi Y, Kondo S. Parvalbumin, somatostatin and cholecystokinin as chemical markers for specific GABAergic interneuron types in the rat frontal cortex. J Neurocytol. 2002; 31: 277-87. doi: 5123783.
33. Kawaguchi SY, Hirano T. Signaling cascade regulating long-term potentiation of GABA(A) receptor responsiveness in cerebellar Purkinje neurons. J Neurosci. 2002; 22: 3969-76. doi: 20026379.
34. Qi Y, Huang L, Ni H, Zhou X, Zhang J, Zhu Y, Ge M, Guan S, Wang JH. Intracellular Ca2+ regulates spike encoding at cortical GABAergic neurons and cerebellar Purkinje cells differently. Biochemical and Biophysical Research Communications. 2009; 381: 129-33.
35. Sugiyama Y, Kawaguchi SY, Hirano T. mGluR1-mediated facilitation of long-term potentiation at inhibitory synapses on a cerebellar Purkinje neuron. Eur J Neurosci. 2008; 27: 884-96. doi: 10.1111/j.1460-9568.2008.06063.x.
36. Wang JH, Kelly PT. Ca2+/CaM signalling pathway up-regulates glutamatergic synaptic function in non-pyramidal fast-spiking neurons of hippocampal CA1. J Physiol (Lond). 2001; 533: 407-22.
37. Sharp BM, Chen H, Gong S, Wu X, Liu Z, Hiler K, Taylor WL, Matta SG. Gene expression in accumbens GABA neurons from inbred rats with different drug-taking behavior. Genes Brain Behav. 2011; 10: 778-88. doi: 10.1111/j.1601-183X.2011.00716.x.
38. Hubner K, Phi-van L. KN-62, a selective inhibitor of Ca(2+)/calmodulin-dependent protein kinase II, inhibits the lysozyme pre-mRNA splicing in myelomonocytic HD11 cells. Biochem Biophys Res Commun. 2004; 319: 405-9. doi: 10.1016/j.bbrc.2004.05.002.
39. Ishii A, Kiuchi K, Kobayashi R, Sumi M, Hidaka H, Nagatsu T. A selective Ca2+/calmodulin-dependent protein kinase II inhibitor, KN-62, inhibits the enhanced phosphorylation and the activation of tyrosine hydroxylase by 56 mM K+ in rat pheochromocytoma PC12h cells. Biochem Biophys Res Commun. 1991; 176: 1051-6.
40. Tokumitsu H, Chijiwa T, Hagiwara M, Mizutani A, Terasawa M, Hidaka H. KN-62, 1-`N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl:-4- phenylpiperazi ne, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1990; 265: 4315-20.
41. Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1990; 172: 993-9.
42. Liu B, Feng J, Wang JH. Protein kinase C is essential for kainate-induced anxiety-related behavior and glutamatergic synapse upregulation in prelimbic cortex. CNS Neuroscience and Therapeutics. 2014; 2:982-90. doi: 10.1111/cns.12313.
43. Stevens CF. Presynaptic function. Current Opinion in Neurobiology. 2004; 14: 341-5.
44. Zucker RS, Regehr WG. Short-term synaptic plasticity. Ann Rev Physiol. 2002; 25: 355-405.
45. Goldman L. Stationarity of sodium channel gating kinetics in excised patches from neuroblastoma N1E 115. Biophysics Journal. 1995; 69: 2364-8.
46. Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952; 117: 500-44.
47. Hodgkin AL, Huxley AF. The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. J Physiol. 1952; 116: 497-506.
48. Chen N, Chen SL, Wu YL, Wang JH. The refractory periods and threshold potentials of sequential spikes measured by whole-cell recordings. Biochemical and Biophysical Research Communications. 2006; 340: 151-7.
49. Huang KP. The mechanism of protein kinase C activation. TINS. 1989; 12: 425-32.
50. Schulman H, Kuret J, Jefferson AB, Nose PS, Spitzer KH. Ca2+/Calmodulin-Dependent Microtubule-Associated Protein 2 Kinase: Broad Substrate Specificity and Multifunctional Potential in Diverse Tissues. Biochem. 1985; 24: 5320-7.
51. Ashpole NM, Herren AW, Ginsburg KS, Brogan JD, Johnson DE, Cummins TR, Bers DM, Hudmon A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J Biol Chem. 2012; 287: 19856-69. doi: M111.322537.
52. Schulman H. Protein phosphorylation in neuronal plasticity and gene expression. Curr Opin Neurobiol. 1995; 5: 375-81.
53. Wang JH, Feng DP. Postsynaptic protein kinase C essential to induction and maintenance of long-term potentiation in the hippocampal CA1 region. Proc Natl Acad Sci U S A. 1992; 89: 2576-80.
54. Benarroch EE. Neuron-astrocyte interactions: partnership for normal function and disease in the central nervous system. Mayo Clin Proc. 2005; 80: 1326-38.
55. Corty MM, Freeman MR. Cell biology in neuroscience: Architects in neural circuit design: glia control neuron numbers and connectivity. J Cell Biol. 2013; 203: 395-405. doi: 10.1083/jcb.201306099.
56. Florian C, Vecsey CG, Halassa MM, Haydon PG, Abel T. Astrocyte-derived adenosine and A1 receptor activity contribute to sleep loss-induced deficits in hippocampal synaptic plasticity and memory in mice. J Neurosci. 2011; 31: 6956-62. doi: 10.1523/JNEUROSCI.5761-10.2011.
57. Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 2003; 26: 523-30. doi: S0166223603002662.
58. Ransom B, Behar T, Nedergaard M. New roles for astrocytes (stars at last). Trends Neurosci. 2003; 26: 520-2. doi: S0166223603002595.
59. Schachner M. Cell surface recognition and neuron-glia interactions. Ann N Y Acad Sci. 1991; 633: 105-12.
60. Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011; 144: 810-23. doi: 10.1016/j.cell.2011.02.018.
61. Zhao J, Wang D, Wang JH. Barrel cortical neurons and astrocytes coordinately respond to an increased whisker stimulus frequency. Mol Brain. 2012; 5: 12. doi: 1756-6606-5-12.
62. Wang JH, Cui S. Multi-target therapy for subcellular incompatibility in brain disorders. Brain Disorders & Therapy. 2015; 4: 1-5. doi: 10.4172/2168-975X.1000200.
63. Chamorro A. Immediate anticoagulation in acute focal brain ischemia revisited: gathering the evidence. Stroke. 2001; 32: 577-8.
64. Dalkara T, Moskowitz MA. Recent developments in the experimental stroke. NeuroScience News. 1999; 2: 20-7.
65. Devasenapathy A, Hachinski VC. Current treatment of acute ischemic stroke. NeuroScience News. 1999; 2: 4-13.
66. Jander S, Schroeter M, Saleh A. Imaging inflammation in acute brain ischemia. Stroke. 2007; 38: 642-5. doi: 38/2/642.
67. Leonardo CC, Pennypacker KR. Neuroinflammation and MMPs: potential therapeutic targets in neonatal hypoxic-ischemic injury. J Neuroinflammation. 2009; 6: 13. doi: 1742-2094-6-13.
68. Macchi L, Sorel N, Christiaens L. Aspirin resistance: definitions, mechanisms, prevalence, and clinical significance. Curr Pharm Des. 2006; 12: 251-8.
69. Muir KW, Roberts M. Thrombolytic therapy for stroke: a review with particular reference to elderly patients. Drugs Aging. 2000; 16: 41-54.
70. Puca A. Thrombolysis in cerebral ischemia. A review of clinical and experimental data. J Neurosurg Sci. 1993; 37: 63-70.
71. Spiess BD. Ischemia--a coagulation problem? J Cardiovasc Pharmacol. 1996; 27: S38-41.
72. Tuttolomondo A, Di Sciacca R, Di Raimondo D, Arnao V, Renda C, Pinto A, Licata G. Neuron protection as a therapeutic target in acute ischemic stroke. Curr Top Med Chem. 2009; 9: 1317-34. doi: CTMC-Abs-023-9-14.
73. Vaughan CJ, Delanty N. Neuroprotective properties of statins in cerebral ischemia and stroke. Stroke. 1999; 30: 1969-73.
74. Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008; 55:363-89.
75. Ginsberg MD. Current status of neuroprotection for cerebral ischemia: synoptic overview. Stroke. 2009; 40: S111-4. doi: STROKEAHA.108.528877.
76. Taoufik E, Probert L. Ischemic neuronal damage. Curr Pharm Des. 2008; 14: 3565-73.
77. Zhang G, Gao Z, Guan S, Zhu Y, Wang JH. Upregulation of excitatory neurons and downregulation of inhibitory neurons in barrel cortex are associated with loss of whisker inputs. Mol Brain. 2013; 6: 2. doi: 1756-6606-6-2.
78. Chen N, Yu J, Qian H, Ge R, Wang JH. Axons amplify somatic incomplete spikes into uniform amplitudes in mouse cortical pyramidal neurons. PLoS ONE. 2010; 5:e11868.
79. Ge R, Qian H, Wang JH. Physiological synaptic signals initiate sequential spikes at soma of cortical pyramidal neurons. Mol Brain. 2011; 4: 19. doi: 1756-6606-4-19.
80. Wang JH, Wei J, Chen X, Yu J, Chen N, Shi J. The gain and fidelity of transmission patterns at cortical excitatory unitary synapses improve spike encoding. Journal of Cell Science. 2008; 121: 2951-60.
81. Yu J, Qian H, Wang JH. Upregulation of transmitter release probability improves a conversion of synaptic analogue signals into neuronal digital spikes. Mol Brain. 2012; 5: 26. doi: 1756-6606-5-26.
82. Zhang F, Liu B, Lei Z, Wang J. mGluR1,5 activation improves network asynchrony and GABAergic synapse attenuation in the amygdala: implication for anxiety-like behavior in DBA/2 mice. Mol Brain. 2012; 5: 20. doi: 1756-6606-5-20.
83. Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996; 6: 347-470.
84. Lu W, Wen B, Zhang F, Wang JH. Voltage-independent sodium channels emerge for an expression of activity-induced spontaneous spikes in GABAergic neurons. Mol Brain. 2014; 7: 38. doi: 10.1186/1756-6606-7-38.
85. McKay BE, Turner RW. Physiological and morphological development of the rat cerebellar Purkinje cell. Journal of Physiology (London). 2005; 567: 829-50.
86. Yu J, Qian H, Chen N, Wang JH. Quantal glutamate release is essential for reliable neuronal encodings in cerebral networks. PLoS ONE. 2011; 6: e25219. doi: 10.1371/journal.pone.0025219PONE-D-11-14578.
87. Gao Z, Chen L, Fan R, Lu W, Wang D, Cui S, Huang L, Zhao S, Guan S, Zhu Y, Wang JH. Associations of unilateral whisker and olfactory signals induce synapse formation and memory cell recruitment in bilateral barrel cortices: cellular mechanism for unilateral training toward bilateral memory. Frontiers in Cellular Neuroscience. 2016; 10: 1-16. doi: 10.3389/fncel.2016.00285.
88. Yan F, Gao Z, Chen P, Huang L, Wang D, Chen N, Wu R, Feng J, Cui S, Lu W, Wang JH. Coordinated plasticity between barrel cortical glutamatergic and GABAergic neurons during associative memory. Neural Plasticity. 2016; 2016: 1-20.
89. Yang Z, Gu E, Lu X, Wang JH. Essential role of axonal VGSC inactivation in time-dependent deceleration and unreliability of spike propagation at cerebellar Purkinje cells. Mol Brain. 2014; 7: 1. doi: 10.1186/1756-6606-7-1.
90. Chen N, Zhu Y, Gao X, Guan S, Wang JH. Sodium channel-mediated intrinsic mechanisms underlying the differences of spike programming among GABAergic neurons. Biochemical and Biophysical Research Communications. 2006; 346: 281-7.
91. Ge R, Qian H, Chen N, Wang JH. Input-dependent subcellular localization of spike initiation between soma and axon at cortical pyramidal neurons. Mol Brain. 2014; 7: 26. doi: 10.1186/1756-6606-7-26.
92. Chen N, Chen X, Wang JH. Homeostasis established by coordination of subcellular compartment plasticity improves spike encoding. Journal of Cell Science. 2008; 121: 2961-71.
93. Wen B, Qian H, Feng J, Ge RJ, Xu X, Cui ZQ, Zhu RY, Pan LS, Lin ZP, Wang JH. A Portion of Inhibitory Neurons in Human Temporal Lobe Epilepsy are Functionally Upregulated: An Endogenous Mechanism for Seizure Termination. CNS Neurosci Ther. 2015; 21: 204-14. doi: 10.1111/cns.12336.
94. Begon S, Pickering G, Eschalier A, Mazur A, Rayssiguier Y, Dubray C. Role of spinal NMDA receptors, protein kinase C and nitric oxide synthase in the hyperalgesia induced by magnesium deficiency in rats. Br J Pharmacol. 2001; 134: 1227-36. doi: 10.1038/sj.bjp.0704354.
95. Keenan C, Goode N, Pears C. Isoform specificity of activators and inhibitors of protein kinase C gamma and delta. FEBS Lett. 1997; 415: 101-8. doi: S0014-5793(97)01104-6.
96. Kukreja RC, Qian YZ, Okubo S, Flaherty EE. Role of protein kinase C and 72 kDa heat shock protein in ischemic tolerance following heat stress in the rat heart. Mol Cell Biochem. 1999; 195: 123-31.
97. Serrano PA, Rodriguez WA, Pope B, Bennett EL, Rosenzweig MR. Protein kinase C inhibitor chelerythrine disrupts memory formation in chicks. Behav Neurosci. 1995; 109: 278-84.