
INTRODUCTION
According to cancer stem-cell theory, malignant tumors are heterogeneous, with a sub-population of tumor cells with stem features [1, 2]. There is growing evidence for the role of these sub-populations of cancer stem-cells in drug resistance [2, 3, 4 ], even though there are few in situ studies on human tumor samples. In human samples of renal cell carcinoma, we recently demonstrated that sunitinib, a tyrosine kinase inhibitor, was able to generate resistance to its own therapeutic effect in cancer stem cells via induced hypoxia [5]. In women with localized breast cancer, resistance to chemotherapy delivered before surgery is associated with larger numbers of cancer stem-cells after treatment [6].
The most severe breast cancer in younger women, associated with poor prognosis even when treated at a localized stage [7], is triple negative breast cancer (TNBC) defined by lack of expression of HER2, estrogen and progesterone receptors. The standard care for localized TNBC, when inflammatory or over 3 cm in diameter, is neoadjuvant chemotherapy before surgical removal of the primary tumor [8]. The absence of residual tumor at the time of surgery defines complete pathological response (pCR) [9], which is a relevant prognostic endpoint in clinical trials evaluating neoadjuvant chemotherapy for breast cancer [10]. The prognosis for women with pCR is excellent [9], but when pCR is not achieved, TNBC patients have a high relapse rate and poor survival [7]. Factors predicting pCR, and thus response to neoadjuvant chemotherapy, are still lacking.
The mechanisms by which cancer stem-cells resist anticancer agents are also not deciphered. Macro-autophagy, here referred to as autophagy, is a lysosomal pathway whereby a cell digests its own cytoplasmic components [11]. Initially described as a cell death mechanism [12], autophagy is also a cell survival pathway to escape programmed cell death and maintain cellular homeostasis, and that can be upregulated in quiescent cells [13]. It can thus be a survival process for cancer cells in response to intrinsic or extrinsic stress conditions, including hypoxic stress [14–16]. BNIP3L, an autophagy related protein, is linked to hypoxia: HIF1α induces its expression, leading to the activation of BECLIN1 and the autophagy pathway [16, 17]. Recent in vitro studies have also demonstrated the critical role of autophagy in the maintenance of breast cancer stem-cells [18, 19].
We investigated here the relationship between complete pathological response after neoadjuvant chemotherapy and breast cancer stem-cell characteristics in pre-treatment biopsies of 78 women with TNBC. Using patient-derived xenografts obtained from women with metastatic TNBC, we further investigated the role of autophagy in the chemoresistance of breast cancer stem-cells.
RESULTS
Patient follow-up, biopsies and pCR
Table 1 shows clinical data for 78 women with a ductal TNBC, prospectively enrolled in a registry and treated with neoadjuvant chemotherapy at Saint-Louis-Hospital between 2005 and 2011.
Table 1: Pretreatment characteristics and univariate associations with pCR
pCR |
non-pCR |
p |
|
|---|---|---|---|
Clinical data |
|||
Median age (years) |
48 |
52 |
ns |
SBR grade |
|||
I–II |
30% |
21% |
ns |
III |
70% |
79% |
ns |
Chemotherapy regimen |
|||
SIM |
25% |
17% |
ns |
4 cycles EC - 4 cycles D |
65% |
69% |
ns |
Taxane-based |
10% |
14% |
ns |
Pathological data |
|||
CD133-expressing cells |
3.5% (± 0.9) |
10.4% (± 4) |
< 0.01 |
ALDH1-expressing cells |
2.6% (± 2.8) |
8.6% (± 5.9) |
< 0.01 |
CD146-expressing cells |
6.3% (± 3.8) |
17.8% (± 6.3) |
< 0.01 |
CD133/ALDH1 |
|||
co-expressing cells |
1.2% (± 1.6) |
6.3% (± 3.6) |
< 0.01 |
CD133/CD146 |
|||
co-expressing cells |
3.1% (± 1.1) |
9.2% (± 3.1) |
< 0.01 |
Necrosis |
|||
Yes |
25% |
74% |
< 0.01 |
No |
75% |
26% |
< 0.01 |
SBR: Scarff, Bloom and Richardson grading system.
SIM: Dose-dense epirubicin-cyclophosphamide-based regimen (Lehmann-Che et al., 2010).
EC = epirubicin (100 mg/m²/cycle) and cyclophosphamide (500 mg/m²/cycle), one cycle every 21 days.
D = docetaxel (100 mg/m²/cycle), one cycle every 21 days.
The 78 pre-treatment biopsies, performed with a 16-gauge needle, provided samples with a mean length of 13.21 mm (± 1.56 mm), mean width 1.14 mm (± 0.12 mm), and mean surface area 15.05 mm² (± 1.88 mm²).
Breast surgery was performed after neoadjuvant chemotherapy for all patients. The same two pathologists (PB, AR) analyzed all surgical pieces. pCR, defined as absence of residual disease at the time of surgery in both breast and axillary lymph nodes, was found for 20 surgical pieces (25.6%). Over a median clinical follow-up of 33.2 months (range 4–75.5), the relapse rate was 14.8% for pCR patients, differing significantly (p < 0.01) from the 59.2% relapse rate for non-pCR patients (Supplementary Figure 1).
Cancer stem-cell characterization and counts in patient tumor samples (Figure 1, Table 1)
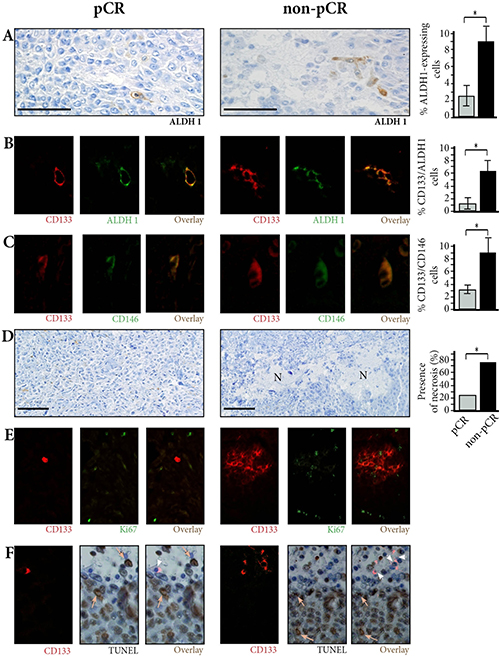
Figure 1: Breast cancer stem-cells in pre-treatment biopsies. (A) ALDH1-expressing cells are few in pCR patients, more numerous in non-pCR patients. Immunoperoxydase ×400. (B) Co-expression of CD133 and ALDH1 markers is found in tumor cells. Double immunofluorescence (IF) ×800. (C) Co-expression of CD133 and CD146 markers is found in tumor cells. Double IF ×800. (D) Small areas of necrosis (N) are found in non-pCR patients. ×200. (E) Ki67-expressing cells do not co-express CD133 except for one cell in the non-pCR patient. Double IF ×400. (F) CD133-expressing cells have blue, negative nuclei on TUNEL assay (arrowheads), contrasting with characteristic brown, apoptotic nuclei (arrows). Combined CD133 fluorescence labeling and TUNEL assay. ×400.
We identified and counted breast cancer stem-cells in pre-treatment biopsies using CD133, CD146 and ALDH1 immunostaining.
Counted on single immunoperoxydase staining (Figure 1A), CD133 expressing cells, ALDH1 expressing cells as well as CD146 expressing cells were significantly more numerous in non-pCR versus pCR patients (10.4% vs. 3.5%, p < 0.01; 8.6% vs. 2.6%, p < 0.01, and 17.8% vs. 6.3%, p < 0.01 respectively).
Since CD133, ALDH1, and CD146 might not precisely identify the same stem-cells, we performed double immunofluorescence stainings and counted the cells co-expressing CD133 and ALDH1 (Figure 1B), and the cells co-expressing CD133 and CD146 (Figure 1C). In the 78 biopsies, the number of CD133/ALDH1 coexpressing cells and the number of CD133/CD146 coexpressing cells were again significantly larger in non-pCR patients than in pCR patients (6.3% ± 1.2 vs. 1.2% ± 0.3 for CD133/ALDH1 coexpressing cells, and 9.2% ± 3.1 vs. 3.1% ± 1.1 for CD133/CD146 coexpressing cells) (p < 0.01 in both cases).
Breast cancer stem-cells are in a quiescent autophagic state related to hypoxia in patient tumor samples
As biopsies had been performed at some distance from necrotic areas detectable on ultrasonography, necrosis, when present, was restricted to small areas of mean 0.37 mm² (± 0.14 mm²) (Figure 1D). Necrosis being an indirect marker of hypoxia [5], we assessed presence or absence of necrosis on the biopsies. Presence of necrosis was significantly associated with non-pCR (p < 0.01), and with the number of CD133/ALDH1 or CD133/CD146 co-expressing cells (p < 0.01).
We then assessed proliferation and apoptosis in patient tumor samples (Figure 1E and 1F, Supplementary Table 1). When all tumor cells were counted, the Ki67 proliferation rate and apoptotic cell numbers were not significantly different in pCR and non-pCR patients (41% versus 32.3%, p = 0.14; and 24% versus 20.5%, p = 0.21 respectively). When only CD133 expressing cells were counted in non-pCR patients, proliferation and apoptosis rates were both significantly lower than in all tumor cells (p < 0.05).
To determine if this quiescent state of breast cancer stem cells was linked to activation of autophagy pathway, we assessed the expression of three autophagy related proteins: BNIP3L, BECLIN1, and LC3B. Particularly, for LC3B marker which enables the detection of autophagic vesicles [20, 21], we observed typical cytoplasmic punctuated staining (Figure 2B), as previously reported [22]. When all tumor cells were counted, numbers of BECLIN1 expressing cells, BNIP3L expressing cells, and LC3B expressing cells were significantly smaller in pCR patients than in non-pCR patients (1.1% vs. 5.8%, 8.5% vs. 18%, and 3.6% vs. 12% respectively, p < 0.05).
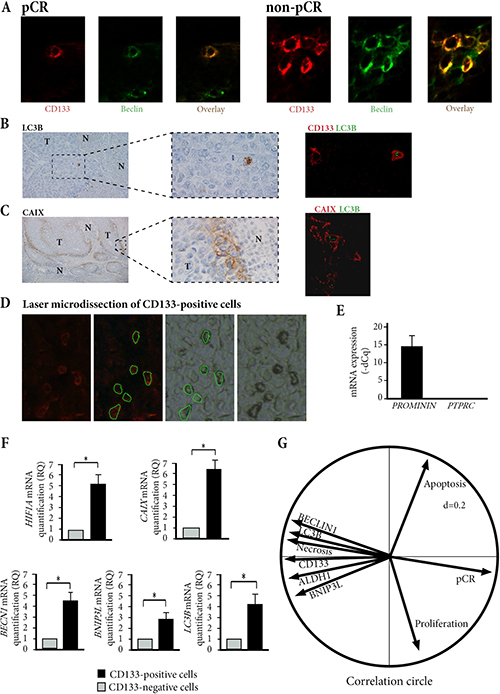
Figure 2: Hypoxia and autophagy markers in breast cancer stem-cells. (A) Co-expression of CD133 and BECLIN1 in pCR and non-pCR patients. Double IF X800. (B) Tumor cells (T) distributed around necrosis (N) express LC3B with typical cytoplasmic punctuated staining, ×1000. Double IF shows that some CD133-positive tumor cells co-express LC3B. ×400. (C) Tumor cells (T) distributed around necrosis (N) express CAIX, on their cytoplasmic membrane, ×1000. Double IF shows a co-expression of LC3B and CAIX in some tumor cells. ×400. (D) Laser microdissection of CD133-positive cells. (E) Laser-microdissected CD133-positive cells express PROMININ and not PTPRC (CD45). (F) mRNA relative quantification of HIF1A, CAIX, LC3B, BECN1, and BNIP3L show a significantly higher expression level in CD133-positive cells versus CD133-negative cells, in the 35 non-pCR biopsies. *p < 0.05. (G) Multivariate analysis of tumor cell characteristics and pCR shows an inverse correlation between pCR and cells bearing markers of stemness or autophagy.
In CD133-expressing cells, the percentage of cells with autophagic activity was significantly higher than in all tumor cells, of 68%, 79% and 75% for BECLIN1, BNIP3L, and for LC3B respectively (p < 0.01) (Figure 2A, 2B and Supplementary Table 1). We checked a possible link between hypoxia and autophagy marker expression in these breast cancer stem-cells. On pre-treatment biopsies, tumor cells expressing carbonic anhydrase IX (CAIX), a hypoxic-related protein [23], were distributed in peri-necrotic areas (Figure 2B). In addition, double immunostaining showed that LC3B-expressing cells co-expressed CAIX (Figure 2C), and were thus preferentially distributed in the peri-necrotic areas. To control these results, we used laser-microdissection of CD133-expressing and CD133-negative tumor cells in pre-treatment biopsies of non-pCR patients (Figure 2D and 2E). We found that CD133-expressing stem-cells had significantly higher mRNA expression levels of BECN1, BNIP3L, MAPLC3B, CAIX and HIF1A than CD133-negative tumor cells (p < 0.05) (Figure 2F).
Multivariate analysis
Cancer stem-cell numbers, presence of necrosis, and numbers of Ki67-, TUNEL-, BECLIN1- BNIP3L-, or LC3B-expressing cells were studied in multivariate analysis for their correlation with pCR. Multivariate forward regression showed that CD133, BECLIN1, BNIP3L and LC3B expression was negatively associated with pCR (p < 0.05) and the principal component analysis also assessed this negative correlation (Figure 2G).
Taken together, breast cancer stem-cells in pre-treatment biopsies of TNBC patients with non-pCR after neoadjuvant chemotherapy were neither proliferative nor apoptotic, but in an autophagic state related to hypoxia.
Patient-derived xenografts are relevant pre-clinical pharmacological models
Four patient-derived xenograft murine models (XBC1 to XBC4), previously obtained from samples of four metastatic TNBC, were classified from transcriptomic analyses as basal-like 1 (BL1), basal-like 2 (BL2), and stem-like (Stm) (Supplementary Excel file, Supplementary Table 2, and Figure 3A). The molecular signature of each xenograft model remained unchanged over the 10 successive passages used in this study.
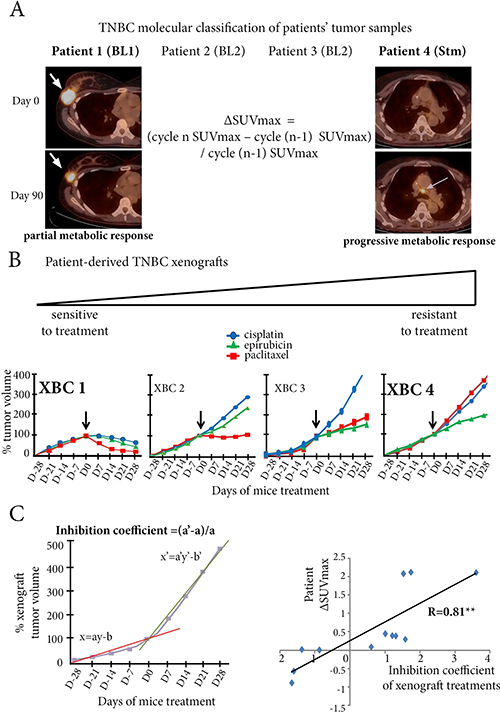
Figure 3: Patient-derived TNBC xenografts are relevant preclinical pharmacological models. (A) Patients 1, 2, 3 and 4 have breast cancers with basal-like 1 (BL1), basal-like 2 (BL2), BL2, and stem-like (Stm) molecular types respectively. Patient 1 (left panel) has a breast lesion (white arrow) on fused PET/CT with intense fluoro-deoxyglucose uptake at Day 0 and a marked decrease in SUVmax at Day 90 after 2 cycles of epirubicin-cyclophosphamide. In contrast (right panel), a subcarinal hypermetabolic lymph node (grey arrow) appears at Day 90 in Patient 4, after 2 cycles of paclitaxel and bevacizumab. (B) Corresponding patient-derived xenografts obtained from tumor samples from these four patients. Treatment with cisplatin, epirubicin, or paclitaxel identifies two opposite models, one chemosensitive (XBC1, derived from Patient 1), the other chemoresistant (XBC4, derived from Patient 4). XBC2 and XBC3 models have an intermediate profile of drug sensitivity. (C) In all xenograft models, the coefficient of inhibition for a drug is calculated as (a’-a)/a, a being the slope of the curve before the start of treatment (Day 0), and a’ the slope of the curve between Day 0 and Day 28 of treatment. The right hand panel shows a significant correlation (R = 0.81) between ΔSUVmax of a patient chemotherapy regimen with the coefficient of inhibition of the corresponding xenograft with the same chemotherapy. **p < 0.01.
Each xenograft model was tested for three cytotoxic drugs used for patients with metastatic TNBC: epirubicin, paclitaxel, and cisplatin. Of the four models, one, XBC1, was chemosensitive to the three drugs, and one, XBC4 corresponding to the stem-like tumor, was chemoresistant. XBC2 and XBC3 xenograft models had intermediate sensitivity to the three drugs (Figure 3B).
Each individual xenograft was also tested for drugs used for the corresponding patient during her clinical course (Supplementary Table 3). For each chemotherapy tested in mice, an inhibition coefficient was calculated (Figure 3C), using a ratio of the slopes (a and a’) of the straight lines before and after treatment. We found a strong correlation between the ΔSUVmax of a chemotherapy in a patient and the inhibition coefficient in the corresponding xenograft with the same chemotherapy (R = 0.81, p < 0.01) (Figure 3C).
Overall, our xenograft models from human metastatic TNBC were relevant pre-clinical pharmacological models to study chemoresistance.
Experimental hypoxia induced autophagy and resistance to drugs in cancer stem-cells derived from the sensitive TNBC xenograft model
Further experiments were performed using XBC1 chemosensitive and XBC4 chemoresistant patient-derived xenograft models.
When we assessed CD133, ALDH1, and CD146 expression in the two models, the numbers of cells expressing CD133 or CD146 were significantly larger in XBC4 than in XBC1. As in patients, CD133-expressing cells did not co-express Ki67 and were negative on TUNEL assay (Supplementary Figure 2).
We grew spheres larger than 100 μm diameter [24] from CD133-positive cells sorted from XBC1 and XBC4 models (Figure 4A). Under normoxic conditions, CD133-expressing cells and CD146-expressing cells were more numerous in XBC4 spheres than in XBC1 spheres (p < 0.05) (Figure 4B).
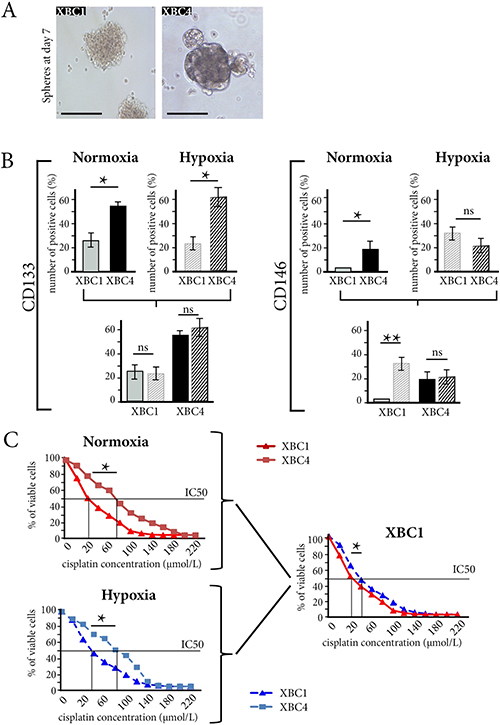
Figure 4: Experimental hypoxia induces autophagy and drug resistance in cancer stem-cells derived from XBC1, the sensitive TNBC xenograft. (A) Spheres from XBC1 and XBC4 at Day 7 after sorting of CD133-expressing cells from tumors. ×40. (B) Under normoxia, the number of CD133- or CD146-expressing cells is larger in XBC4 spheres than in XBC1spheres. Hypoxia increases the number of CD146-expressing cells in XBC1, but not in XBC4. *p < 0.05, **: p < 0.01, ns = not significant. (C) Under normoxia, XBC1spheres have a higher sensitivity to cisplatin than XBC4 spheres. Hypoxia increases drug resistance of XBC1 spheres. *p < 0.05, IC = inhibitory concentration
When XBC1 spheres were cultured under hypoxic conditions, the number CD146-expressing cells increased from 3 to 31% (p < 0.01), a high percentage comparable to the 20% of CD146-expressing cells in XBC4 spheres (Figure 4B).
For autophagy markers, in normoxia, mRNA BECN1 and BNIP3L levels were overexpressed in XBC4 spheres compared to XBC1 spheres (p < 0.05). Experimental hypoxia increased BECN1 and BNIP3L mRNA levels in both XBC1 and XBC4 spheres (p < 0.05) (Supplementary Figure 3).
Under normoxic conditions, resistance to cisplatin, epirubicin and paclitaxel was higher for XBC4 spheres than for XBC1 spheres. Experimental hypoxia increased resistance to the three drugs in XBC1 spheres, but not in XBC4 spheres (Supplementary Table 4, Figure 4C for cisplatin).
In vitro autophagy inhibition reversed chemoresistance in cancer stem-cells derived from the resistant TNBC xenograft model
To understand the role of autophagy in chemoresistance of breast cancer stem cells, we invalidated BECN1 or BNIP3L expression in XBC4 spheres. To knock out these two genes, we used the innovative CRISPR-CAS9 technology, particularly suited to spheres with brief viability (Supplementary Figure 4). 48 h after transfection and antibiotic selection, we obtained significant decreases in mRNA copy numbers: 71% (from 1798 to 525, p < 0.05) for BECN1 and 65% (from 683 to 239, p < 0.05) for BNIP3L (Figure 5A).
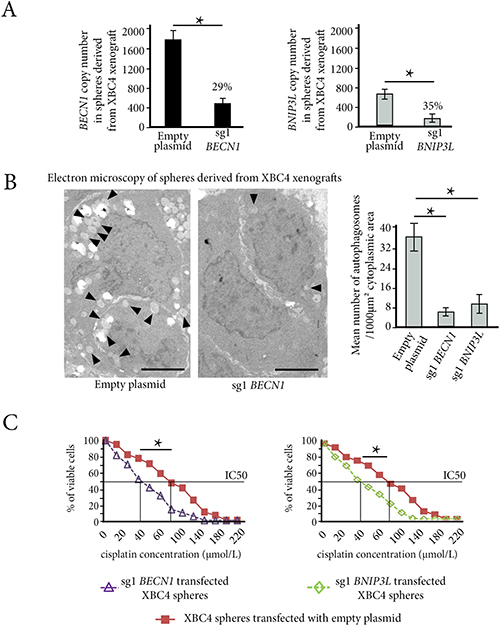
Figure 5: In vitro autophagy inhibition reverses chemoresistance in cancer stem-cells derived from XBC4, the resistant TNBC xenograft model. (A) Invalidation of BECN1 or BNIP3L genes with CRISPR-Cas9 technology in XBC4 spheres. Bar graphs show a decrease of 71% in BECN1 copy number in sg1BECN1 transfected cells (left panel), and a decrease of 65% in BNIP3L copy number in sg1BNIP3L cells (right panel). (B) The mean number of autophagosomes per 1000 μm² of cytoplasmic area on electron microscopy is lower in XBC4 spheres transfected with sg1BECN1 or with sg1BNIP3L than in XBC4 spheres transfected with empty plasmids. (C) Under normoxia, spheres from XBC4 transfected with sg1BECN1 or with sg1BNIP3L have a higher sensitivity to cisplatin than spheres transfected with empty plasmids. *p < 0.05.
Electron microscopic study focused on cytoplasmic areas of tumor cells (see Methods and Supplementary Figure 5). It showed a striking difference between tumor cells transfected with empty plasmid, which had large numbers of cytoplasmic autophagosomes, and sg1BECN1 or sg1BNIP3L transfected tumor cells (Figure 5B).
Counts performed on cytoplasmic areas of 25 cells in each experimental condition showed that the mean number of autophagosomes per 1000 μm² of cytoplasmic area was 35.8 in cells transfected with empty plasmid versus 6.5 and 9.8 in sg1BECN1 and sg1BNIP3L transfected cells respectively (p < 0.05 in both cases).
Resistance to cisplatin, epirubicin and paclitaxel significantly decreased in XBC4 spheres transfected either with sg1BECN1 or sg1BNIP3L when compared to XBC4 spheres transfected with empty plasmid (illustrated for cisplatin in Figure 5C, Supplementary Table 5).
In vivo autophagy inhibition reversed chemoresistance in resistant TNBC xenograft model
We assessed the tumorigenicity of XBC4 spheres co-transfected with GFP and sg1BECN1 or sg1BNIP3L. Using laser micro-dissection in liquid medium, GFP-positive cells were selected 48 h after transfection and antibiotic selection (Figure 6A). We checked that these cells were viable and that they also had a more than 65% decrease in BECN1 or BNIP3L copy numbers, but when 200 cells were injected sub-cutaneously in 8 mice, there was no engraftment (Figure 6B). Conversely, when we selected GFP-negative cells, they had no significant decrease in BECN1 or BNIP3L copy numbers, and 200 cells were tumorigenic in 6/8 and 7/8 grafted mice respectively (Figure 6B).
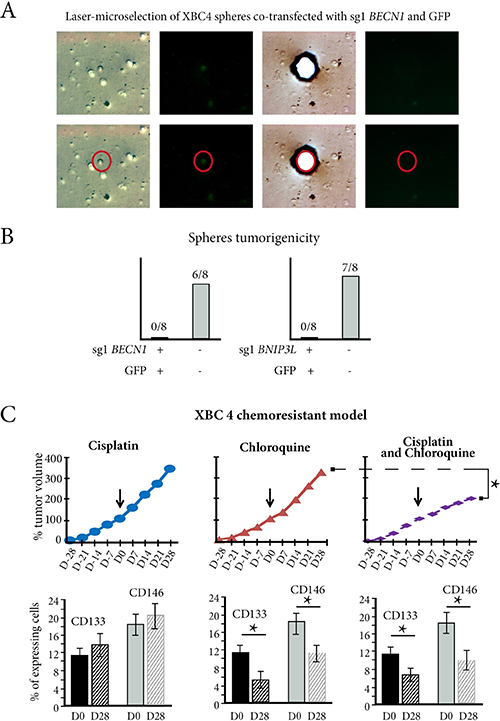
Figure 6: In vivo autophagy inhibition reverses chemoresistance in resistant TNBC xenograft. (A) Cells from XBC4 spheres co-transfected with sgBECN1 and GFP have green fluorescence, enabling a single cell laser-microdissection (red cycle). (B) GFP positive-cells (GFP+) co-transfected with sgBECN1 or with sg1BNIP3L are not tumorigenic, while GFP-negative cells are. (C) Treatment of the XBC4 chemoresistant model with chloroquine does not inhibit tumor growth at Day 28, but the numbers of CD133 or CD146-expressing cells decrease. Chloroquine added to cisplatin inhibits tumor growth when compared to chloroquine alone. It also induces a decrease in CD133 or CD146-expressing cell numbers when Day28 is compared to Day 0. *p < 0.05.
When we used an autophagy inhibitor, chloroquine [25], to treat the three cisplatin-resistant xenografts, XBC2, XBC3 and XBC4 (Figure 6C and Supplementary Figure 6), there was no change in tumor growth at Day 28, but the numbers of CD133 or CD146-expressing tumor cells significantly decreased (p < 0.05) (illustrated for XBC4 in Figure 6C).
When chloroquine was added to cisplatin, there was significant tumor growth inhibition compared to treatment with chloroquine alone in the three cisplatin-resistant xenografts: the percentage of tumor volume at Day28 was 267% versus 365% for chloroquine alone (p < 0.01) for XBC2; 208% versus 432% for chloroquine alone (p < 0.01) for XBC3; 202% versus 345% for chloroquine alone (p < 0.01) for XBC4 (Supplementary Figure 6).
The combination of cisplatin and chloroquine in the three cisplatin-resistant xenografts also induced a significant decrease in the numbers of CD133 or CD146-expressing cells when Day28 was compared to Day 0 (p < 0.01) (illustrated for XBC4 in Figure 6C).
We thus demonstrated that the molecular or chemical inhibition of the autophagic pathway was able to reverse chemoresistance in triple negative breast carcinoma.
DISCUSSION
In this study, we demonstrated the role of hypoxia and autophagy in the resistance to chemotherapy of cancer stem-cells in TNBC.
In pre-treatment biopsies of 78 patients with TNBC, we first showed that chemoresistance was associated with large numbers of cancer stem-cells. These cancer stem-cells were hypoxic, and this was not due to tissue processing since all pre-treatment biopsies were performed under ultrasonography to avoid large necrotic areas. Biopsies were calibrated and processed identically, enabling us to analyze comparable areas of tumor tissue. [26] After chemotherapy, pathological response was assessed on breast surgery pieces. The pCR rate we found was 25.6%, close to the 27.9% pCR rate reported in 663 TNBCs treated with neoadjuvant chemotherapy. [10]
For in situ characterization of breast cancer stem cells, we did not use the combination of markers CD44+CD24-/low, [18, 27] since this combination of positive and negative markers, currently used in cytometry, is more difficult to translate into in situ tissue analyses and cell counts. [28] CD133, ALDH1 and CD146 are three positive markers used for studying triple negative breast cancer stem-cells. [29–32] In addition, CD133 and ALDH1 cancer stem cells are preferentially distributed in hypoxic areas [5, 33] while CD44+CD24-/low breast cancer stem cells are mainly found on the tumor invasive edge. [28] In the pre-treatment biopsies studied here, whether counted on single staining or on combined double staining, breast cancer stem-cell numbers were significantly larger in non-pCR patients than in pCR patients. This could result from changes in proliferation and/or apoptosis rates. But the Ki67 proliferation rate was significantly lower for stem-cells than for tumor cells as a whole, a result coherent with in vitro studies showing that ALDH1-positive breast cancer stem-cells are not in a proliferative state. [34] In the same pre-treatment biopsies, the apoptosis rate was also significantly lower for stem-cells than for tumor cells as a whole. This fact, not hitherto reported, could contribute to the maintenance of a large number of cancer stem-cells in non-pCR patients.
The role of autophagy in the quiescent state of cancer stem-cells [2] and in the maintenance of breast cancer stem-cells [18, 19, 27, 35] has recently been demonstrated. When we looked for autophagy markers in our pre-treatment biopsies of non-pCR TNBCs, we found that CD133-expressing cancer stem cells co-expressed BECLIN1, LC3B, and BNIP3L. When we used another independent method based on laser-microdissection of CD133-expressing cells combined with quantitative PCR, we also found an expression of the three autophagy markers in cancer stem-cells. A multivariate analysis based on cell counts found an inverse correlation between the number of cells expressing markers of stemness or autophagy and pCR, a bio-clinical correlation hitherto not demonstrated.
On the basis of our experience with patient-derived renal cancer xenograft for the study of cancer stem-cells and resistance to drugs, [5] we implemented patient-derived xenografts of TNBC. Patient-derived xenografts are more relevant pharmacological tools than xenografts from human cancer cell lines, which have poor reliability in predicting response to chemotherapy in patients. [36–38] Using four patient-derived xenograft models with genomic stability over passages, we found that three of them had resistance to cisplatin. Two of the four models had opposed sensitivity to three drugs used to treat women with metastatic TNBC. These two models reproduced the clinical response to drugs, with a large number of quiescent cancer stem-cells in the chemoresistant model. We focused our pre-clinical studies on breast cancer stem-cells derived from these two opposite models to study the mechanisms of drug resistance in TNBC stem-cells.
We first demonstrated that hypoxia increased drug resistance of autophagic TNBC stem-cells. Using spheres derived from the chemosensitive model, we showed that experimental hypoxia increased the number of autophagic cancer stem-cells, a result in accordance with in vitro studies using human cancer cell lines. [19, 39, 40]
In the same chemosensitive model, experimental hypoxia also increased resistance to cisplatin, epirubicin, and paclitaxel. So far, a link between hypoxia and drug resistance of cancer stem-cells has only been demonstrated for anti-angiogenic drugs. Sunitinib generates tumor hypoxia and increases the numbers of cancer stem-cells in xenografts obtained from human TNBC cell lines. [33] In human renal carcinomas, we demonstrated that sunitinib was able to generate resistance to its own therapeutic effect via induced hypoxia in cancer stem cells. [5] Here, on TNBC cancer stem-cells, we studied three cytotoxic drugs with main effects not on angiogenic vessels but on tumor cells. Cisplatin forms platinum adducts with DNA, which impedes DNA replication and transcription, thus leading to apoptotic cell death. [41] Paclitaxel is a taxoid that binds to tubulin polymers and inhibits microtubule depolymerisation, thus disrupting tumor cell microtubule dynamics and leading to apoptotic cell death. [42] Epirubicin is an anthracycline that inhibits the activity of topoisomerase II, thus impairing tumor cell DNA replication. However, a recent study demonstrated that paclitaxel is able to induce the HIF pathway in TNBC cell lines. [43] Thus, although it does not target angiogenic vessels, paclitaxel is able to induce hypoxia in tumoral tissue. The originality of our results is that they link hypoxia, autophagy and drug resistance in TNBC stem-cells. We used experimental hypoxia on TNBC stem-cells to reproduce the results found in biopsies of TNBC patients resistant to chemotherapy who had a large number of stem-cells expressing autophagic markers around necrotic areas.
This link between hypoxia, autophagy and drug resistance in TNBC stem-cells raises a new issue for the translational relevance for women with metastatic TNBC. Is it possible to modulate autophagy in breast cancer stem-cells to reverse resistance to chemotherapy? In recent years, autophagy has been proposed as a potential target for cancer therapy. [25] There are pre-clinical [44, 45] and clinical [46, 47] data on the potential benefit of autophagy inhibition as an anticancer therapy. Particularly, in a pediatric brain tumor with BRAF mutation, hydroxycholoroquine was able to reverse acquired resistance to BRAF inhibitor. [48] In our study, we demonstrated both in vitro and in vivo that autophagy inhibition was able to reverse resistance to drugs. In vitro, using spheres derived from the chemoresistant patient-derived xenograft model, the molecular invalidation of autophagy markers enabled us to reverse chemoresistance to cisplatin, epirubicin and paclitaxel. In vivo, we used chloroquine, a chemical inhibitor of autophagy, and showed that chloroquine alone induced a decrease in numbers of cancer stem cells, but with limited cytotoxic effect and thus no change in tumor growth inhibition, two effects previously described in in vitro models [49, 50]. However chloroquine added to cisplatin was able to reverse resistance to cisplatin in the three cisplatin-resistant patient-derived xenograft TNBC models, probably as a result of autophagy inhibition in the breast cancer stem cells but also in the whole tumor.
This study also affects existing knowledge in the field of response to therapy in TNBC, since it identifies a marker predictive of response to chemotherapy in biopsies performed before any treatment, thus at the optimal moment to design a therapeutic strategy.
The clinical relevance of our results is a major strength of this work, and this has been obtained through rigorous clinical, radiological and pathological methodologies. However, the generalisation of the conclusions requires a validation cohort.
MATERIALS AND METHODS
Patients and tumor samples
In the Centre-Maladies-du-Sein, Hôpital-St-Louis, Paris, a registry of patients treated with neoadjuvant chemotherapy for localized breast carcinoma prospectively enrolled 361 patients between 2005 and 2011. For diagnostic purposes, all patients underwent core needle biopsies before chemotherapy; 107 had ductal triple negative adenocarcinoma (TNBC).
Ultrasonography-guided pre-treatment biopsies were similarly performed, using a 16-gauge needle, at least 1 cm from necrotic areas detected on ultrasonography. Tissue samples were similarly processed, one formaldehyde-fixed and paraffin-embedded, and one snap-frozen.
In compliance with French Bioethics law (2004–800, 06/08/2004), all patients had been informed of the research use of the part of their samples remaining after diagnosis had been established, and did not oppose it. Informed consent was obtained for each patient. Tumor tissue was available for this study for 78 of the 107 TNBC patients.
Surgical pieces obtained after neoadjuvant chemotherapy were independently analyzed by two pathologists (AR, PB) unaware of response to treatment. pCR was defined, according to [10], by absence of residual tumor at time of surgery both in breast and axillary lymph node samples.
Radiotherapy was delivered after surgery to patients who had breast-conserving surgery or mastectomy for initial advanced stages. The patients’ follow-up was clinical, biological and radiological according to institutional guidelines, and the relapse rate was calculated considering local and metastatic relapse, or cancer-related death.
Necrosis assessment in human tumor samples
The 78 pre-treatment biopsies were reviewed by two pathologists unaware of response to treatment (AJ, CL) for the presence or absence of necrotic areas. When present in any field of the two different sections analyzed for each biopsy, necrosis was delineated on virtual slides created on a Nanozoomer2.0H scanner (Hamamatsu/Japan), and quantified using DotSlide2 software. Results were expressed as the sum of necrotic areas for each biopsy, and the mean ± SEM.
Cancer stem-cell characterization and counts in human tumor samples
CD133, ALDH1 and CD146 expression was assessed and cells counted on TNBC pre-treatment biopsies; we also counted ALDH1/CD133 and CD146/CD133-coexpressing cells (Supplementary Methods).
Proliferation, apoptosis, autophagy and cancer stem-cells in human tumor samples
On pre-treatment biopsies, i) tumor cell and cancer stem-cell proliferation rates were assessed; ii) in situ apoptosis was assessed using TUNEL assay, and CD133-expressing/TUNEL-expressing cells were counted; iii) the expression of three cellular markers of autophagy, BECLIN1, BNIP3L, and LC3B was assessed (Supplementary Methods).
Autophagy and hypoxia in cancer stem-cells of human tumor samples
To characterize hypoxic areas on non-pCR pre-treatment biopsies, we assessed the in situ expression of a hypoxic-related protein, CAIX.
To check any possible co-expression of hypoxic and autophagy markers in cancer cells, we performed double immunofluorescent stainings using anti-human LC3B and CAIX (NB100-417, Novus Biologicals, France) antibodies, labelled with Apex-alexa-Fluor 488 nm and 647 nm (A10475, Life-Technology) respectively.
To control these immunostaining results, we used a completely different method combining morphological analyses and molecular markers. On frozen pre-treatment biopsies, CD133-expressing cells were laser-microdissected and their BNIP3L, BECN1, MAP1LC3B, CAIX and HIF1A-expression levels assessed using quantitative PCR (Supplementary Methods).
Patients with metastatic TNBC and gene expression profiling
Four women with metastatic TNBC, different from the 78 other patients, participated in this study.
For each patient, five samples of a metastasis were obtained during the procedure of imaging-guided biopsy at the time of relapse, before any medical treatment. Informed written consent was obtained from the patients. The Clinical Research Board Ethics Committee approved this study (CPP Ile-de-France N°13218).
Among the five tumor samples, i) two were formaldehyde-fixed and paraffin-embedded for histological analyses, ii) two were immediately snap-frozen in liquid nitrogen and stored in Hôpital-St-Louis Tumorbank for molecular analysis, iii) and one was set aside in culture medium for xenografting.
Total RNA was extracted from the frozen tumor sample as above, and transcriptomic analyses were performed using MiltenyiBiotec Microarray (Supplementary Methods). Classification was provided by correlating gene expression profiles with the centroids for each of the 6 TNBC subtypes described by Lehmann et al [51], and with Parker centroids for PAM50 classification ([52], https://genome.unc.edu/pubsup/breastGEO/pam50_centroids.txt).
Patient-derived breast cancer xenografts and treatments
Four patient-derived xenografts of human TNBC were studied (XBC1 to XBC4). They had been established for a pilot study on personalized treatment for women with metastatic triple negative breast carcinoma [53], before any medical treatment of the metastatic disease (Supplementary Methods).
The University Institute Board Ethics Committee for experimental animal studies approved this study (N°2012-15/728-0115).
For each xenograft model, the day when tumors reached a volume of 300mm3 – i.e. 100% tumor volume – was considered as Day0. Mice (n = 5 mice per treatment group) were then treated over 28 days with three types of chemotherapy (Supplementary Table 6 for drugs tested in TNBC xenografted mice). A daily clinical score was recorded and tumor growth was measured weekly until tumor weight reached the ethically recommended limit of less than 10% of mouse weight (Directive 2010/63/EU of European Parliament and Council of 22 September 2010 on the protection of animals used for scientific purposes; Official Journal of European Union L 276/33).
Assessment of tumor response in patients
For each line of chemotherapy, the patient response under treatment was characterized (Supplementary Methods).
Cancer stem-cells from xenografted human TNBC: spheres and cytotoxicity
We studied spheres obtained from untreated tumor samples of XBC1 and XBC4 xenograft models.
Cells expressing CD133 were obtained from dissociated sections using magnetic sorting, with anti-CD133 micro-bead antibody (Miltenyi-Biotech, Germany), using the manufacturer’s procedures. We controlled CD133 cell purity by flow cytometry. CD133 positive cells were placed in a low-attachment six-well plate at a density of 2000 to 5000 cells/well, and cultured in a serum-free, high-glucose medium (DMEM-F12, Gibco, France) supplemented with 2% B27-NeuroMix and 0.4% fetal-bovine-serum (PAA, France), 5 mg/mL insulin (Sigma, France), 20 ng/mL epidermal-growth-factor (R&D systems, France), and 0.5 ng/mL hydrocortisone (Sigma, France). Spheres were obtained after 48 h of culture in a humidified chamber (37°C, 5% CO2) under normoxia (20% O2) for further in vitro functional experiments and in vivo tumorigenicity experiments (Supplementary Figure 4). For cytotoxicity tests, see Supplementary Methods.
Experimental hypoxia, assessment of stem-cell markers and of mammosphere sensitivity to drugs
For the two xenograft models, XBC1 or XBC4, spheres were separated into two groups maintained in a humidified chamber for 48 h, one under experimental hypoxia (1% O2) and the other under normoxia (20% O2). For experimental hypoxia, we chose a pO2 level of 1% as it is now accepted that stem-cell niches are hypoxic with oxygen tension as low as 1% [54].
The relative number of CD133 and CD146 expressing cells in hypoxic and normoxic spheres was assessed by flow cytometry, and for cytotoxicity, MTT test was performed (Supplementary Methods).
Experimental hypoxia and autophagy in spheres
Total RNA was extracted from normoxic and hypoxic spheres. TaqMan RT-qPCR was performed to determine BECN1 and BNIP3L gene expression levels.
Knock-out of autophagy gene expression in spheres
Because of a 16-day lifetime of spheres derived from patient-derived xenografts (XBC1 and XBC4 models), CRISPR-CAS9 technology was chosen to invalidate BECN1 and BNIP3L gene expression. A dedicated algorithm (see http://crispr.mit.edu website) enabled us to choose the target sequence of 20 bases around the active sites of phosphorylation for each of the two genes. Oligos designed to build the vectors containing the sgRNA1 targeting BECN1 or BNIP3L genes are detailed in Supplementary Table 7, and the method in Supplementary Methods.
Droplet digital PCR
Droplet digital PCR was performed to assess the copy number of BECN1 or BNIP3L genes in spheres transfected with empty plasmid and spheres transfected with sg1BECN1 or sg1BNIP3L (Supplementary Methods).
Electron microscopy of spheres KO for BECN1 or BNIP3L
Spheres transfected with empty plasmid and spheres transfected with sg1BECN1 or sg1BNIP3L were dissociated and cells in suspension were counted. After centrifugation, a pellet of 106 cells was further processed for electron microscopy (Supplementary Methods). Ultrastructural analysis focused on the cytoplasms of tumor cells to detect characteristic autophagosomes according to the “Guidelines for the use and interpretation of assays for monitoring autophagy” [11].
A quantitative study was performed comparing the numbers of autophagosomes per cytoplasmic area in cells transfected with empty plasmid versus sg1BECN1 or sg1BNIP3L transfected cells (Supplementary Figure 5).
Tumorigenicity of spheres KO for BECN1 or BNIP3L
48 h after co-transfection with GFP and after antibiotic selection, a volume of 10μL of culture medium containing 2 × 105 cells dissociated from spheres was transferred and spread on a specific plate dedicated to laser-microdissection of living cells. Laser-microdissection was performed using a PALM-Microbeam/Zeiss-system equipped with a 480 nm wavelength filter. GFP-positive fluorescent cells were catapulted one by one into a cap with culture medium. GFP-negative cells were also selected using the same process. For a given set of cells, the procedure did not exceed 30 minutes. Selected cells were rinsed, and a minimum of 200 GFP-expressing cells were grafted subcutaneously in nude mice to assess the tumorigenicity of cells transfected with sg1BECN1 or sg1BNIP3L. Cells transfected with empty plasmid served as the control. Mice were followed up over a period of 6 months in dedicated animal housing.
Chemical inhibition of autophagy in XBC4 xenograft
When tumors reached a volume of 300mm3, the mice were treated for 28 days with cisplatin alone, chloroquine alone (50 mg/kg/day intra-peritoneally, Sigma-Aldrich, France), or a combination of cisplatin and chloroquine (n = 5 mice per treatment group) (Supplementary Table 8 for drugs tested in TNBC xenografted mice). A daily clinical score was recorded and tumor growth was measured weekly.
Statistics
The statistical analyses were performed using R 2.15.2 statistical software (R-Foundation for Statistical Computing, Vienna/Austria) (Supplementary methods).
CONCLUSIONS
Our results support breast cancer stem-cell evaluation in pre-treatment biopsies of TNBC patients, and further research on autophagy inhibition to reverse resistance to chemotherapy.
Authors’ contributions
Guilhem Bousquet: Conception and design, Provision of study material or patient, Data analysis and interpretation, Manuscript writing, Final approval of manuscript; Morad El Bouchtaoui: Collection and/or assembly of data, Data analysis and interpretation, Final approval of manuscript; Sophie Tan: Collection and assembly of data, Final approval of manuscript; Christophe Leboeuf: Collection and assembly of data, Manuscript writing, Final approval of manuscript; Cédric de Bazelaire: Performed tumor biopsies, Final approval of manuscript; Jean-Paul Feugeas: Performed the statistical analyses, Final approval of manuscript; Philippe Ratajczak: Data analysis and interpretation, Final approval of manuscript; Sylvie Giacchetti: Provision of study material or patient, Final approval of manuscript; Anne de Roquancourt: Assessed pathological response, Final approval of manuscript; Philippe Bertheau: Assessed pathological response, Final approval of manuscript; Laurence Verneuil: Data analysis and interpretation, Final approval of manuscript; Marc Espié: Provision of study material or patient, Final approval of manuscript; Anne Janin: Conception and design, Financial support, Administrative support, Provision of study material, Data analysis and interpretation, Manuscript writing, Final approval of manuscript.
ACKNOWLEDGMENTS
We thank Pr.Muh-HwaYang for manuscript critical review; M.ElBouchtaoui, Tumorothèque-hôpital-Saint-Louis; Mrs A.Swaine for English language.
CONFLICTS OF INTEREST
None.
FUNDING
Inserm, University Paris 7.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Ren SC, Qu M, Sun YH. Investigating intratumour heterogeneity by single-cell sequencing. Asian journal of andrology. 2013; 15:729–734.
2. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, Kost-Alimova M, Muller F, Colla S, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014; 514:628–632.
3. Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, Tamaki Y, Noguchi S. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin Cancer Res. 2009; 15:4234–4241.
4. Guan JL, Simon AK, Prescott M, Menendez JA, Liu F, Wang F, Wang C, Wolvetang E, Vazquez-Martin A, Zhang J. Autophagy in stem cells. Autophagy. 2013; 9:830–849.
5. Varna M, Gapihan G, Feugeas JP, Ratajczak P, Tan S, Ferreira I, Leboeuf C, Setterblad N, Duval A, Verine J, Germain S, Mongiat-Artus P, Janin A, et al. Stem cells increase in numbers in perinecrotic areas in human renal cancer. Clin Cancer Res. 2015; 21:916–924.
6. Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008; 100:672–679.
7. Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML, Perou CM. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007; 13:2329–2334.
8. Rastogi P, Anderson SJ, Bear HD, Geyer CE, Kahlenberg MS, Robidoux A, Margolese RG, Hoehn JL, Vogel VG, Dakhil SR, Tamkus D, King KM, Pajon ER, et al. Preoperative chemotherapy: updates of National Surgical Adjuvant Breast and Bowel Project Protocols B-18 and B-27. J Clin Oncol. 2008; 26:778–785.
9. Fisher B, Redmond CK, Fisher ER. Evolution of knowledge related to breast cancer heterogeneity: a 25-year retrospective. J Clin Oncol. 2008; 26:2068–2071.
10. von Minckwitz G, Eidtmann H, Rezai M, Fasching PA, Tesch H, Eggemann H, Schrader I, Kittel K, Hanusch C, Kreienberg R, Solbach C, Gerber B, Jackisch C, et al. Neoadjuvant chemotherapy and bevacizumab for HER2-negative breast cancer. N Engl J Med. 2012; 366:299–309.
11. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016; 12:1–222.
12. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012; 19:107–120.
13. Calabretta B, Salomoni P. Inhibition of autophagy: a new strategy to enhance sensitivity of chronic myeloid leukemia stem cells to tyrosine kinase inhibitors. Leuk Lymphoma. 2011; 52:54–59.
14. Tan Q, Wang M, Yu M, Zhang J, Bristow RG, Hill RP, Tannock IF. Role of Autophagy as a Survival Mechanism for Hypoxic Cells in Tumors. Neoplasia. 2016; 18:347–355.
15. Hu YL, DeLay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS, Aghi MK. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012; 72:1773–1783.
16. Schlie K, Spowart JE, Hughson LR, Townsend KN, Lum JJ. When Cells Suffocate: Autophagy in Cancer and Immune Cells under Low Oxygen. Int J Cell Biol. 2011; 2011:470597.
17. Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Molecular and cellular biology. 2009; 29:2570–2581.
18. Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA. Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle. 2011; 10:3871–3885.
19. Gong C, Bauvy C, Tonelli G, Yue W, Delomenie C, Nicolas V, Zhu Y, Domergue V, Marin-Esteban V, Tharinger H, Delbos L, Gary-Gouy H, Morel AP, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. 2013; 32:2261–2272, 2272e 2261–2211.
20. Schlafli AM, Berezowska S, Adams O, Langer R, Tschan MP. Reliable LC3 and p62 autophagy marker detection in formalin fixed paraffin embedded human tissue by immunohistochemistry. Eur J Histochem. 2015; 59:2481.
21. Zhao H, Yang M, Zhao J, Wang J, Zhang Y, Zhang Q. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med Oncol. 2013; 30:475.
22. Ladoire S, Penault-Llorca F, Senovilla L, Dalban C, Enot D, Locher C, Prada N, Poirier-Colame V, Chaba K, Arnould L, Ghiringhelli F, Fumoleau P, Spielmann M, et al. Combined evaluation of LC3B puncta and HMGB1 expression predicts residual risk of relapse after adjuvant chemotherapy in breast cancer. Autophagy. 2015; 11:1878–1890.
23. Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nature cell biology. 2008; 10:295–305.
24. Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY, Yang WH, Huang CH, Kao SY, Tzeng CH, Tai SK, Chang SY, Lee OK, Wu KJ. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nature cell biology. 2010; 12:982–992.
25. Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nature reviews Clinical oncology. 2011; 8:528–539.
26. de Kerviler E, Benet C, Briere J, de Bazelaire C. Image-guided needle biopsy for diagnosis and molecular biology in lymphomas. Best Pract Res Clin Haematol. 2012; 25:29–39.
27. Maycotte P, Jones KL, Goodall ML, Thorburn J, Thorburn A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Molecular cancer research. 2015; 13:651–658.
28. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, Hong S, Adams A, D’Angelo R, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports. 2014; 2:78–91.
29. Park SY, Lee HE, Li H, Shipitsin M, Gelman R, Polyak K. Heterogeneity for stem cell-related markers according to tumor subtype and histologic stage in breast cancer. Clin Cancer Res. 2010; 16:876–887.
30. Zhao P, Lu Y, Jiang X, Li X. Clinicopathological significance and prognostic value of CD133 expression in triple-negative breast carcinoma. Cancer Sci. 2011; 102:1107–1111.
31. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007; 1:555–567.
32. Zeng Q, Li W, Lu D, Wu Z, Duan H, Luo Y, Feng J, Yang D, Fu L, Yan X. CD146, an epithelial-mesenchymal transition inducer, is associated with triple-negative breast cancer. Proc Natl Acad Sci USA. 2012; 109:1127–1132.
33. Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG, Wicha MS. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA. 2012; 109:2784–2789.
34. Liu S, Patel SH, Ginestier C, Ibarra I, Martin-Trevino R, Bai S, McDermott SP, Shang L, Ke J, Ou SJ, Heath A, Zhang KJ, Korkaya H, et al. MicroRNA93 regulates proliferation and differentiation of normal and malignant breast stem cells. PLoS Genet. 2012; 8:e1002751.
35. Chaterjee M, van Golen KL. Breast cancer stem cells survive periods of farnesyl-transferase inhibitor-induced dormancy by undergoing autophagy. Bone Marrow Res. 2011; 2011:362938.
36. Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, Korn RL, Desai N, Trieu V, Iglesias JL, Zhang H, Soon-Shiong P, Shi T, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011; 29:4548–4554.
37. Caponigro G, Sellers WR. Advances in the preclinical testing of cancer therapeutic hypotheses. Nat Rev Drug Discov. 2011; 10:179–187.
38. Peterson JK, Houghton PJ. Integrating pharmacology and in vivo cancer models in preclinical and clinical drug development. Eur J Cancer. 2004; 40:837–844.
39. Song YJ, Zhang SS, Guo XL, Sun K, Han ZP, Li R, Zhao QD, Deng WJ, Xie XQ, Zhang JW, Wu MC, Wei LX. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013; 339:70–81.
40. Zhu H, Wang D, Liu Y, Su Z, Zhang L, Chen F, Zhou Y, Wu Y, Yu M, Zhang Z, Shao G. Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer cell international. 2013; 13:119.
41. Mehmood RK. Review of Cisplatin and oxaliplatin in current immunogenic and monoclonal antibody treatments. Oncology reviews. 2014; 8:256.
42. Zhang D, Yang R, Wang S, Dong Z. Paclitaxel: new uses for an old drug. Drug design, development and therapy. 2014; 8:279–284.
43. Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci USA. 2014; 111:E5429–5438.
44. Ojha R, Jha V, Singh SK, Bhattacharyya S. Autophagy inhibition suppresses the tumorigenic potential of cancer stem cell enriched side population in bladder cancer. Biochimica et biophysica acta. 2014; 1842:2073–2086.
45. Fukuda T, Oda K, Wada-Hiraike O, Sone K, Inaba K, Ikeda Y, Miyasaka A, Kashiyama T, Tanikawa M, Arimoto T, Kuramoto H, Yano T, Kawana K, et al. The anti-malarial chloroquine suppresses proliferation and overcomes cisplatin resistance of endometrial cancer cells via autophagy inhibition. Gynecologic oncology. 2015; 137:538–545.
46. Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, McAfee Q, Fisher J, Troxel AB, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014; 10:1359–1368.
47. Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, Schuchter LM, Torigian DA, Panosian JT, Troxel AB, Tan KS, Heitjan DF, DeMichele AM, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014; 10:1391–1402.
48. Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, Morgan MJ, Mirsky DM, Handler MH, Foreman NK, Thorburn A. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 2014; 4:773–780.
49. De Mei C, Ercolani L, Parodi C, Veronesi M, Lo Vecchio C, Bottegoni G, Torrente E, Scarpelli R, Marotta R, Ruffili R, Mattioli M, Reggiani A, Wade M, et al. Dual inhibition of REV-ERBbeta and autophagy as a novel pharmacological approach to induce cytotoxicity in cancer cells. Oncogene. 2015; 34:2597–2608.
50. Ojha R, Jha V, Singh SK. Gemcitabine and mitomycin induced autophagy regulates cancer stem cell pool in urothelial carcinoma cells. Biochimica et biophysica acta. 2016; 1863:347–359.
51. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. The Journal of clinical investigation. 2011; 121:2750–2767.
52. Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009; 27:1160–1167.
53. Bousquet G, Feugeas JP, Ferreira I, Vercellino L, Jourdan N, Bertheau P, de Bazelaire C, Barranger E, Janin A. Individual xenograft as a personalized therapeutic resort for women with metastatic triple-negative breast carcinoma. Breast cancer research. 2014; 16:401.
54. Mohyeldin A, Garzon-Muvdi T, Quinones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell. 2010; 7:150–161.