
Introduction
The identification of cancer susceptibility genes has provided for a greater understanding of the mechanisms of tumor biology. Furthermore, genetic associations are likely to prove increasingly valuable via the functional links they reveal, either endorsing current etiological hypotheses or suggesting novel ones that merit testing via gene-environment specific hypotheses.
While the familial clustering of B-cell malignancies was well recognized over forty years ago and a strong association between HLA and Hodgkin lymphoma (HL) risk established in 1967 [1], it is only recently that concerted efforts have been directed to understanding inherited susceptibility to hematological malignancy.
Here we discuss the impact findings from genome-wide association studies (GWAS) are having on our understanding of B-cell chronic lymphocytic leukemia (CLL) and acute lymphoblastic leukemia (ALL) development.
Inherited genetic susceptibility to CLL and ALL
B-cell CLL (MIM 151400) accounts for ~ 25% of all leukemia and is the most common form of adult lymphoid malignancy in Western countries. Evidence for inherited genetic predisposition to CLL is provided by a seven-fold elevated risk of CLL seen in first-degree relatives of CLL patients [2, 3]. Genome-wide linkage scans of CLL families have, however, consistently failed to identify a high risk disease locus for CLL locus making a polygenic model of inherited predisposition based on the co-inheritance of multiple low-risk alleles more likely [4, 5].
Acute lymphoblastic leukemia is the major pediatric cancer in economically developed countries; precursor B-cell (BCP-ALL; MIM 613065) accounting for ~ 70% of childhood ALL. In contrast to CLL, evidence for a familial risk to ALL is weak. Data from the Swedish family-cancer database does, however, lend some support to an excess risk in relatives of patients, independent of the high concordance in monozygotic twins [6], which has a non-genetic, in-utero explanation. Although rare (<5% of ALL), direct evidence for inherited genetic susceptibility is provided by the high risk of ALL associated with Bloom’s syndrome, neurofibromatosis, ataxia telangiectasia and constitutional trisomy 21. While evidence linking an environmental exposure to risk of childhood ALL has largely been inconsistent, epidemiological data for an infectious etiology is persuasive, albeit indirect [7]. Implicit in a model of ALL having an infectious etiological basis is that ALL represents a rare sequelae [7] of infection with germline variation influencing host response.
Candidate gene polymorphism studies
It has long been speculated that common polymorphic variation contributes to the susceptibility of hematological malignancy but evidence for common low risk alleles has emerged only recently. Numerous associations have been proposed from candidate gene analyses conducted throughout the past 30 years. Despite much research, no definitive susceptibility alleles have been unequivocally identified through these studies. As with many other diseases, positive associations have been reported for various polymorphisms but few of the initial positive results have been replicated in subsequent studies. The inherent statistical uncertainty of case-control studies involving just a few hundred cases and controls seriously limits the power of such studies to reliably identify genetic determinants conferring modest but potentially important risks. Furthermore, without a clear understanding of the biology of predisposition, the definition of suitable genes for the disease is inherently problematic making an unbiased approach to loci selection highly desirable.
The advent of genome-wide association studies
Recently, GWAS have proved a powerful approach for the identification of common, low-penetrance loci for cancer without prior knowledge of location and function. Most of these GWA studies are based on tag SNPs which capture a high proportion of common variation in the genome through linkage disequilibrium (LD). This approach is unbiased and does not depend upon prior knowledge of function or presumptive involvement of any gene in disease causation. Moreover, this strategy avoids the possibility of missing the identification of important variants in hitherto unstudied genes.
Genome-wide association studies of CLL and ALL
To identify common disease causing alleles for CLL and ALL we have recently conducted GWA studies of these diseases using a two-stage strategy [8-11]. In the first stage we compared the frequency of ~ 300,000 tag SNP genotypes in a discovery set of cases and controls and the SNPs showing the strongest associations were subsequently genotyped in multiple independent case-control series. The two-stage strategy is highly efficient and use of multiple independent case-control series guarded against false positives being reported. Only P-values of ~ 1.0x10-7 are conventionally regarded as providing evidence of significance at the genome-wide level.
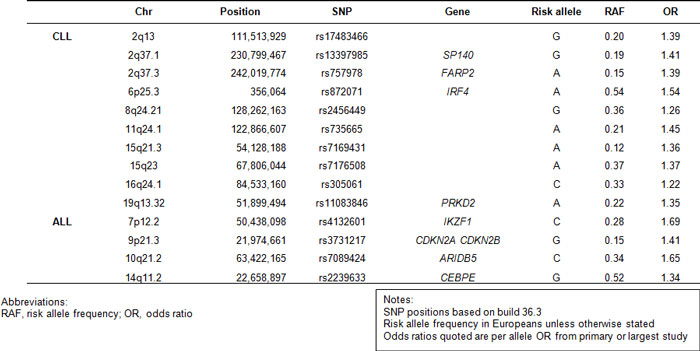
To date we have identified 10 novel CLL risk loci at 2q13 (rs17483466), 2q37.1 (rs13397985, SP140), 6p25.3 (rs872071, IRF4), 11q24.1 (rs735665), 15q23 (rs7176508), and 19q13.32 (rs11083846, PRKD2) 2q37.3 (rs757978), 8q24.21 (rs2456449), 15q21.3 (rs7169431) and 16q24.1 (rs305061, IRF8) [8, 10] and four risk loci for ALL at 7p12.2 (IKZF1, rs4132601), 10q21.2 (ARID5B, rs7089424) and 14q11.2 (CEBPE, rs2239633) [9, 11, 12]. Intriguingly, excluding CDKN2A, none of the genes implicated by these GWA scans have previously been evaluated in targeted association studies, emphasizing that the candidate gene approach was severely limited by inadequate knowledge of tumor biology.
Impact of low-risk susceptibility alleles to individual risk of CLL and ALL
While the risks of CLL and ALL associated with each of the 14 SNPs we identified are modest, relative risks 1.2-1.7 per allele (Table 1) their respective contribution to disease incidence is significant as a high proportion of the population are carriers of these risk alleles. Furthermore, the risk increases with increasing numbers of variant alleles and for the 2% of the population who possess 13 or more risk alleles the risk of CLL is increased ~ 8-fold [8]. The present data, however, provide only crude estimates of the overall effect on susceptibility attributable to variation at these loci. The effect of the actual common causal variant responsible for the association, once identified, will typically be larger. Furthermore, many of the loci may carry additional risk variants, potentially including low-frequency variants with larger influences on disease risk.
Implications of GWAS findings to our understanding of B-cell tumor development
The SNP genotyped in GWAS are not generally candidates for causality, and enumeration of the causal variant at a specific locus can poise a significant challenge. While fine-mapping and resequencing is required to identify functional variant(s) the associations identified for CLL and ALL implicate a number of genes in the etiology of B-cell tumors.
On the assumption of cis-effects GWAS findings implicate variation in SP140, IRF4, PRKD2 and IRF8 in CLL development. IRF4 is a strong candidate for a CLL susceptibility gene a priori being a key regulator of lymphocyte development, and proliferation (Figure 1) [13-15]. Through interaction with transcription factors including PU.1, IRF4 controls the termination of pre-B-cell receptor signaling and promotes the differentiation of pro-B cells to small B-cells. Furthermore, via BLIMP1 and BCL6, IRF4 controls the transition of memory B-cells, thought to be the precursor cell type for CLL, to plasma cells. A model of disease etiology based on the causal variant influencing risk by arresting transition of memory B-cells through decreased IRF4 expression is supported by a relationship between risk genotype and mRNA expression level; expression being significantly associated with genotype in a dose-dependent fashion with lower levels being associated with risk alleles [10] (Figure 2). IRF8 is also part of the transcriptional network which governs B-cell lineage, specification and differentiation, regulating α- and β-interferon response, immunoglobulin rearrangement and regulation of germinal center reaction [16]. Over-expression of IRF8 in differentiated B-cell progenitors is associated with growth inhibition and apoptosis with IFR8 knockout mice displaying reduced numbers of pre- and pro-B cells and increased numbers of myeloid cells.
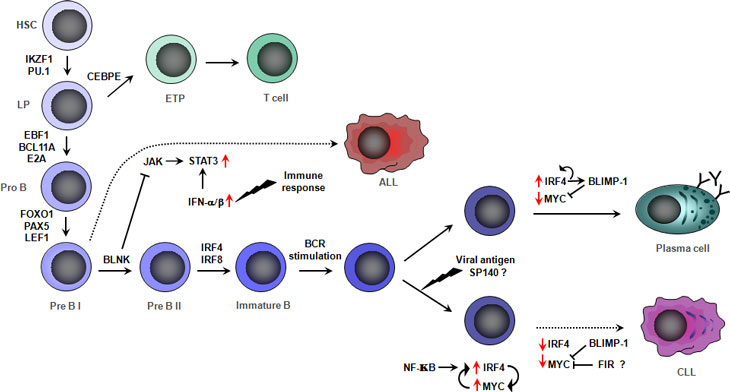
Figure 1: Genes regulating lymphoid development
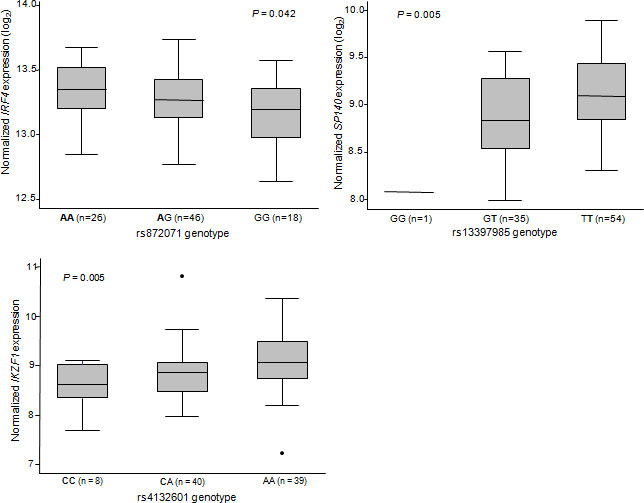
Figure 2: Relationship between lymphocyte mRNA expression levels of IRF4 and rs872071 genotype, SP140 and rs13397985 genotype, and IKZF1 and rs4132601 genotype. Risk alleles emboldened [10, 11]
SP140/LYSp100B is the lymphoid-restricted homolog of SP100 expressed in all mature B cells and plasma cell lines, as well as some T-cells. SP100 is a major mediator of EBV-encoded nuclear antigen leader protein co-activation which is important for establishment of latent viral infections and B-cell immortalization [17]. Since SP140 expression is implicated in host response to immunodeficiency virus type 1 [18] it is possible that SP140 genotype influences CLL risk by affecting response to antigenic challenge.
While a role for PRKD2 (Protein Kinase D2) in CLL is limited, low levels of PRKD2 expression and autophosphorylation have been reported to be a feature of a number of B-cell tumors including mantle cell and Burkitt’s lymphoma, and ~ 50% of CLL/small lymphocytic lymphomas [19].
A number of the SNPs associated with CLL risk map to non-coding regions of the genome, most notably those which define the 8q24.21 association. GWAS of other cancers have shown that the 128– 130 Mb genomic interval at 8q24.21 harbors multiple independent loci with different tumor specificities which are distinct from the CLL association [20-25]. The 8q24.21 region to which these cancer associations map is bereft of genes and predicted transcripts. The colorectal and prostate cancer SNP rs6983267 has been shown to affect TCF4 binding to an enhancer for MYC, providing a mechanistic basis for an 8q24.21 association [26, 27]. If the 8q24.21 CLL locus influences risk through a similar cis-effect on differential MYC expression, the association is intriguing because MYC is a direct target of IRF4 in activated B-cells.
The strongest association signal for ALL in our GWAS and a contemporaneous GWAS conducted by other researchers was attained at 7p12.2 with rs4132601, which maps to the 3’ region of the Ikaros family zinc finger 1 (IKZF1) gene. Ikaros proteins are master regulators of lymphocyte development (Figure 1) and differentiation plays a pivotal role in CD4 versus CD8 T-cell lineage commitment decisions [28, 29]. In homozygous mutant mice deleted for the N-terminal zinc finger DNA binding domain of IKZF1 loss of expression leads to arrest of lymphocyte development at its earliest recognizable stage followed by rapid development of leukemia. The observation of a strong relationship between rs4132601 genotype and IKZF1 mRNA expression level in EBV-transformed lymphocytes is thus consistent with a model in which the causal variant influences risk by impacting on early B-cell differentiation [11] (Figure 2).
The association at 10q21.2 implicates the AT rich interactive domain 5B (ARID5B) gene in the etiology of ALL. While ARID5B has not been extensively studied evidence for ARID5B having a role in defining B-cell lineage is supported by data from homozygous knockout mice, which display decreased bone marrow cellularity and reduced numbers of B-cell progenitors [30].
The 14q11.2 association with ALL annotates the gene encoding CCAAT/enhancer-binding protein, epsilon (CEBPE). CEBP is a suppressor of myeloid leukemogenesis. CEBPE and along with other CEBP family members is occasionally targeted by recurrent IGH translocations in BCP-ALL [31] suggesting opposing functions of CEBP dysregulation in myeloid and lymphoid leukemogenesis and a role in susceptibility to ALL.
The primary impact of variation defined by the 7p12.2, 10q21.2 and 14q11.2 risk variants on ALL risk is for B-cell disease. The 10q21.2 (ARID5B) risk association however, appears to be highly selective for the subset of BCP-ALL with hyperdiploidy [11, 12].
The region of LD defining the 9p21.3 association encompasses the CDKN2A and CDKN2B tumor-suppressor genes and the noncoding antisense RNA encoded by CDKN2BAS. CDKN2A encodes both p16 (INK4A), a negative regulator of cyclin-dependant kinases, and p14 (ARF1), an activator of p53. CDKN2A and CDKN2B are frequently inactivated in multiple hematological malignancies. Moreover, mono- or biallelic deletion of CDKN2A is one of the most frequent genetic events in both childhood BCP and T-ALL. Perhaps not surprisingly the association between 9p21.3 risk genotype and ALL is generic and not confined to a specific form of ALL.
Conclusions and future challenges
GWAS have provided the first unambiguous evidence that common genetic variation contributes to the risk of developing CLL and ALL and implicate genes involved in transcriptional regulation and differentiation of B-cell progenitors as the biological basis of predisposition to B-cell malignancy. Furthermore, their identification provides novel insight into disease causation of these two major haematological malignancies. The cell lineage of CLL coupled with the reciprocal familial risks between CLL and other B-cell LPDs including HD and non-Hodgkin lymphoma suggests that the variants may also influence the risk of other related B-cell LPDs. As the risk variants identified for CLL account for <10% of the familial risk of CLL, the prospects for identifying additional variants for this disease through additional GWAS should be high, and such studies are likely to provide further insights in the biology of B-cell tumors.
Acknowledgments
The Leukaemia Lymphoma Research Fund and the Kay Kendal Leukaemia Fund provide support to the authors’ laboratory.
References
1. Amiel JL. 1967 Study of the leucocyte phenotypes in Hodgkin’s disease. In Histocompatibiity Testing, P.I. Teraski, ed., (Munksgaard: Copenhagen), pp. 79-81.
2. Sellick GS, Catovsky D, and Houlston RS. Familial chronic lymphocytic leukemia. Semin Oncol 2006; 33:195-201.
3. Jonsson V, Houlston RS, Catovsky D, Yuille MR, Hilden J, Olsen JH, Fajber M, Brandt B, Sellick G, Allinson R, and Wiik A. CLL family ‘Pedigree 14’ revisited: 1947-2004. Leukemia 2005; 19:1025-8.
4. Sellick GS, et al. A high-density SNP genome-wide linkage search of 206 families identifies susceptibility loci for chronic lymphocytic leukemia. Blood 2007; 110:3326-33.
5. Fuller SJ, Papaemmanuil E, McKinnon L, Webb E, Sellick GS, Dao-Ung LP, Skarratt KK, Crowther D, Houlston RS, and Wiley JS. Analysis of a large multi-generational family provides insight into the genetics of chronic lymphocytic leukemia. Br J Haematol 2008; 142:238-45.
6. Hemminki K and Jiang Y. Risks among siblings and twins for childhood acute lymphoid leukaemia: results from the Swedish Family-Cancer Database. Leukemia 2002; 16:297-8.
7. Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer 2006; 6:193-203.
8. Crowther-Swanepoel D, et al. Common variants at 2q37.3, 8q24.21, 15q21.3 and 16q24.1 influence chronic lymphocytic leukemia risk. Nature Genetics 2010; 42:132-U59.
9. Prasad RB, et al. Verification of the susceptibility loci on 7p12.2, 10q21.2, and 14q11.2 in precursor B-cell acute lymphoblastic leukemia of childhood. Blood 2009; 115:1765-7.
10. Di Bernardo MC, et al. A genome-wide association study identifies six susceptibility loci for chronic lymphocytic leukemia. Nat Genet 2008; 40:1204-10.
11. Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, Kinsey SE, Lightfoot T, Roman E, Irving JAE, Allan JM, Tomlinson IP, Taylor M, Greaves M, and Houlston RS. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet 2009; 41:1006-10.
12. Trevino LR, Yang W, French D, Hunger SP, Carroll WL, Devidas M, Willman C, Neale G, Downing J, Raimondi SC, Pui CH, Evans WE, and Relling MV. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet 2009; 41:1001-5.
13. Busslinger M. Transcriptional control of early B cell development. Annu Rev Immunol 2004; 22:55-79.
14. Klein U and Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol 2008; 8:22-33.
15. Shapiro-Shelef M and Calame K. Regulation of plasma-cell development. Nat Rev Immunol 2005; 5:230-42.
16. Wang H, Lee CH, Qi C, Tailor P, Feng J, Abbasi S, Atsumi T, and Morse HC, 3rd. IRF8 regulates B-cell lineage specification, commitment, and differentiation. Blood 2008; 112:4028-38.
17. Ling PD, Peng RS, Nakajima A, Yu JH, Tan J, Moses SM, Yang WH, Zhao B, Kieff E, Bloch KD, and Bloch DB. Mediation of Epstein-Barr virus EBNA-LP transcriptional coactivation by Sp100. EMBO J 2005; 24:3565-75.
18. Madani N, Millette R, Platt EJ, Marin M, Kozak SL, Bloch DB, and Kabat D. Implication of the lymphocyte-specific nuclear body protein Sp140 in an innate response to human immunodeficiency virus type 1. J Virol 2002; 76:11133-8.
19. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, and Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999; 94:1848-54.
20. Amundadottir LT, et al. A common variant associated with prostate cancer in European and African populations. Nat Genet 2006; 38:652-8.
21. Easton DF, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087-93.
22. Gudmundsson J, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet 2007; 39:631-7.
23. Kiemeney LA, et al. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat Genet 2008; 40:1307-12.
24. Tomlinson I, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nature Genetics 2007; 39:984-988.
25. Yeager M, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet 2007; 39:645-9.
26. Pomerantz MM, et al. Evaluation of the 8q24 prostate cancer risk locus and MYC expression. Cancer Res 2009; 69:5568-74.
27. Tuupanen S, et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat Genet 2009; 41:885-90.
28. Georgopoulos K, Bigby M, Wang JH, Molnar A, Wu P, Winandy S, and Sharpe A. The Ikaros gene is required for the development of all lymphoid lineages. Cell 1994; 79:143-56.
29. Klug CA, Morrison SJ, Masek M, Hahm K, Smale ST, and Weissman IL. Hematopoietic stem cells and lymphoid progenitors express different Ikaros isoforms, and Ikaros is localized to heterochromatin in immature lymphocytes. Proc Natl Acad Sci U S A 1998; 95:657-62.
30. Wilsker D, Patsialou A, Dallas PB, and Moran E. ARID proteins: A diverse family of DNA binding proteins implicated in the control of cell growth, differentiation, and development. Cell Growth & Differentiation 2002; 13:95-106.
31. Akasaka T, et al. Five members of the CEBP transcription factor family are targeted by recurrent IGH translocations in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). Blood 2007; 109:3451-61.