
Introduction
The discovery of mutated “cancer genes”, has provided key insights into the mechanisms of tumorigenesis, which has been useful for developing targeted cancer therapies. Most recent examples include ALK inhibitors in non-small cell lung cancer with an ALK rearrangement or BRAF inhibitors in melanoma with a BRAF mutation.[1, 2]
Yet, hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide and the third leading cause of death after lung and gastric cancers, but lacks molecular targets or signatures.[3] HCC comprises several distinct entities defined by etiology, and often arises in the wake of cirrhosis and liver dysfunction. Most cases of HCC are associated with hepatitis B or hepatitis C, as well as metabolic derangements such as alcoholic liver disease and nonalcoholic steatohepatitis. It is plausible that each entity may have a different molecular genetic profile that determines biological behavior, prognosis and response to molecularly targeted therapy.[4] Currently, complete resection or liver transplant are the only therapeutic approaches offering long-term survival. Most patients (>85%) with HCC present with advanced disease and about 12 months of survival on standard therapies.[5, 6] HCC is a highly heterogeneous tumor for which molecular signatures or druggable targets remain to be identified. According to the COSMIC database, which includes data from cell lines and tumor samples, there have been 1,709 oncogenes with oncogenic mutations identified in HCC to date (accessed 2/12/2013).[7] Mutations are primarily prevalent in TP53 (31%), CTNNB1 (19%), AXIN1 (16%), NFE2L2 (14%), ARID2 (13%), and PIK3CA (7%).[7] However, systematic efforts to delineate the molecular profile of HCC in a large group of tumor samples from patients are still underway (http://cancergenome.nih.gov/).[8] Here we report the results of targeted next generation sequencing (NGS) and treatment outcomes in a case series of 14 patients with advanced/metastatic HCC.
Results
Patients
A total of 14 patients with advanced metastatic HCC were analyzed with NGS, of whom 10 (71%) were men and 4 (29%) women. Their median age at diagnosis was 58 years (range, 27 to 79 years) and 10 (71.5%) patients were White, 3 (21.5%) African American, and 1 (7%) Asian. In addition, 3 (21.5%) patients had hepatitis C-related HCC, 1 (7%) hepatitis B-related HCC, 2 (14.5%) hepatitis B and C-related HCC and 8 (57%) had HCC without identified predisposing factors. Patients received a median of 1 (range, 1-4) therapies prior to being referred for experimental therapies to the Clinical Center for Targeted Therapy (CCTT).
Molecular profiling and treatment outcomes
Patient 1, an Asian male, was diagnosed at the age of 56 years with hepatitis B-related HCC and was initially treated with a liver transplant (Table 1). At the time of disease recurrence, the patient received firstline therapy with sorafenib and progressed after 4 months. NGS performed on a tumor sample from the epidural biopsy obtained after the firstline therapy revealed an ATR W463* mutation and MDM2 amplification (Figure 1). ATR encodes the protein ataxia telangiectasia, which plays a key role in maintaining genomic integrity via regulation of DNA repair and replication (Figure 2).[9] The ATR W463*mutation truncates the ATR protein and is likely to lead to a loss of protein function. In HCC ATR mutations have been reported anecdotally.[7] Based on preclinical evidence, ATR-deficient tumors may be sensitive to PARP inhibitors.[9] MDM2 acts to prevent the activity of the tumor suppressor p53; therefore, amplification of MDM2 may be oncogenic (Figure 2). MDM2 amplification has been identified according to some data in up to 44% of patients with HCC.[10] It is assumed that MDM2 amplification may predict sensitivity to MDM2 inhibitors; however, currently available evidence is inconclusive.[11] At the time of referral to the CCTT the patient had metastatic disease to lungs and bones and subsequently received experimental therapy with sorafenib (200 mg orally twice a day), temsirolimus (15 mg intravenously weekly), and bevacizumab (10 mg/kg intravenously every 3 weeks), which did not match any molecular target. Although the patient demonstrated a 25% improvement per RECIST after 8 weeks of therapy, he ultimately progressed in his bones after 2.8 months of therapy.
Patient 2, an African American male, was diagnosed at the age of 51 years with hepatitis C and cirrhosis-related HCC (Table 1). He progressed after 1.8 months of firstline therapy with sorafenib. NGS performed on a tumor sample from the chest wall biopsy obtained at the time of diagnosis revealed PIK3CA mutation H1047R, CTNNB1 mutation S37C, and protein tyrosine phosphatase delta (PTPRD) mutation S1845fs*2 (Figure 1). PIK3CA encodes the active subunit of phosphatidylinositol 3-kinase (PI3K), which regulates cell growth, proliferation, differentiation, motility and survival (Figure 2).[12] Mutations in PIK3CA have been reported in 7% of liver cancer cases.[7] Preclinical and early clinical data suggest that activating mutations in PIK3CA may predict sensitivity to inhibitors of the PI3K/AKT mTOR pathway.[12-14] CTNNB1 encodes beta-catenin, a key component of the Wnt signaling pathway (Figure 2). CTNNB1 exon 3 mutations, such as S37C, are considered to be activating and lead to activation of the Wnt pathway.[15] CTNNB1 mutations have been reported in 19% of HCC.[7] PTPRD, a tumor suppressor, plays an essential role in dephosphorylating STAT3 (Figure 2).[16] Mutations in PTPRD occur only sporadically in HCC (1%).[7] S1845fs*2 is a frameshift mutation that truncates the PTPRD protein within the second tyrosine phosphatase domain (amino acids 1644-1903) only 1 amino acid from the phosphatase active site, possibly resulting in an inactive protein. At the time of referral to the CCTT the patient had metastatic disease to the mediastinum, bones, and liver and subsequently received experimental therapy with a mTOR inhibitor sirolimus (4 mg orally daily) in combination with a histone deacetylase inhibitor (HDAC) vorinostat (300 mg orally daily), which was matching a molecular abnormality in the PI3K/AKT/mTOR pathway (PIK3CA H1047R mutation) and attained 15% shrinkage per RECIST. He progressed after 3.8 months.[17] Then he received a mTOR inhibitor everolimus (10 mg orally every other day) and multikinase inhibitor pazopanib (600 mg orally every other day), which again targeted the PI3K/AKT/mTOR pathway and induced 11% shrinkage per RECIST after 2 months, but he progressed after a total of 2.9 months. The patient then received the EGFR inhibitor erlotinib (100 mg orally daily) and antimetabolite praletrexate (15 mg/m2 intravenously weekly for 3 weeks followed by a week off), which did not match any of the detected abnormalities and progressed after 2.1 months with no initial response.
Patient 3, a White male, was diagnosed at the age of 59 years with hepatitis C-related HCC and history of hepatitis B. He was initially treated with a liver transplant. At the time of the second disease recurrence he progressed on firstline therapy with sorafenib after 5.9 months. NGS of a tumor sample obtained from a lung resection performed at the time of the first recurrence after the initial liver transplantation revealed RB1 loss (Figure 1). RB1 encodes the retinoblastoma protein, a tumor suppressor and negative regulator of the cell cycle (Figure 2).[18] The loss of RB1 protein expression has been linked to poor survival and may prognosticate a decreased response to kinase inhibitors.[18] RB1 mutations have been reported sporadically in HCC.[7] At the time of referral to the CCTT the patient had metastatic disease to the right pterygoid fossa and liver and his clinical condition precluded experimental therapy.
Patient 4, a White male, was diagnosed at the age of 57 years with hepatitis C, hepatitis B, and alcohol-related HCC. He was initially treated with liver transplantation and at the time of disease recurrence he received firstline therapy with sorafenib and progressed after 7 months. NGS performed on the tumor sample from the liver transplant after diagnosis revealed CTNNB1 mutation I35S, and TP53 V143M mutation (Figure 1). The CTNNB1 gene plays a role in the Wnt signaling pathway and is characterized in detail above and the I35S mutation is considered to be activating (Figure 2).[15] Functional loss of tumor suppressor p53, which is encoded by the TP53 gene, is common in advanced cancers (Figure 2).[19] TP53 V143M mutation is located within the DNA-binding domain of the p53 protein, which is thought to result in loss of function.[20] Mutations of TP53 have been reported in 31% of cases of HCC.[7] A recent meta-analysis reported that HCC patients with TP53 alterations experienced significantly shorter recurrence-free survival and overall survival.[19] There are no approved therapies to address TP53 mutations; however, tumors with TP53 mutations may be sensitive to Wee1 inhibitors, which are in clinical trials. At the time of referral to the CCTT the patient had metastatic disease to the lungs, liver, retroperitoneum, and bones. He did not receive experimental therapy because of insurance denial.
Patient 5, a White male, was diagnosed at the age of 66 years with hepatitis C-related HCC. He was initially treated with sorafenib and progressed after 15.5 months; however, during that period of time he also underwent transarterial chemoembolization of the liver. NGS of a tumor sample obtained from a liver biopsy performed at the time of diagnosis revealed CDKN2A mutation H83Y and CTNNB1 mutation T41A (Figure 1). The CDKN2A gene encodes the tumor suppressor p16(Ink4a), which plays a vital role in cell cycle G1 checkpoint regulation and its deletion contributes to inactivation of the p16-CDK4/Cyclin/Rb pathway and loss of cell cycle control (Figure 2).[21] The CDKN2A H83Y mutation leads to a reduced ability to induce cell cycle arrest. CTNNB1, a key component of the Wnt signaling pathway, is characterized above (Figure 2).[15] CTNNB1 exon 3 mutations, such as T41A, are considered to be activating the Wnt pathway.[22] At the time of referral to the CCTT the patient had metastatic disease to the liver and decided to pursue therapeutic options outside of MD Anderson.
Patient 6, a White male, was diagnosed at the age of 61 years with hepatitis C-related HCC (Table 1). He was initially treated with sorafenib and progressed after 4.5 months. NGS of a tumor sample obtained from a bone biopsy performed at the time of diagnosis revealed CTNNA1 mutation K889* and CTNNB1 mutation T41A (Figure 1). The CTNNA1 gene plays a role in cell adhesion downstream in the Wnt signaling pathway (Figure 2).[15] The CTNNA1 K889* mutation is expected to be inactivating; however, its functional consequencies are not fully understood. CTNNB1, a key component of the Wnt signaling pathway, is characterized above (Figure 2).[15] CTNNB1 exon 3 mutations, such as T41A, are considered to be activating the Wnt pathway.[22] At the time of referral to the CCTT the patient had metastatic disease to his liver, adrenal glands and bones and subsequently received experimental therapy with hepatic arterial infusion oxaliplatin (140 mg/m2 every 3 weeks), intravenous bevacizumab (10 mg/kg every 3 weeks) and oral; capecitabine (1500 mg/m2 on days 1-4 every 3 weeks) and progressed after 2.2 months. The experimental therapies did not match any detected molecular abnormality.
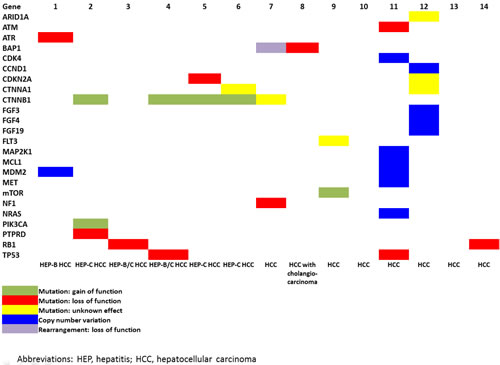
Figure 1: Overview of mutations in 14 patients with hepatocellular carcinoma. The heatmap shows gain of function mutations (green), loss of function mutations (red), mutations with unknown effects (yellow), copy number variations (blue), and rearrangements (purple).
Patient 7, an African American female, was diagnosed at the age of 46 years with HCC (Table 1). She progressed on firstline therapy with sorafenib after 2.1 months. Then she progressed after 1.3 months of experimental therapy with an oncolytic virus and ultimately responded very well to a combination of bevacizumab and erlotinib, which she received twice with a PFS of 16.8 months and 10.1 months, respectively. NGS performed on a tumor sample from a liver biopsy obtained at the time of diagnosis revealed NF1 mutation R1241*, BAP1 truncation (exon 12), and CTNNB1 mutation N387K (Figure 1). NF1 encodes neurofibromin 1, a GTPase-activating protein (GAP) that is a key negative regulator of the RAS and PI3K signaling pathway (Figure 2).[23] The NF1 R1241* mutation leads to a nonsense codon, resulting in truncation of the NF1 protein within the GAP-related domain, which is likely to negate tumor suppressing activity. NF1 mutations have been reported in 10% of HCC samples and such tumors may thus be sensitive to PI3K/AKT/mTOR and MAPK inhibitors.[7, 24] BAP1 rearrangement results in a truncation of the BAP1 protein (Figure 2). The resulting truncated protein product is believed to be nonfunctional.[25] BAP1 rearrangements have not been previously reported in HCC.[7] While there are no therapies that directly target BAP1 loss, HDAC inhibitors are being explored based on the results of a preclinical study.[26] CTNNB1, a key component of the Wnt signaling pathway, is characterized above (Figure 2).[15] N387K is a rare mutation, located in the region responsible for binding to APC.[27] N387K has not been characterized, but other mutations in this region were shown to abrogate APC binding, although beta catenin was still degraded in the presence of these mutations.[27] The functional effect of N387K is unknown. At the time of referral to the CCTT the patient had metastatic disease to her lungs, mediastinum, peritoneum and liver and subsequently received the experimental therapy with mTOR inhibitor everolimus (7.5 mg orally every other day) and multikinase inhibitor pazopanib (600 mg orally every other day), which was matched to a molecular abnormality activating the PI3K/AKT/mTOR pathway (NF1 R1241*mutation). Restaging CT scans revealed a PR (-30% per RECIST), which was maintained for the total of 8.3 months. Then the patient received mTOR inhibitor sirolimus (5 mg orally daily) and HDAC inhibitor vorinostat (300 mg orally daily), which again targeted the PI3K/AKT/mTOR pathway and was due to have her first reassessment after 2 months at the time of analysis.
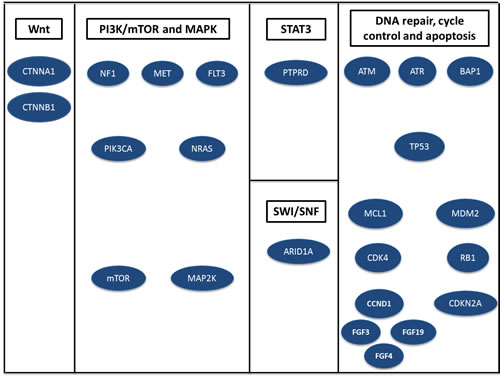
Figure 2: Major pathways altered by somatic mutations, copy number variations or rearrangements in 14 patients with hepatocellular carcinoma
Table 1: Characteristics of patients with molecular aberrations who received experimental therapies
Patient number |
Sex |
Ethnicity |
Age at diagnosis (years) |
Subtype |
Number of prior systemic therapies |
Metastatic spread |
Mutations (expected consequence) |
Copy number variations |
Rearrangements |
Experimental therapy (target) |
Therapy matching target |
RECIST (%) |
Time to progression (months) |
1 |
Male |
Asian |
56 |
HCC, hepatitis B |
1 |
Lungs, bones |
ATR (impaired DNA repair and cell cycle control) |
MDM2 (reduced apoptosis) |
None |
Sorafenib, temsirolimus, bevacizumab |
No |
-25 |
2.8 |
2 |
Male |
African-American |
51 |
HCC, hepatitis C, cirrhosis |
1 |
Mediastinum, bones, liver |
PIK3CA (PI3K/mTOR activation), CTNNB1 (Wnt activation), PTPRD (STAT3 activation) |
None |
None |
Sirolimus (mTOR), vorinostat (HDAC) |
Yes |
-15 |
3.8 |
Everolimus (mTOR), pazopanib (multikinase) |
Yes |
-11 |
2.9 |
||||||||||
Erlotinib (EGFR), praletrexate (antifolate analog) |
No |
+11 (PD in non-target lesions) |
2.1 |
||||||||||
6 |
Male |
White |
61 |
HCC, hepatitis C, cirrhosis |
1 |
Liver, adrenal glands, bones |
CTNNA1 (Wnt activation), CTNNB1 (Wnt activation) |
None |
None |
Oxaliplatin (DNA damage), bevacizumab (VEGF), capecitabine (antimetabolite) |
No |
+13 |
3.2 |
7 |
Female |
African-American |
46 |
HCC |
4 |
Lungs, mediastinum, liver, peritoneum |
NF1 (PI3K/mTOR and MAPK activation), CTNNB1 (Wnt activation) |
None |
BAP1 (loss of interaction with BRCA1) |
Everolimus (mTOR), pazopanib (multikinase) |
Yes |
-30 |
8.3 |
8 |
Female |
African-American |
56 |
HCC with cholangio-carcinoma |
3 |
Lungs, mediastinum, liver, peritoneum, porta hepatis |
BAP1 (loss of interaction with BRCA1) |
None |
None |
Sirolimus (mTOR) |
No |
+130 |
1.4 |
Nab-paclitaxel (mitotic inhibitor), bevacizumab (VEGF), gemcitabine (antimetabolite) |
No |
+15 (clinical PD) |
1.6 |
||||||||||
12 |
Male |
White |
66 |
HCC |
2 |
Liver |
CDKN2A (loss of p16 function), ARID1A (SWI/SNF chromatin remodeling complex) |
CCND1 (impaired cell cycle control), FGF3,4,19 (impaired cell cycle control) |
None |
MET kinase inhibitor |
No |
+14 |
4 |
14 |
Female |
White |
53 |
HCC |
2 |
Lungs, mediastinum, liver |
Rb1 loss (loss of cell cycle control) |
None |
None |
Oxaliplatin (DNA damage), bevacizumab (VEGF), capecitabine (antimetabolite) |
No |
-18 |
2.9 |
Abbreviations: mTOR, mammalian target of rapamycin; HDAC, histone deacetylase; PI3K, phosphoinositide 3-kinase; EGFR, epidermal growth factor receptor
Patient 8, an African American female, was diagnosed at the age of 56 years years with HCC with islands of cholangiocarcinoma (Table 1). She underwent a left hepatectomy and at the time of disease recurrence was treated with firstline therapy sorafenib and progressed after 10.7 months. Then she received a combination of oxaliplatin and gemcitabine and progressed after 6.6 months, followed by treatment with 5-fluorouracil, leucovorin, irinotecan, and bevacizumab, but progressed after 1.6 months. NGS performed on a tumor sample from the liver resection obtained at the time of diagnosis revealed BAP1 mutation L65* (Figure 1). A BAP1 L65* mutation in this tumor results in a truncation of the BAP1 protein near the amino terminus. Details about BAP1 are outlined above (Figure 2) [25]. At the time of referral to the CCTT the patient had metastatic disease to her lungs, mediastinal and hilar nodes, porta hepatis, peritoneum, and liver and subsequently received experimental therapy with mTOR inhibitor sirolimus (4 mg orally daily). She progressed after 1.4 months and then received hepatic arterial infusion nab-paclitaxel (180 mg/m2 every 3 weeks) and intravenous bevacizumab (10 mg/kg every 3 weeks) and gemcitabine (800 mg/m2 on days 1 and 8 every 3 weeks) and progressed after 1.6 months. None of the experimental therapies was matched to any detected molecular abnormalities.
Patient 9, a White male, was diagnosed at the age of 65 years with HCC, which was initially treated with a right hepatectomy and radiofrequency ablation. At the time of disease recurrence he was treated on firstline therapy with cisplatin, interferon, adriamycin, and 5-fluorouracil and remained progression-free for 23.7 months. He was next treated with capecitabine and progressed after 1.8 months, then treated on a clinical trial with regorafenib for 49.3 months and then with sorafenib, but progressed after 1.7 months. NGS performed on a tumor sample from the liver resection obtained at the time of diagnosis revealed mTOR mutation S2215Y and FLT3 mutation splice site 1418+2A>G (Figure 1). mTOR acts downstream of multiple pathways, including the PI3K/AKT/mTOR pathway (Figure 2). This mTOR S2215Y mutation is located in the kinase domain.[28] The S2215Y mutation has been reported to result in constitutive activation of mTOR complex 1 (mTORC1) but not mTORC2 and to retain sensitivity to sirolimus.[29] Mutations in mTOR have not yet been reported in HCC.[7] FLT3 encodes a receptor tyrosine kinase. Signaling through the FLT3 pathway leads to phosphorylation of SHC1 and AKT1 and activation of mTOR, as well as RAS activation and phosphorylation of ERK1 and 2 (Figure 2).[30] The 1418+2A>G splice site mutation seen in this tumor disrupts the canonical GT splice-donor site of the 5’ end of the intron separating exon 11 and exon 12; however, the resulting GC dinucleotide sequence has been reported as a non-canonical splice site in the human genome.[31] Mutation of the conserved 5’ GT splice site often results in exclusion of the neighboring exon, but may also result in truncation of the encoded protein.[32] This alteration has not been previously reported and its consequences are difficult to predict. Among FDA approved therapies sorafenib is known to be a FLT3 inhibitor and regorafenib also has weak anti-FLT3 activity.[33] At the time of referral to the CCTT the patient had metastatic disease to the liver and spleen. The patient did not receive experimental therapy due to his worsening performance status.
Patient 10 (White female diagnosed with HCC at the age of 67 years) had no molecular abnormality revealed through NGS.
Patient 11, a White male, was diagnosed at the age of 70 years with HCC, which was initially treated with a right partial hepatectomy. At the time of disease recurrence he was treated on firstline therapy with sorafenib and was taken off therapy after 4.6 months because of squamous cell carcinomas related to sorafenib therapy. NGS performed on a tumor sample from the liver resection performed at the time of disease recurrence revealed ATM mutation K53fs*3 and a TP53 A161S and M160I mutations. In addition, MET, CDK4, MAP2K1, MCL1, MDM2, NRAS amplifications were found (Figure 1). ATM encodes the protein ataxia telangiectasia mutated, which plays a key role in sensing double-strand DNA breaks and activating cellular checkpoint pathways, arresting the cell cycle when DNA damage is present (Figure 2).[34] ATM K53fs*3 mutation is predicted to be inactivating due to loss of most of the ATM protein, including the kinase domain. According to the COSMIC database ATM mutations were found in all 4 tested HCC samples.[7] Preclinical evidence suggested that ATM deficient tumors may be sensitive to PARP inhibitors.[35] Functional loss of TP53 is common in aggressive advanced cancers and is described in detail above (Figure 2).[19] Both A161 and M160 are located within the DNA-binding domain of TP5. While the A161S mutation is predicted to be severe, the M160I mutation is predicted to be non-severe.[36] DNA-binding domain mutations are thought to result in loss of function.[20] MET encodes a receptor tyrosine kinase that is activated by the ligand HGF, and MET activation promotes angiogenesis, resistance to apoptosis, proliferation, and invasion of cancer cells (Figure 2).[37] MET amplification has been reported in 2% of HCC.[38] MET amplification may predict sensitivity to MET inhibitors, some of which are FDA approved (crizotinib, cabozantinib).[39] CDK4 encodes cyclin-dependent kinase 4, which, along with functional homolog CDK6 and family member CDK2, regulates cell cycle G1 phase progression and the G1/S transition (Figure 2).[40] CDK4 amplification has been reported in 2% of HCC samples.[38] There are currently no approved therapies that directly target CDK4; however a number of drugs are under investigation in clinical trials. MAP2K1 (also known as MEK1) encodes the signaling protein mitogen-activated protein kinase kinase 1. MEK1 phosphorylates the ERK1/2 proteins in the RAS/RAF/MEK pathway (Figure 2).[41] MAP2K1 amplification has been reported in 2% of HCC samples.[38] The relationship between MEK amplification and sensitivity to MEK inhibitors is unknown. MCL1 encodes the protein MCL1, which is a member of the BCL2 family and can result in antiapoptotic activity (Figure 2).[42] MCL1 amplification has been reported in 12% of HCC.[38] There are no FDA approved therapies to address MCL1 copy number amplification at this time, but investigations focused on small molecule inhibitors of MCL1 are underway. In addition, the multikinase inhibitor sorafenib has been shown to down-regulate MCL1 and thereby induce apoptosis in preclinical studies.[43] Also, preclinical studies of patient-derived tumor cells suggest that increased MCL1 levels may confer resistance to antitubulin therapies such as paclitaxel.[44] MDM2 acts to prevent the activity of the tumor suppressor p53 and its function is described in detail above.[10] NRAS encodes a member of the RAS family of small GTPases that mediate transduction of growth signals (Figure 2).[41] Activation of RAS signaling causes cell growth, differentiation, and survival by activating the RAF/MAPK/ERK, PI3K, and other pathways. NRAS amplification has not been reported in HCC.[38] There are no approved therapies to address cancers associated with NRAS amplification or activating mutations. NRAS putatively leads to activation of the RAF/MEK/ERK pathway. Therefore, NRAS activation might theoretically predict sensitivity to MEK inhibitors. At the time of referral to the CCTT the patient had metastatic disease to the liver, but decided not to receive experimental therapy.
Patient 12, a White male, was diagnosed at the age of 79 years with idiopathic cirrhosis-related HCC (Table 1). He progressed after 2.7 months of firstline therapy with sorafenib. Then he received experimental therapy with bevacizumab and erlotinib and progressed after 3.8 months. NGS performed on a tumor sample from the liver biopsy obtained at the time of diagnosis revealed CDKN2A mutation A102P, ARID1A mutation I1485fs*5, CCND1 amplification, FGF19 amplification, FGF3 amplification and FGF4 amplification (Figure 1). CDKN2A regulates the p16-CDK4/Cyclin/Rb pathway and is characterized above (Figure 2).[21] The CDKN2A A102P mutation has not been characterized, but it is likely to be inactivating.[45] Overall, CDKN2A mutations has been reported in 8% of HCC.[7] ARID1A encodes the AT-rich interactive domain-containing protein 1A, a member of the SWI/SNF chromatin remodeling complex. ARID1A is believed to function as a tumor suppressor and deletion of ARID1A can lead to tumor formation.[46] The I1485fs*5 mutation results in truncation of the protein and similar truncations have been predicted to be inactivating.[46] ARID1A mutations have been reported in HCC in the COSMIC database.[7] CCND1 encodes Cyclin D1, which interacts with the cyclin-dependent kinases CDK4 and Cdk6, resulting in inactivation of RB1 and progression of the cell cycle. Amplification of CCND1 may therefore lead to increased proliferation. CCND1 amplification has been reported in 4% of HCC.[47] FGF3 encodes fibroblast growth factors 19, an FGFR4 ligand involved in regulation of hepatic protein and glycogen metabolism.[48] FGF3 plays a central role in development of the inner ear and FGF3 germline mutations give rise to an autosomal recessive syndrome characterized by microdontia, deafness and complete lack of inner ear structures.[49] FGF4 plays a central role in tooth development.[50] At the time of referral to the CCTT the patient had metastatic disease to the liver and subsequently received experimental therapy with an oral daily dose of an experimental MET kinase inhibitor, which did not match any detected molecular abnormality. He had stable disease (+14% per RECIST) and progressed after 4.0 months.
Patient 13 (White male diagnosed with HCC at the age of 27 years) had no molecular abnormality revealed through NGS.
Patient 14, a White female, was diagnosed at the age of 53 years with HCC (Table 1). She progressed after 6 months of firstline therapy with sorafenib. Then she received experimental therapy with bevacizumab and erlotinib and progressed after 1.8 months. NGS performed on a tumor sample from the liver biopsy obtained at the time of diagnosis revealed RB1 loss (Figure 1). RB1 encoding the Rb protein is characterized above (Figure 2).[18] At the time of referral to the CCTT the patient had metastatic disease to her lungs, mediastinum and liver and subsequently received experimental therapy with hepatic arterial infusion oxaliplatin (140 mg/m2 every 3 weeks), intravenous bevacizumab (10 mg/kg every 3 weeks) and oral capecitabine (1500 mg/m2 on days 1-4 every 3 weeks) and was taken off therapy after 2.9 months because of poor tolerance. The experimental therapies did not match any detected molecular abnormality.
DISCUSSION
Targeted NGS analysis of tumor samples from 14 consecutive patients with advanced HCC refractory to standard therapies referred to the CCTT for experimental therapies with targeted agents revealed that 12 (86%) patients had at least one somatic molecular aberration (median, 2; range, 0-8) in their tumor samples (activating mutations, n=6; loss of function mutations, n=10; mutations with unknown effects, n=6; copy number variations, n=11; rearrangements, n=1) and 9 (64%) had more than one molecular aberration. Of interest, molecular aberrations were found in the PI3K/AKT/mTOR pathway (mTOR, n=1; PIK3CA, n=1; NF1, n=1), Wnt pathway (CTNNA1, n=1, CTNNB1, n=5), the MAPK pathway (MAP2K1, n=1; NRAS, n=1), DNA repair mechanisms, cell cycle control and apoptosis (ATM, n=1, ATR, n=1; BAP1, n=2, CCND1, n=1; CDKN2A, n=2; CDK4, n=1; FGF3, n=1; FGF4, n=1, FGF19, n=1; MCL1, n=1; MDM2, n=2; RB1, n=2; TP53, n=2). Six (43%) patients with HCC in the context of hepatitis B (n=1), hepatitis C (n=3) or both (n=2) had molecular abnormalities related to the PI3K/AKT/mTOR pathway (PIK3CA, n=1), Wnt pathway (CTNNA1, n=1; CTNNB1, n=4), STAT3 signaling (PTPRD, n=1), DNA repair mechanisms, cell cycle control and apoptosis (ATR, n=1; CDKN2A, n=1; RB1, n=1; MDM2, n=1; TP53, n=1). Despite the fact that several patients had aberrations in some genes such as CTTNB1 and CDKN2A, the specific combination of aberrations and their subtypes were unique to each individual patient, important distinctions for selecting personalized cancer therapies.
Patients with hepatitis B or C-related HCC had a median of 2 aberrations (range, 1-3), which was similar to a median of 1.5 aberration in patients without hepatitis (range, 0-8; p = 0.74). To date, the molecular landscape of HCC has not been clearly defined and major initiatives such as The Cancer Genome Atlas are in progress. Only limited data on copy number variations are available, demonstrating that MCL1, and BAP1 are the most frequent copy number abnormalities (12%, 4%, respectively).[38] According to the COSMIC database, the genes most frequently mutated include TP53 (31%), CTNNB1 (19%), AXIN1 (16%), NFE2L2 (14%), ARID2 (13%), and PIK3CA (7%).[7] An integrated analysis of somatic mutations and focal copy number variations with whole exome sequencing of 24 HCC cases revealed recurrent mutations in CTNBB1 (46%), AXIN1 (21%), TP53 (13%), CDKN2A (8%), and ARID2 (8%).[51] CTTNB1 was also the most frequently mutated gene in our study (5/14, 36%). Of interest mTOR mutation S2215Y has never been reported before in HCC.
Of the 14 patients in our study, 3 (21%) had genomic alterations putatively activating the PI3K/AKT/mTOR pathway and 2 such patients received therapies that included an mTOR inhibitor. Both patients had some therapeutic benefit ranging from a PR to minor shrinkage per RECIST (-30%, -15%; respectively), which was maintained for 3.8 months and 8.3 months. According to the COSMIC database, with the exception of PIK3CA, none of the mutations putatively leading to PI3K/AKT/mTOR activation are commonly found in HCC; however, preclinical models and early clinical data generally suggest that targeting the PI3K/AKT/mTOR pathway can be an effective strategy in subsets of patients with HCC.[52, 53] However, none of the studies investigated these therapies in HCC with PI3K/AKT/mTOR activation.[54, 55] Revisiting such studies with gating of HCC patient for alterations in the PI3K/AKT/mTOR pathway may yield better evidence of the efficacy of inhibitors of the PI3K/mTOR pathway in this difficult to treat disease.[56-58]
In conclusion, we have identified multiple molecular abnormalities in a small data set of patients with advanced HCC. Interestingly, a proportion of our patients had molecular aberrations putatively associated with PI3K/AKT/mTOR pathway activation and these patients attained some benefit (although not always durable) from targeting the PI3K/AKT/mTOR pathway. Despite several limitations such as small patient numbers, absence of randomization and control groups, our data suggest that comprehensive molecular profiling can increase our knowledge of HCC biology to an unprecedented level, which can be translated to patient care.
PATIENTS AND METHODS
Patients
We reviewed the electronic medical records of consecutive patients with advanced/metastatic HCC (excluding fibrolamellar type given its distinct entity and natural history) referred to the CCTT at MD Anderson Cancer Center. Data were collected from transcribed notes and radiology reports in the electronic medical record and other source documentation. Registering patients in the database, clinical, pathologic, laboratory and pathology assessment were performed at MD Anderson. The study and all treatments were conducted in accordance with the guidelines of the MD Anderson Institutional Review Board.
Molecular analysis
Archival tumor samples obtained from standard diagnostic or therapeutic procedures were tested with Clinical Laboratory Improvement Amendment-certified targeted NGS (Foundation One, Foundation Medicine, MA) using the Illumina HiSeq 2000 platform. Genomic libraries were captured for 3,230 exons representing the complete coding sequence of 182 cancer-related genes plus 37 introns from 14 genes often rearranged in cancer and sequenced to an average median depth of 734X with 99% of bases covered >100X. On January 1, 2013, the platform was upgraded to cover 3,769 exons of 236 cancer-related genes and 47 introns from 19 genes to an average depth of 1000X.
Treatment
Patients were enrolled in clinical trials according to the discretion of treating physicians and availability of clinical trials at the time of enrollment. Treatment continued until disease progression or unacceptable toxicity. Treatment was carried out according to the specific requisites in the treatment protocols selected.
Assessments, including history, physical examination, and laboratory evaluations, were performed as specified in each protocol, typically before the initiation of therapy, weekly during the first cycle, and then, at a minimum, at the beginning of each new treatment cycle. Response was assessed from computed tomography (CT) scans and/or magnetic resonance imaging (MRI) at baseline before treatment initiation and then every 2 cycles (6-8 weeks). All radiographs were read in the Department of Radiology at MD Anderson and reviewed in the Department of Investigational Cancer Therapeutics tumor measurement clinic. Responses were categorized per RECIST criteria and categorized as complete response (CR); partial response (PR), stable disease (SD) or progressive disease (PD).[17]
Statistical analysis
Mann-Whitney test was used to assess the association between continuous variables. All tests were two-sided, and P values less than 0.05 were considered statistically significant. All statistical analyses were carried out using SPSS 19 computer software (SPSS Chicago, IL).
ACknowledgementS
We thank Ms. Joann Aaron for scientific review and editing of this article. This study was supported in part by Grant Number RR024148 from the National Center for Research Resources, a component of the NIH Roadmap for Medical Research (http://nihroadmap.nih.gov/clinicalresearch/overview-translational.asp).
References
1. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010; 363(18):1693-1703.
2. Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC, O’Day SJ, Blackman SC, Curtis CM, Lebowitz P, Ma B, Ouellet D, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012; 379(9829):1893-1901.
3. El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011; 365(12):1118-1127.
4. Ganapathy-Kanniappan S, Kunjithapatham R and Geschwind JF. Glyceraldehyde-3-phosphate dehydrogenase: a promising target for molecular therapy in hepatocellular carcinoma. Oncotarget. 2012; 3(9):940-953.
5. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008; 359(4):378-390.
6. Cervello M, McCubrey JA, Cusimano A, Lampiasi N, Azzolina A and Montalto G. Targeted therapy for hepatocellular carcinoma: novel agents on the horizon. Oncotarget. 2012; 3(3):236-260.
7. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR and Futreal PA. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011; 39(Database issue):D945-950.
8. Totoki Y, Tatsuno K, Yamamoto S, Arai Y, Hosoda F, Ishikawa S, Tsutsumi S, Sonoda K, Totsuka H, Shirakihara T, Sakamoto H, Wang L, Ojima H, Shimada K, Kosuge T, Okusaka T, et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011; 43(5):464-469.
9. Peasland A, Wang LZ, Rowling E, Kyle S, Chen T, Hopkins A, Cliby WA, Sarkaria J, Beale G, Edmondson RJ and Curtin NJ. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer. 2011; 105(3):372-381.
10. Jablkowski M, Bocian A, Bialkowska J and Bartkowiak J. A comparative study of P53/MDM2 genes alterations and P53/MDM2 proteins immunoreactivity in liver cirrhosis and hepatocellular carcinoma. J Exp Clin Cancer Res. 2005; 24(1):117-125.
11. Liu W, He L, Ramirez J and Ratain MJ. Interactions between MDM2 and TP53 Genetic Alterations, and Their Impact on Response to MDM2 Inhibitors and Other Chemotherapeutic Drugs in Cancer Cells. Clin Cancer Res. 2009; 15(24):7602-7607.
12. Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, Hong DS, Naing A, Falchook GS, Moroney JW, Piha-Paul SA, Wheler JJ, Moulder SL, Fu S and Kurzrock R. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Molecular cancer therapeutics. 2011; 10(3):558-565.
13. Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008; 14(12):1351-1356.
14. Janku F, Wheler JJ, Naing A, Falchook GS, Hong DS, Stepanek VM, Fu S, Piha-Paul SA, Lee JJ, Luthra R, Tsimberidou AM and Kurzrock R. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013; 73(1):276-284.
15. Tejpar S, Nollet F, Li C, Wunder JS, Michils G, dal Cin P, Van Cutsem E, Bapat B, van Roy F, Cassiman JJ and Alman BA. Predominance of beta-catenin mutations and beta-catenin dysregulation in sporadic aggressive fibromatosis (desmoid tumor). Oncogene. 1999; 18(47):6615-6620.
16. Veeriah S, Brennan C, Meng S, Singh B, Fagin JA, Solit DB, Paty PB, Rohle D, Vivanco I, Chmielecki J, Pao W, Ladanyi M, Gerald WL, Liau L, Cloughesy TC, Mischel PS, et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc Natl Acad Sci U S A. 2009; 106(23):9435-9440.
17. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D and Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009; 45(2):228-247.
18. Edamoto Y, Hara A, Biernat W, Terracciano L, Cathomas G, Riehle HM, Matsuda M, Fujii H, Scoazec JY and Ohgaki H. Alterations of RB1, p53 and Wnt pathways in hepatocellular carcinomas associated with hepatitis C, hepatitis B and alcoholic liver cirrhosis. Int J Cancer. 2003; 106(3):334-341.
19. Liu J, Ma Q, Zhang M, Wang X, Zhang D, Li W, Wang F and Wu E. Alterations of TP53 are associated with a poor outcome for patients with hepatocellular carcinoma: evidence from a systematic review and meta-analysis. Eur J Cancer. 2012; 48(15):2328-2338.
20. Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R and Ishioka C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100(14):8424-8429.
21. Yarbrough WG, Buckmire RA, Bessho M and Liu ET. Biologic and biochemical analyses of p16(INK4a) mutations from primary tumors. J Natl Cancer Inst. 1999; 91(18):1569-1574.
22. Tanaka Y, Kato K, Notohara K, Hojo H, Ijiri R, Miyake T, Nagahara N, Sasaki F, Kitagawa N, Nakatani Y and Kobayashi Y. Frequent beta-catenin mutation and cytoplasmic/nuclear accumulation in pancreatic solid-pseudopapillary neoplasm. Cancer Res. 2001; 61(23):8401-8404.
23. Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messaien L, Bronson RT, McMahon M, Granter S, Flaherty KT, Wargo JA, Marais R and Cichowski K. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discovery. 2012.
24. Rubin JB and Gutmann DH. Neurofibromatosis type 1 - a model for nervous system tumour formation? Nature reviews Cancer. 2005; 5(7):557-564.
25. Ventii KH, Devi NS, Friedrich KL, Chernova TA, Tighiouart M, Van Meir EG and Wilkinson KD. BRCA1-associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer research. 2008; 68(17):6953-6962.
26. Landreville S, Agapova OA, Matatall KA, Kneass ZT, Onken MD, Lee RS, Bowcock AM and Harbour JW. Histone deacetylase inhibitors induce growth arrest and differentiation in uveal melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012; 18(2):408-416.
27. von Kries JP, Winbeck G, Asbrand C, Schwarz-Romond T, Sochnikova N, Dell’Oro A, Behrens J and Birchmeier W. Hot spots in beta-catenin for interactions with LEF-1, conductin and APC. Nat Struct Biol. 2000; 7(9):800-807.
28. Hardt M, Chantaravisoot N and Tamanoi F. Activating mutations of TOR (target of rapamycin). Genes Cells. 2011; 16(2):141-151.
29. Sato T, Nakashima A, Guo L, Coffman K and Tamanoi F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene. 2010; 29(18):2746-2752.
30. Chen W, Drakos E, Grammatikakis I, Schlette EJ, Li J, Leventaki V, Staikou-Drakopoulou E, Patsouris E, Panayiotidis P, Medeiros LJ and Rassidakis GZ. mTOR signaling is activated by FLT3 kinase and promotes survival of FLT3-mutated acute myeloid leukemia cells. Mol Cancer. 2010; 9:292.
31. Burset M, Seledtsov IA and Solovyev VV. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic acids research. 2000; 28(21):4364-4375.
32. Venables JP. Aberrant and alternative splicing in cancer. Cancer research. 2004; 64(21):7647-7654.
33. Zhang W, Konopleva M, Shi YX, McQueen T, Harris D, Ling X, Estrov Z, Quintas-Cardama A, Small D, Cortes J and Andreeff M. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. Journal of the National Cancer Institute. 2008; 100(3):184-198.
34. Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nature reviews Cancer. 2003; 3(3):155-168.
35. Peng G and Lin SY. Exploiting the homologous recombination DNA repair network for targeted cancer therapy. World J Clin Oncol. 2011; 2(2):73-79.
36. Joerger AC and Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008; 77:557-582.
37. Gherardi E, Birchmeier W, Birchmeier C and Vande Woude G. Targeting MET in cancer: rationale and progress. Nature reviews Cancer. 2012; 12(2):89-103.
38. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C and Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery. 2012; 2(5):401-404.
39. Lennerz JK, Kwak EL, Ackerman A, Michael M, Fox SB, Bergethon K, Lauwers GY, Christensen JG, Wilner KD, Haber DA, Salgia R, Bang YJ, Clark JW, Solomon BJ and Iafrate AJ. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011; 29(36):4803-4810.
40. Wu A, Wu B, Guo J, Luo W, Wu D, Yang H, Zhen Y, Yu X, Wang H, Zhou Y, Liu Z, Fang W and Yang Z. Elevated expression of CDK4 in lung cancer. J Transl Med. 2011; 9:38.
41. Schubbert S, Shannon K and Bollag G. Hyperactive Ras in developmental disorders and cancer. Nature reviews Cancer. 2007; 7(4):295-308.
42. Moore MJ, Wang Q, Kennedy CJ and Silver PA. An alternative splicing network links cell-cycle control to apoptosis. Cell. 2010; 142(4):625-636.
43. Yu C, Bruzek LM, Meng XW, Gores GJ, Carter CA, Kaufmann SH and Adjei AA. The role of Mcl-1 downregulation in the proapoptotic activity of the multikinase inhibitor BAY 43-9006. Oncogene. 2005; 24(46):6861-6869.
44. Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, Belmont LD, Kaminker JS, O’Rourke KM, Pujara K, Kohli PB, Johnson AR, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011; 471(7336):110-114.
45. Milde-Langosch K, Ocon E, Becker G and Loning T. p16/MTS1 inactivation in ovarian carcinomas: high frequency of reduced protein expression associated with hyper-methylation or mutation in endometrioid and mucinous tumors. Int J Cancer. 1998; 79(1):61-65.
46. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, Yang W, Heravi-Moussavi A, Giuliany R, Chow C, Fee J, Zayed A, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010; 363(16):1532-1543.
47. Takahashi Y, Kawate S, Watanabe M, Fukushima J, Mori S and Fukusato T. Amplification of c-myc and cyclin D1 genes in primary and metastatic carcinomas of the liver. Pathol Int. 2007; 57(7):437-442.
48. Kir S, Beddow SA, Samuel VT, Miller P, Previs SF, Suino-Powell K, Xu HE, Shulman GI, Kliewer SA and Mangelsdorf DJ. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science. 2011; 331(6024):1621-1624.
49. Tekin M, Hismi BO, Fitoz S, Ozdag H, Cengiz FB, Sirmaci A, Aslan I, Inceoglu B, Yuksel-Konuk EB, Yilmaz ST, Yasun O and Akar N. Homozygous mutations in fibroblast growth factor 3 are associated with a new form of syndromic deafness characterized by inner ear agenesis, microtia, and microdontia. Am J Hum Genet. 2007; 80(2):338-344.
50. Kratochwil K, Galceran J, Tontsch S, Roth W and Grosschedl R. FGF4, a direct target of LEF1 and Wnt signaling, can rescue the arrest of tooth organogenesis in Lef1(-/-) mice. Genes Dev. 2002; 16(24):3173-3185.
51. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clement B, Balabaud C, Chevet E, Laurent A, Couchy G, Letouze E, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012; 44(6):694-698.
52. Simioni C, Martelli AM, Cani A, Cetin-Atalay R, McCubrey JA, Capitani S and Neri LM. The AKT inhibitor MK-2206 is cytotoxic in hepatocarcinoma cells displaying hyperphosphorylated AKT-1 and synergizes with conventional chemotherapy. Oncotarget. 2013; 4(9):1496-1506.
53. Yi KH, Axtmayer J, Gustin JP, Rajpurohit A and Lauring J. Functional analysis of non-hotspot AKT1 mutants found in human breast cancers identifies novel driver mutations: implications for personalized medicine. Oncotarget. 2013; 4(1):29-34.
54. Thomas HE, Mercer CA, Carnevalli LS, Park J, Andersen JB, Conner EA, Tanaka K, Matsutani T, Iwanami A, Aronow BJ, Manway L, Maira SM, Thorgeirsson SS, Mischel PS, Thomas G and Kozma SC. mTOR inhibitors synergize on regression, reversal of gene expression, and autophagy in hepatocellular carcinoma. Sci Transl Med. 2012; 4(139):139ra184.
55. Zhu AX, Abrams TA, Miksad R, Blaszkowsky LS, Meyerhardt JA, Zheng H, Muzikansky A, Clark JW, Kwak EL, Schrag D, Jors KR, Fuchs CS, Iafrate AJ, Borger DR and Ryan DP. Phase 1/2 study of everolimus in advanced hepatocellular carcinoma. Cancer. 2011; 117(22):5094-5102.
56. Floc’h N and Abate-Shen C. The promise of dual targeting Akt/mTOR signaling in lethal prostate cancer. Oncotarget. 2012; 3(12):1483-1484.
57. Garrett JT, Chakrabarty A and Arteaga CL. Will PI3K pathway inhibitors be effective as single agents in patients with cancer? Oncotarget. 2011; 2(12):1314-1321.
58. Markman B, Dienstmann R and Tabernero J. Targeting the PI3K/Akt/mTOR pathway--beyond rapalogs. Oncotarget. 2010; 1(7):530-543.