
INTRODUCTION
Basal metabolic activity as well as environmental factors lead to more than 20,000 DNA alterations per cell every day [1]. Since structural DNA damage may interfere with transcription and, consequently, disarray proteome homeostasis, maintenance of genomic integrity is critical for cellular function. Genomic maintenance primarily depends on a network of different repair mechanisms, collectively termed DNA damage response (DDR). Decades of intense research revealed that one of the DDR pillars is the poly-adenosine diphosphate-ribose polymerase (PARP). Based on its central role in the nuclear repair machinery, PARP has been considered as a potential target in cancer cells with compromised repair machinery. According to this concept, inhibition of PARP acts as a “second hit” on the malfunctioning cellular repair armament of, for instance, BRCA1/2-negative cancers resulting in fatal chromosome instability, cell cycle arrest and apoptosis. This led to the development of therapeutic PARP inhibitors and the idea of their use to potentiate antineoplastic alkylating agents.
Although PARP inhibitors, like Olaparib or Veliparib, failed to deliver expected results in clinical trials, they led to the discovery of a number of PARP-mediated extranuclear effects including mitochondrial functions or crosstalk with canonical signaling pathways [2–4]. Here, we overview our current understanding on the mechanism of action of PARP and its pharmacological inhibitors with particular attention to their clinical relevance.
PARP
Poly(ADP-ribose) polymerases play pivotal role in the intricate network of intracellular processes counteracting genetoxic stress in higher eukaryotic cells [5]. PARP was first identified in 1963 as a nuclear enzyme responsible for the majority of poly(ADP-ribosyl)ation activity [6]. Although PARP-1 is the most abundantly expressed isoform, multiple PARP-encoding sequences have been identified in the human genome [7]. Analyses of transcript abudance in epithelial cell lines of various tissues revealed that, apart from PARP-15, all PARPs are expressed [8]. According to the currently available structural data, all 17 known PARP polypeptides, with the exception of PARP-4, have a conserved C-terminal catalytic domain. In contrast, they display great variability of various domains and functional motifs in their N terminus. These include regulatory and binding motifs (e.g. the PARP alpha helical motif or PRD), localization signals, specific regions responsible for interaction with partner molecules (e.g. the WWE domains in PARP-7, the Myc-binding region in PARP-10 or the HIF1AN-binding region in PARP-5b), regions of compositional bias (e.g. the poly-Histidine, Proline- or Serine-rich regions in PARP-5a) or the zinc finger and ubiquitin-binding domains. Although the relevance of the N-terminal domain structure variance is still not clear, it suggests differential physiologic roles and, potentially, spatio-temporal control of the PARP isoforms and represents the basis of their classification into one of the five subgroups of the family (Figure 1).
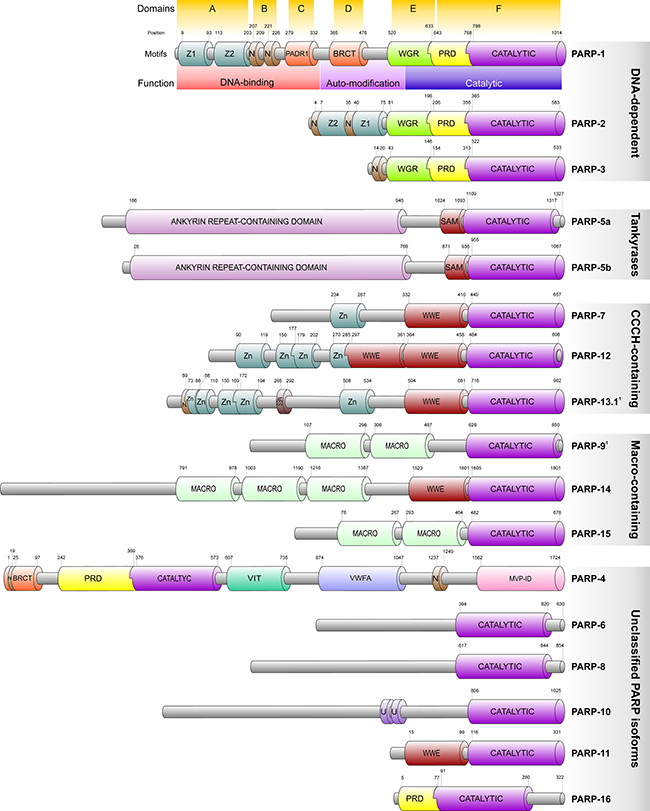
Figure 1: Domain structure and classification of the human PARP polypeptides. Figure is based on the Uniprot, RCSB and InterPro databases. Members are related on the basis of the presence of the conserved PARP catalytic domain (CATALYTIC) typically located at the C-terminus of the polypeptides and is characteristic of all PARP protein family members. Within the catalytic domain, the active site is formed by a block of 50 amino acids which is strictly conserved among the vertebrates and highly conserved among all species. Proteins are not drawn to scale but typical structural limits and positions are indicated. 1PARP-9 and -13.1 are believed to be catalytically inactive. The alternative isoform of PARP-13 (PARP-13.2) is not indicated in the figure due to its complete lack of the PARP catalytic domain. Labelled structures are follows: Z, Z1 and Z2: PARP-type zinc finger domains; N: Nuclear localization signal; NES: Nuclear export signal; BRCT: breast cancer susceptibility gene associated C-terminal domain; WGR: It is a domain with unspecified functions present in a number of PARPs and named after its most conserved central motif.; PRD: PARP regulatory domain, consists of a duplication of two helix-loop-helix structural repeats and is typically associated with the C-terminal catalytic domain.; SAM: Sterile Alpha Motif, also known as helix-loop-helix domain, exhibits a conserved structure involved in interactions with proteins, DNA and RNA.; WWE: The WWE domain is named after three of its conserved residues and believed to serve as an interaction module.; MACRO: The Macro domain is a 180 amino acids long region that mediate ADP-ribose binding associated with catalytic domains of PARP or sirtuins.; VIT: Vault Protein inter-alpha-trypsin domain; VWFA: von Willebrand factor type A domain that mediates metal ion-dependent adhesion of partner proteins.; MVP-ID: Major Vault Protein interaction domain; U: ubiquitin-binding motif.
Poly(ADP-ribosyl)ation is catalyzed by the DNA-dependent and tankyrase PARPs as well as the unclassified PARP-4. The majority of the other isoforms, however, perform mono(ADP-ribosyl)ation with the exceptions of PARP-10 which has poly(ADP) transferase activity, and PARP-9 and -13 that are believed to be catalytically inactive [9, 10]. While intracellular localisation of the PARP family members varies during cell cycle, PARP-1 was shown to be predominantly nuclear. PARP-1, -2 and -3 are ubiquitously expressed in mammalian tissues and are the only DNA strand break-activated isoforms identified so far [11–13].
PARP-1, the best characterised member of the PARP family, is a 116 kDa protein composed of 6 main domains (domain A to F) each with distinct functions (Figure 1) [14]. Domain A functions as part of the DNA-binding module (DBM) and is responsible for the recognition of damaged DNA loci via two zinc-finger motifs. Domain B spans a bipartite nuclear localisation signal that directs PARP-1 into the nucleus and serves as a caspase-3 cleavage site [15]. Similar to domain A, domain C contains a zinc-binding motif (PADR1) that facilitates formation of the DNA-activated conformation of PARP-1 via interdomain contacts. Although not required for physical DNA binding, its absence compromises the catalytic activity of PARP-1 upon DNA binding. Domain C together with Domain A and B are believed to form the N-terminal DNA-binding module of PARP-1 [16]. Domain D mediates negative auto-regulatory post-translational modifications via glutamate, aspartate and lysine residues that reduce enzymatic activity leading to relaxed DNA binding [17, 18]. Another region of Domain D, termed Breast Cancer-Associated 1 C-terminal domain (BRCT), acts as a binding interface for various nuclear partners [19]. Domain E and F together form the catalytic site that binds nicotinamide adenine dinucleotide (NAD+) and catalyses poly(ADP-ribosyl)ation also termed as PARylation [20]. In addition, the catalytic domain promotes localized compaction of chromatin into supranucleosomal structures through interaction with the DNA binding domain independently of its enzymatic activity [21].
Although DNA single-strand breaks are the primary stimuli for PARP-1, its activity has also been reported in the absence of DNA lesions. Indeed, PARP-1 recognizes unusual DNA conformations including cruciformed or supercoiled structures as well as bent or stably un-paired DNA regions [22–24]. In support of these observations, PARP-1 is activated in response to a wide range of stimuli that potentially affect DNA conformation including oxidative agents, ethanol, DNA alkylation, excitotoxic injury, lipopolysaccharides (LPS), elevated extracellular glucose concentration or vitamin A depletion [25–31]. Since PARP-2 accounts for approximately 15% of the cellular poly(ADP)-ribose only, PARP-1 is believed to be responsible for the majority of cellular PARylation. Similar distribution of their cellular targets further supports the idea that PARP-1 is the primary PARylating entity [32].
Upon activation, the DBM scans and directs PARP-1 to damaged DNA loci by recognizing aberrant DNA conformations or disruptions of the sugar-phosphate backbone [12]. Activated PARP-1 cleaves NAD+ into nicotinamide and ADP-ribose and covalently attaches 50–200 ADP-ribose units to target molecules through glutamate, aspartate or lysine residues [6, 18]. At damaged DNA loci, auto-PARylation triggers recruitment of repair enzimes and, consequently, initiates DNA repair [33]. Being negatively charged, poly(ADP-ribose) alters the biochemical properties of the acceptor molecules resulting in the modulation of their structure, function or localization [34]. Indeed, due to their physico-chemical properties, initially synthesized long and branched PAR chains repell chromatin from the vicinity of the damaged locus preventing accidental homologous recombination between the broken backbone and neighbouring chromatin sections. These initially formed PAR chains, however, are trimmed back to short ones by poly(ADP-ribose) glycohydrolase (PARG), that persist on auto-modified PARP-1 even after its release from DNA. This persistent auto-PARylation prevents PARP-1 to rejoin damaged DNA that might inhibit the execution of the repair process [35].
Elevated NAD+ consumption and PAR levels are both observed upon treatments with DNA alkylating agents or ionizing irradiation supporting the idea that PARP-1-mediated PARyaltion is part of the DDR [5]. Indeed, a number of repair molecules harbour PAR-binding motifs including elements of the base-excision repair (BER) [36]. The BER acts on single nucleotide lesions catalysing excision and replacement of damaged or incorrectly incorporated nucleotides [37]. As part of the BER, PARP is believed to bind DNA strand-breaks and by simultaneous auto- and target PARylation facilitates the recognition and repair of affected loci. The nature of the interplay between PARP-1 and elements of the BER, however, is still obscure due to apparently conflicting experimental results. Although PARylation facilitates recruitment of XRCC1, the scaffold molecule that recruits additional BER enzymes to damaged foci, BER remains intact even in PARP-1 negative cells [38–40]. In addition, while they show increased sensitivity toward PARP inhibitors, XRCC1-depleted cells exhibit intact BER activity suggesting a BER-independent role of the PARP-1-XRCC1 interaction [41]. To date, the most widely accepted scenario is that PARP-1, perhaps via XRCC1, contributes to certain types of the BER depending on the nature of the insult or intermediates formed but it does not play an indispensible role in the process reflecting the existing redundancy in the BER system [42].
In the nucleotide excision repair (NER), PARP-1 PARylates DDB2, an essential protein for recognition and removal of UV-induced DNA lesions. PARylation of DDB2 prolongs its chromatin retention time and recruitment of the chromatin remodeler ALC1 that facilitates nucleosome sliding and recruitment of further NER proteins like XPC [43]. Interaction of PARP-1 with various elemets of the mismatch repair has also been reported. These include replication protein A, replication factor C and the proliferating cell nuclear antigen (PCNA) as well as the MutSα-exonuclease 1 (EXO1) complex. The interplay between PARP-1 and the MutSα-activated EXO1 complex results in enhanced 5′-directed excision activities by, possibly, repressing EXO1-mediated hydrolysis, preferentially, on homoduplex DNA [44].
In addition, pharmacological inhibition of PARP-1 was found to result aberrant activation of the non-homologous end joining repair (NHEJ) as well suggesting its role in the repair of double-strand breaks. In support of this concept, not only DNA-dependent protein kinases were identified in the PARP-1 interactome but PARylation was also found to increase their kinase activity [36, 45].
A critical prerequisite for the catalytic activity of recruited repair enyzmes is termination of PARylation and dissociation of PARP-1 from affected loci. In this process first, PARP-1 auto-PARylates within domain D that results in the release of PARP-1 from target molecule, a critical step in the abortion of target PARylation. Apparently, one of the critical regulators of this step is the recently described Histone PARylation factor 1 (HPF1) that controls retention time of PARP-1 via regulation of both ADP-ribosylation of histones and auto-modification of PARP-1 [46]. Once released, PARP-1 is ubiquitinated by the PAR-dependent E3 ubiquitin ligase RING finger protein 146 rendering PARP-1 for proteasomal degradation [47]. Finally, removal of polymerized PAR flags are mediated by dedicated enzymes PARG and poly(ADP-ribose) hydrolase 3 that catalyse degradation of PAR within minutes [48, 49] (Figure 2).
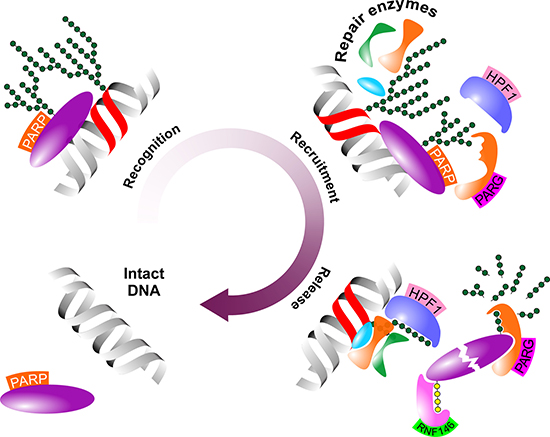
Figure 2: Schematic figure of the role of PARP-1 in the DNA demage response. PARP contributes to the DDR in three major steps upon DNA damage. First, PARP scans and identifies dmaged DNA loci (Recognition). Second, PARP-catalysed PARylation attracts repair elements to the damaged DNA locus (Recruitment). Finally, de-PARylation detaches PARP from partner molecules and facilitates its proteasomal degradation (Release). Abbreviated elements are: PARP: Poly(adenosine diphosphate-ribose) polymerase-1; HPF1: Histone PARylation factor 1; PARG: poly(ADP-ribose) glycohydrolase; RNF146: RING finger protein 146. Green and yellow polymers represent PAR and polyubiquitin chains, respectively.
Intensive research on PARP-1’s mechanism of action in DNA repair led to the concept that defective PARP-1 functions result in genomic instability and cell death fueling the idea of the use of pharmacological PARP-1 inhibitors in anti-cancer therapies [50].
PARP inhibition in cancer treatment
The first in vitro experiments with distinct PARP inhibitors concluded that these compounds are more potent in BRCA-deficient cellular models and exert synergistic effects when used in combination with chemotherapeutics like the alkylating agent temozolomide or the topoisomerase inhibitor topotecan [51–53]. Considering the central role of BRCA-1 and -2 in DNA repair, these observations underpinned the idea that PARP inhibition primarily targets repair capacity. It is noteworthy however, that other studies reported no or only cell type-dependent effects of PARP inhibitors when tested in combination of platinum agents [54, 55]. More interestingly, some studies reported that excessive PARylation and sustained existence of PARP-DNA complexes are more cytotoxic than genetic depletion of PARP suggesting a more versatile role of PARP-1-mediated PARylation [56].
In vivo studies using PARP inhibiting compounds in combination with chemotherapeutics led to similar conclusions. Using the PARP inhibitor compound AG14361 in a human colon tumor xenograft model, improved therapeutic index was reported when used in combination with temozolomide, irinotecan or irradiation [57]. Veliparib, also known as ABT-888, was found to enhance the effect of temozolomide in various xenograft models in a dose-dependent manner leading to increased survival rates [58]. Similarly, Olaparib, also known as compound AZD2281, showed synergistic effects with cisplatin and carboplatin in a BRCA1-deficient mammary tumor model resulting in prolonged survival [59, 60]. In the theet of encouraging preliminary experimental data on pharmacological inhibition of PARP-1, clinical trials delivered rather inconsistent results. While a phase I trial reported significant antitumor activity in Olaparib-treated patients suffering from BRCA mutation-harbouring breast, ovarian and prostate cancers, another study found no synergistic effect between Olaparib and dacarbazine in chemonaive melanoma patients [61, 62]. Similarly, while Olaparib was reported effective in ovarian cancer patients in a phase II trial, the same study declared it inefficient in breast cancer patients independently of their BRCA status [63].
Controversal clinical results raised the question whether the capacity of PARP inhibitors to sensitize cells to chemotherapeutic agents is dependent on the therapeutic context and restricted to certain cell types or chemotherapeutic compounds. Indeed, to date, PARP inhibition has only been approved for monotherapy of BRCA mutation-harbouring neoplasms [64]. Contradicting data on the use of PARP inhibitors, however, raised the possiblity of more intricate underlying functions of PARP that might influence cellular physiology beyond repair.
PARP-1 as a mitochondrial regulator
While the role of PARP-1 in various repair machineries is established, only 60% of PARP-1 complexes are detected on DNA breaks suggesting the existence of repair-independent PARP-1 functions [65]. Indeed, PARylation-mediated regulation of chromatin remodeling and transcription has been demonstrated in Drosophila representing a PARP-1-mediated epigenetic regulatory system [66]. In human models, this involves histone H3 and H4 that both bind PARP-1 physically. In addition to their co-localization, Histone H4 has also been shown to enhance PARP-1 activity [67]. DDR-independent recruitment of PARP-1 to chromatin and the post-translational modification of histones and DNA have been shown to induce various genes highlighting the complexity of PARP-1-mediated functions. Indeed, NAD-consuming PARP-1 contributes to the trans-activation of nuclear-encoded mitochondrial genes like the cytochrome c oxidase COX1, COX2 and complex I subunit ND2, critical components of the mitochondrial electron transport chain. Moreover, pharmacological inhibition of PARP-1 leads to repression of the nuclear-encoded mitochondrial DNA repair factors UNG1, MYH1 and APE1 as well as mitochondrial transcription factors TFB1M and TFB2M [68]. Thus, by epigenetic marking, PARP-1 controls integrity and function of mitochondria, a critical source of PARP-1’s co-enzyme NAD+ (Figure 3).
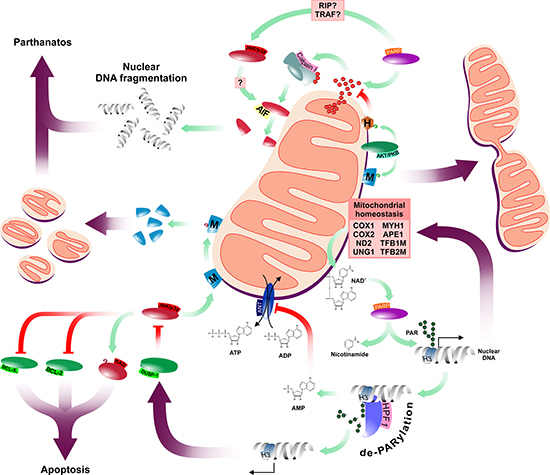
Figure 3: Schematic summary of PARP-1’s mitochondrial functions. PARP has multiple connections to the mitochondrial network affecting both mitochondrial function and morphology. Abbreviated elements are: HPF1: Histone PARylation factor 1; M: Mitofusins; H: Hexokinase II; ANT: mitochondrial adenine nucleotide translocator; H3: Histone H3; AMP, ADP and ATP: adenosine mono-, di- and triphosphate, respectively; AIF: Apoptosis Inducing Factor. Green and purple arrows represent catalysis and overall effects, respectively. Red connectors represent inhibitory interactions.
Besides histones, a growing number of additional molecules have also been recognized as potential targets for PARylation [69]. Systemic analysis of the PARylated proteome revealed links between PARylation and fundamental cellular processes including DNA and RNA metabolism, cell-cycle regulation, apoptosis or canonical cell signaling pathways (Supplementary Table 1). These findings suggest that the overall therapeutic effect of the pharmacological inhibition of PARP-1 may be dependent on the cellular context and further insight of PARP-1-mediated functions, in particular those affecting cellular viability, is critical for the development of safe and efficient therapeutic modalities.
PARP-1 as a regulator of cell death
Increased PARP-1 activity leads to elevated consumption of cellular NAD+ that may accompanied by increased ATP consumption and, consequently, compromised energy balance, a known factor that may facilitate cell death [70, 71]. Depleted intracellular ATP pools are direct results of increased AMP levels generated by the hydrolysis of poly(ADP-ribose) which is accompanied by inhibition of the mitochondrial adenine nucleotide translocator (ANT) and reduction of mitochondrial ADP uptake and ATP release (Figure 3) [72]. Consequently, the adenylate kinase bypass, which recharges ATP from ADP, is also limited due to depleted ADP resources upon PARP-1 up-regulation [73]. Accordingly, it was hypothesized that, upon persistent PARP-1 activation, decreased mitochondrial ATP production inhibits NAD+ re-synthesis forming a feed-forward loop in ATP-consuming processes that, eventually, results in metabolic catastrophy and cell death. The relevance of the relationship between intracellular metabolism and PARP-1 activaton is endorsed by reports that PARP-1 activation down-regulates Hexokinase II contributing to metabolic imbalance and consequent cellular demise [74, 75].
Alternatively, PARP-1 may also reduce cellular viability via parthanatos. The term parthanatos originates from the abbreviation of poly(ADP-ribose) and Thanatos, the name of the God of Death in Greek mythology, and is used for a process accompanied by the loss of cell membrane integrity, large scale DNA fragmentation, phosphatidylserine externalization, dissipation of mitochondrial membrane potential, chromatin condensation and shrinkage, characteristics of a cell dying in consequence of excessive PARP-1 activation. One of the key mediators of parthanatos is the Apoptosis Inducing Factor (AIF), a caspase-independent mitochondrial death effector protein that has three putative PAR binding domains. In normal mitochondria, it shows oxido-reductase activity and functions as an anti-apoptotic factor [76]. Although the majority of AIF is anchored to the inner mitochondrial membrane, an estimated 30% is associated with the cytosolic side of the outer mitochondrial membrane [77, 78]. Upon PARP-1 activation, PAR polymers activate calpain I through mitochondrial Ca2+ dysregulation which leads to truncation of AIF, a critical step of its mitochondrial release and nuclear translocation. In the nucleus, PARylated AIF facilitates caspase-independent parthanatos via chromatin condensation and DNA fragmentation (Figure 3) [78–82]. Insights of the potential role of PARylation in processes like parthanatos, however, placed PARP-1, which was previously considered as a nuclear element of the DNA repair systems, into a wider cellular context and fueled further investigations on PARP-1’s effects in the context of complex extranuclear systems like mitochondria.
PARP-1 as a regulator of mitochondrial functions
PARP-1 hyperactivation is characterized by excessive nuclear NAD+ consumption and accompanied cytosolic NAD+ depletion compromising NAD+-dependent metabolic pathways including the glycolysis and TCA cycle that may culminate in respiratory chain malfunctions and accumulation of reactive oxygen species (ROS) [83]. It has also been documented that ROS-induced mitochondrial permeability transition may lead to intra-mitochondrial loss of NAD+ [84]. Thus, hyperactivation of PARP may initiates events that not only aggravates mitochondrial damage but also contribute to the ROS-mediated generation of additional single-strand DNA breaks [85]. These findings inspired the idea that PARP inhibitors might exert their cytoprotective effects by limiting the PARP-1-induced depletion of cellular NAD+ and, consequently, preserving mitochondrial bioenergetics. This hypothesis is underpinned by the beneficial effects of these compounds in various ischemia-reperfusion injury models, where the protective effects of PARP inhibitors are not mediated by direct antioxidant properties of the compounds used [86]. In support of this concept, PARP inhibitors were found to be effective in multiple pathophysiologic conditions including myocardial or neuronal ischemia, inflammation, oxidative stress-related cellular injury, Diabetes mellitus or traumatic brain injury [87–92].
Follow-up studies on PARP inhibitors using isolated mitochondria, however, suggested the existence of additional mitochondrial-resident targets with capacity to regulate mitochondrial homeostasis [93]. A potential candidate emerged from studies investigating benefits of the use of PARP inhibitor PJ-34 in septic mouse models. Using lipopolysaccharide (LPS) on PJ-34 pre-treated animals, activating phosphorylation of AKT/PKB was detected, raising the possibility that the protective effects of PARP inhibition are mediated by the AKT/PKB pathway [94]. Although the relationship between metabolism and AKT/PKB pathway has been known for decades, the mitochondrial-resident fraction of AKT/PKB was first described in 2003 [95]. Cytosolic AKT/PKB, a critical element of a highly conserved pathway activated by various stimuli in a phosphatidylinositol 3 kinase (PI3K)-dependent manner, phosphorylates a wide range of substrates including signaling pathways elements, apoptosis regulators and transcription factors involved in the regulation of cellular metabolism [96–98]. Indeed, activation of the AKT/PKB pathway leads to the induction of a number of genes involved in the glucose metabolsm including glucose transporters or the hexokinase while, directly, AKT/PKB catalyses post-translational activating phosphorylation of the phosphofructokinase [99–101]. The AKT/PKB pathway also regulates lipogenic genes by the induction of the transcription factor SREBP1c [102]. Accordingly, AKT/PKB-mediated biological responses are pleiotropic, ranging from cell survival to proliferation, intracellular trafficking or complex processes like angiogenesis [103, 104].
Similar to its cytoplasmic counterpart, the mitochondrial AKT/PKB generally exerts pro-survival effects. Indeed, translocation and activation of AKT/PKB to myocardial mitochondria enhances cardiac bioenergetics by influencing mitochondrial oxidative phosphorylation, preserves mitochondrial integrity and prevents cytochrome c release upon induction of the intrinsic apoptotic pathway [105–107]. Interestingly, however, unlike the BCL family members-mediated canonical mitochondrial pro-survival mechanisms, the anti-apoptotic function of mitochondrial AKT/PKB is glucose-dependent [108, 109]. AKT/PKB mediates its mitochondrial effects through phosphorylation of Hexokinase II, the same target that is down-regulated by PARP-1 activation. Hexokinase II phosphorylation promotes its recruitment to the mitochondrial outer membrane voltage-dependent anion channel (VDAC) [75, 108]. Upon binding to VDAC, Hexokinase II converts glucose to glucose-6-phosphate consuming mitochondrial ATP thus facilitating the carbon supply of both glycolysis and the pentose phosphate pathway [110, 111]. AKT/PKB-mediated phosphorylation of Hexokinase II also counteracts oxidant or Ca2+-stimulated opening of mitochondrial permeability transition pore (PTP) (Figure 3) [112].
Although detailed mechanisms are yet to be determined, observations that the angiotensin II-induced PARP-1 activation is Ca2+-mediated in primary culture of newborn cardiomyocytes and that PARP-1 activation follows Ca2+ release from perinuclear stores in depolarized primary culture of rat brain cortical neurons suggest that, at least in certain cellular contexts, Ca2+ may be one of the candidate mediators of PARP-1-activating stimuli [113, 114]. Thus, one might speculate that PARP inhibitors also reduce intracellular Ca2+ levels, possibly by utilizing or mimicking the effects of the AKT/PKB pathway thereby contributing to mitochondrial integrity and cell survival.
PARP-1 as a regulator of mitochondrial morphology
Mitochondrial function and morphology are intimately linked. Indeed, over-expression of the mitochondrial fission proteins DRP1 or hFIS1 increases susceptibility to Ca2+-induced PTP opening [115]. Conversely, over-expression of the mitochondrial fusion GTP-ase Mitofusin-2 (MFN2) inhibits PTP opening [116]. Intriguingly, AKT/PKB also promotes mitochondrial fusion by activating phosphorylation of Mitofusin-1, a mitochondrial fusion-mediator transmembrane GTPase, leading to delayed onset of PTP opening and reduced cell death following ischaemia-reperfusion injury [117, 118]. OPA1, a nuclear-encoded mitochondrial dynamin-related GTPase that controls mitochondrial morphology and ultrastructure, might also be a downstream mediator of the AKT/PKB pathway [119, 120]. OPA1 is reduced under conditions associated with mitochondrial fragmentation in myocardiocytes while, upon insulin stimulation which is a known activator of the AKT/PKB pathway in the myocardium, mitochondrial fusion and OPA1 levels appear to be increased [121, 122]. The concept that AKT/PKB controls mitochondrial morphology via OPA1 is also supported by the observations that expression of both MFN2 and PARL, a protease involved in the anti-apoptotic function of OPA1, is decreased in obese and insulin-resistant patients [123, 124]. Moreover, this effect does not seem to be restricted to the myocardium since coronary endothelial cells also show reduced OPA1 levels and elevated mitochondrial fragmentation in diabetic murine models [125].
The complex effects of the AKT/PKB pathway on mitochondria is further illustrated by recent findings that AKT/PKB induces trafficking of energetically active mitochondria to the cortical cytoskeleton of tumor cells leading to lamellipodia formation, supports turnover of fatty acid complexes and random cell migration [126]. Intriguingly, this phenomenon is similar to the accumulation of mitochondria at synapses, active growth cones and branches in neuronal cells [127]. In both cases, mitochondria might provide a “regional” ATP source to fuel energy-demanding processes [126]. In support of this concept, inhibition of cellular respiration by mitochondrial DNA depletion or direct blockage of the respiratory chain prevents mitochondrial trafficking to the cortical cytoskeleton, abolishes membrane dynamics of cell motility and suppresses cell invasion [126]. Interference with MFNs suppresses mitochondrial repositioning to the cortical cytoskeleton and tumor cell invasion mediated by PI3K inhibitor therapy indicating the interplay between the AKT/PKB and MFN pathways [126]. Hence, one can speculate that alterations in mitochondrial morphology and function promoted by the AKT/PKB pathway represent another mechanism of the observed mitoprotective effects of PARP inhibitors.
The AKT/PKB pathway, however, is not the only signaling mechanism associated with the regulation of mitochondrial morphology and PARP-1. Activation of JNK and p38 MAP kinases has also been reported in response to PARP-1 and both kinases are involved in the regulation of mitochondrial dynamics and function (Figure 3) [128–130]. The potential mediator of this effect is the dual specific protein phosphatase-1 (DUSP-1). Indeed, PARP-1 inhibition attenuates JNK and p38 through the increased expression of DUSP-1 enhancing cells survival [131]. JNK phosphorylates BCL-2 and BCL-XL, attenuating their pro-survival activity that facilitates cytochrome c release and collapse of the mitochondrial membrane potential [132]. It can also directly trigger the intrinsic apoptotic pathway by phosphorylating the pro-apoptotic BCL-2 family member BAD [133]. Apart from the regulation of the canonical intrinsic apoptotic pathway, JNK actively contributes to the mitochondrial morphology as well. JNK-mediated phosphorylation of MFN2 promotes its proteasomal degradation by the E3 ubiquitin ligase HUWE1 leading to mitochondrial fragmentation and enhanced apoptotic cell death [134]. Conversely, blockade of the Transferrin Receptor 1 (TFR1)-JNK pathway reduces HUWE1-mediated MFN2 ubiquitination preserving the fused mitochondrial network and function [135]. Similarly, pharmacological inhibition of p38 biases mitochondrial dynamics toward fusion and maintains mitochondrial functions [136].
Despite the apparent contribution of JNK and p38 to the regulatation of mitochondrial morphology and functions, both proximal and distal elements of the putative PARP-1-JNK/p38 axis remain to be identified. As for proximal mediators, RIP and TRAF were suggested to convey signals between PARP-1 and JNK while the observation that PARP-1-induced JNK activation is indispensable for mitochondrial depolarization, AIF translocation and subsequent cell death suggests that AIF might act as an effector of the PARP-1-TRAF/RIP-JNK/p38 pathway [128, 137].
This model predicts cytoprotective effects of PARP inhibitors in tissues like skeletal muscle or neural ones that highly dependent on mitochondrial functions. In support of this concept, PARP inhibition is apparently beneficial in a number of muscle dysfunction models [138, 139]. Moreover, PARP inhibition attenuated the mitochondrial toxin cuprizone-induced oligodendrocyte depletion and demyelination in experimental models. These mitoprotective effects are believed to be mediated by suppression of JNK and p38 phosphorylation, increased activation of the AKT/PKB pathway and repression of apoptosis [140].
PARP-1 as a regulatior of inflammation
Protective effects of PARP inhibitors in disease models of acute lung inflammation or septic shock predict a role for PARP-1 in inflammation as well [29, 141]. Indeed, analysis of PARP-1-/- mice revealed increased resistance to LPS-induced endotoxic shock and failed induction of NF-κB-regulated inflammatory genes due to the missing co-activator function of PARP-1 [142–144]. Interestingly however, although both lung and liver equally responsive to LPS-mediated NF-κB activation, PARP-1 inhibition disrupts the NF-κB-mediated response to LPS in the liver only suggesting the existence of tissue-specific elements in the PARP-1/NF-κB interactome [94]. One of the candidate interactors is Sir2alpha (SIR2α) that directly targets NF-κB (Figure 4). SIR2α is a primarily nuclear-resident, NAD+-dependent protein deacetylase with a wide range of intracellular targets including signaling molecules like AKT/PKB, protein components of the chromatin or transcription factors and their co-regulators. Since the Km of PARP-1 for NAD+ falls in the low micromolar range, PARP-1 may influence SIR2α activity by reducing NAD+ bioavailability [145–147]. Indeed, depletion of cellular NAD+ levels upon PARP-1 activation reduces SIR2α deacetylase activity. Conversely, reduced PARP-1 activity increases intracellular NAD+ levels and enhances SIR2α activity leading to SIR2α-mediated deacetylation and induction of mitochondrial biogenesis [148, 149]. Intriguingly, PARP-2 also interacts with SIR2α directly down-regulating the SIR2α promoter [150].
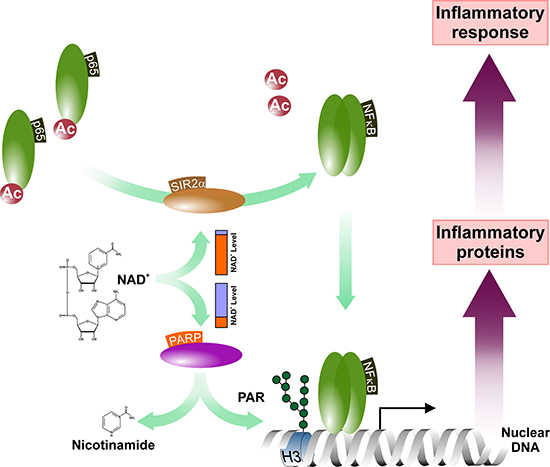
Figure 4: PARP activity influences the NF-κB-mediated inflammatory signaling. The PARP activation-mediated depletion of the NAD+ pools attenuates cellular NAD-dependent systems including the sirtuins. This may affect the rate of deacetylation of, among others, the NF-κB subunit p65 and, thus, influence the NF-κB-mediated inflammatory gene expression. Abbreviated elements are: Ac: Acetyl-group; H3: Histone H3; PAR: poly(ADP-ribose) polymer; SIR2α: NAD-dependent protein deacetylase Sirtuin-1.
Based on the Human Integrated Protein Expression Database, the SIR2α polypeptide is most enriched in lung tissues. It has also been reported that acetylation reduces the DNA-binding affinity of RELA/p65 [151]. Thus, one can speculate that elevated SIR2α activity may activate NF-κB upon LPS treatment in PARP-inhibited lung tissues while, conversely, the low relative abundance of SIR2α in hepatocytes leads to disruption of the inflammatory responses in the liver. This hypothesis also suggests that PARP inhibition-based anti-inflammatory modalities may not be efficient in SIR2α over-expressing tissues like prostate, fetal heart, testis or lymphocytes but may be potent in inflammed organs in which the putative SIR2α-mediated bypass mechanism is not present. In support of this concept, PARP inhibition was found to reduce TNFα induced inflammatory responses of synovial fibroblasts suggesting the potential use of PARP inhibitors in conditions like rheumatoid arthritis [152]. The idea that SIR2α may rescue NF-κB activity upon PARP inhibitions also raises the question whether PARP-1 functions as a transcriptional co-factor of the inflammatory pathways by regulating deacetylation of NF-κB via recruitment of deacetylases or preventing the interaction between acetylases and NF-κB.
Although these questions need further investigations, detailed analysis of the PARP-1/NF-κB interactome holds out the promise to discover novel, potentially tissue-specific targets of future anti-inflammatory therapies. Indeed, pharmacological inhibition of PARP reverted elevated colonic permeability and reduced water absorption in a chronic colitis model using IL-10-deficient mice [153]. Down-regulation of pro-inflammatory gene expression observed in a PARP-1-depleted mouse enterocolitis model upon exposure to Salmonella typhimurium suggests that these effects might be related to the NF-κB mediated gene regulatory functions of PARP-1 [154]. Reduction of the NF-κB activation and pro-inflammatory gene expression observed in a PARP-1-/- negative mouse model of contact dermatitis further supports this concept [155]. Similarly, repression of pro-inflammatory cytokines upon PARP-1 inactivation by either genetic ablation or pharmacologic inhibition might also explain compromised recruitment of inflammatory cells observed in a pulmonary inflammation model. These data may also open the door to the use of PARP inhibitors in pathophysiologic conditions like Diabetes mellitus. Indeed, PARP inhibitors efficiently reduce hyperglycemia-induced NF-κB activation and in vivo podocyte depletion, a hallmark of diabetic glomerulopathy [156].
Conclusions and perspectives
On the ground of discovery of PARP-1 and its role in the DNA repair machinery, PARP inhibition emerged as a novel therapuetic concept to eradicate cancer cells. This concept was further fueled by the discovery of the mitoprotective nature of PARP inhibitors, raising the possibility of reduced side effects on the non-transformed surrounding tissue [86, 157]. What we have learned from both in vitro and in vivo studies on PARP inhibition, however, revealed a more complex picture and prompts reconsideration of the possible and effective use of PARP inhibitors in the clinical practice. Indeed, the periodic increase of PAR activity during cell cycle highligths the importance of PARylation not only in the S-phase but also in the M-G1 transition and might serve as one of the critical factors in the regulation of migration and, potentially, formation of metastases [158, 159]. In addition, the typical mitochondrial effects of PARP inhibition including membrane potential maintenance, reduced oxygen/glucose consumption and lower intracellular concentrations of ROS and ATP are similar to that of cancer cells raising concerns about the systemic use of PARP inhibitors [160].
Data showing activation of the AKT/PKB pathway upon PARP inhibition may also be alarming. By inactivation of glycogen synthase kinase-3, caspase-9, BAD or the forkhead homologue rhabdomyosarcoma transcription factor, AKT/PKB has been shown to be a critical factor of cytostatic resistance of transformed cells [161]. Indeed, AKT/PKB promotes chemo-resistance of cells by regulating the ABC transporter BCRP1 activity which enhances drug efflux. In addition, in prostate cancer cells, AKT/PKB was shown to mediate effects of the metastasis-associated gene 1 that promotes cellular transformation and metastasis generation via regulation of E-cadherin [162–165]. Similar findings have been reported in terms of JNK and SIR2α showing their role in drug resistance and induction of metastases, respectively [166, 167]. Thus, one may be concerned that PARP inhibitors may also exert similar effects by activating the AKT/PKB, JNK or SIR2α pathways. Indeed, PARP inhibition-induced taxol resistance has already been reported to be independent of the intracellular NAD+ level but mediated by the AKT/PKB pathway [161]. Although these findings may be worrying, they also suggest that simultaneous pharmacologic inhibition of PARP-1 and AKT/PKB may be a more efficient and, more importantly, a safer strategy for targeting PARP-1 in future antineoplastic chemotherapies. This concept is supported by the recent report that the use of pharmacologic inhibitors of the repair kinase ATM apparently bypasses chemoresistance for PARP-inhibitors in BRCA-ablated cells [168].
Insight into the extranuclear effects of PARP-1 raised the possibility of the use of PARP inhibitors in a wider range of human pathologies as well. Inhibition of PARP-1 leads to tyrosine phosphorylation of the vascular endothelial growth factor receptor-2 (VEGFR2), activation of AKT/PKB and disruption of NF-κB-mediated inflammation adding new aspects to the therapeutic use of these compounds in chronic inflammatory conditions like vascular disease or Diabetes mellitus [143, 169].
The unexpectedly tangled cellular effects of PARP clearly call for further investigations. These efforts not only hold out the promise of enhancement of the therapeutic concept of PARP inhibition but a better understanding of the complex PARP-1 interactome as well.
Abbreviations
ABC transporter - ATP-binding cassette transporters; ABC transporter BCRP1 - ATP-binding cassette sub-family G member 2 Breast cancer resistance protein; AIF - Apoptosis Inducing Factor; APE1 - DNA-(apurinic or apyrimidinic site) lyase 1; BAD - BCL-2-associated agonist of cell death; BCL-2 - B Cell Lymphoma-2 ; BER - Base-excision repair; BRCA1/2 - Breast cancer type 1/2 susceptibility protein; BRCT - Breast Cancer-Associated 1 C-terminal domain; COX1 - Cytochrome C oxidase 1; COX2 - Cytochrome C oxidase 2; DBM - DNA-binding module of PARP-1; DDB2 - DNA damage-binding protein 2; DDR - DNA damage response; DRP1 - Dynamin-1-like protein; DUSP-1 - Dual specific protein phosphatase-1; EXO1 - MutSα-exonuclease 1; hFIS1 - Human mitochondrial fission 1 protein; HPF1 - Histone PARylation factor 1; HUWE1 - Huwe1 E3 ubiquitin ligase; JNK - c-Jun N-terminal kinase 1; LPS - Lipopolysaccharide; LPS - Lipopolysaccharides; mANT - Mitochondrial adenine nucleotide translocator; MAP kinase - Mitogen-activated protein kinase; MFN2 - Mitofusin-2; mPTP - Mitochondrial permeability transition pore; MYH1 - Adenine DNA glycosylase; NAD+ - Nicotinamide adenine dinucleotide; NER - Nucleotide excision repair; NF-κB - Nuclear factor NF-kappa-B; NHEJ - Non-homologous end joining repair; NLS - Nuclear localisation signal; OPA1 - Optic Atrophy Protein 1; PARG - Poly(ADP-ribose) glycohydrolase; PARP - Poly(ADP-ribose) polymerases; PARP - Poly-adenosine diphosphate-ribose polymerase; PCNA - Proliferating cell nuclear antigen; PI3K - Phosphatidylinositol 3-kinase; PSARL - Presenilins-associated rhomboid-like protein; RELA/p65 - V-Rel Reticuloendotheliosis Viral Oncogene Homolog, Nuclear factor NF-kappa-B p65 subunit; RIP - Receptor-interacting serine/threonine-protein kinase 1; RNF146 - E3 ubiquitin-protein ligase RNF146, Iduna; ROS - Reactive oxygen species; SIR2α - Sir2alpha; SREBP1c - Sterol regulatory element binding protein 1c; TFB1M - Dimethyladenosine transferase 1, mitochondrial ; TFB2M - Dimethyladenosine transferase 2, mitochondrial; TFR1 - Transferrin Receptor 1; TNFα - Tumor necrosis factor alpha; TRAF - Tumor necrosis factor type 2 receptor-associated protein 3; UNG1 - Uracil-DNA glycosylase; VDAC - Voltage-dependent anion channel; VEGFR2 - Vascular endothelial growth factor receptor-2; XRCC1 - X-ray repair cross complementing protein 1.
Authors’ contributions
All authors contributed equally to this work. A. M. C. and Z. F. perpared the manuscript, B.S. and L.S. performed critical revisions of the article.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
A. M. C. was funded by the Campus Hungary Fellowship program TAMOP-4.2.4B/2-11/1-2012-0001 and the EMBO Short Term Fellowship Program.
REFERENCES
1. Lodish HF. Molecular cell biology. New York: W.H. Freeman and Co. 2013.
2. Leichman L, Groshen S, O’Neil BH, Messersmith W, Berlin J, Chan E, Leichman CG, Cohen SJ, Cohen D, Lenz HJ, Gold P, Boman B, Fielding A, et al. Phase II Study of Olaparib (AZD-2281) After Standard Systemic Therapies for Disseminated Colorectal Cancer. Oncologist. 2016; 21:172–177.
3. Kummar S, Fleming GF, Oza AM, Sullivan DM, Gandara DR, Naughton M, Villalona-Calero MA, Morgan R, Szabo PM, Youn A, Chen A, Ji J, Allen D, et al. Randomized trial of oral cyclophosphamide and veliparib in high-grade serous ovarian, primary peritoneal, or fallopian tube cancers, or BRCA-mutant ovarian cancer. Clinical Cancer Research. 2015; 21:1574–82.
4. Kummar S, Wade JL, Oza AM, Sullivan D, Chen AP, Gandara DR, Ji J, Kinders RJ, Wang L, Allen D, Coyne GOS, Steinberg SM, Doroshow JH. Randomized phase II trial of cyclophosphamide and the oral poly (ADP-ribose) polymerase inhibitor veliparib in patients with recurrent, advanced triple-negative breast cancer. Investigational New Drugs. 2016; 34:355–363.
5. Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair. Nature. 1980; 283:593–596.
6. Chambon P, Weill JD, Mandel P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochemical and biophysical research communications. 1963; 11:39–43.
7. Otto H, Reche PA, Bazan F, Dittmar K, Haag F, Koch-Nolte F. In silico characterization of the family of PARP-like poly(ADP-ribosyl)transferases (pARTs). BMC genomics. 2005; 6:139.
8. Vyas S, Chesarone-Cataldo M, Todorova T, Huang YH, Chang P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat Commun. 2013; 4:2240.
9. Kleine H, Poreba E, Lesniewicz K, Hassa PO, Hottiger MO, Litchfield DW, Shilton BH, Lüscher B. Substrate-Assisted Catalysis by PARP10 Limits Its Activity to Mono-ADP-Ribosylation. Molecular cell. 32:57–69.
10. Aguiar RC, Takeyama K, He C, Kreinbrink K, Shipp MA. B-aggressive lymphoma family proteins have unique domains that modulate transcription and exhibit poly(ADP-ribose) polymerase activity. The Journal of biological chemistry. 2005; 280:33756–33765.
11. Shieh WM, Amé JC, Wilson MV, Wang ZQ, Koh DW, Jacobson MK, Jacobson EL. Poly(ADP-ribose) Polymerase Null Mouse Cells Synthesize ADP-ribose Polymers. Journal of Biological Chemistry. 1998; 273:30069–30072.
12. Menissier-de Murcia J, Molinete M, Gradwohl G, Simonin F, de Murcia G. Zinc-binding domain of poly(ADP-ribose)polymerase participates in the recognition of single strand breaks on DNA. Journal of molecular biology. 1989; 210:229–233.
13. Langelier MF, Riccio AA, Pascal JM. PARP-2 and PARP-3 are selectively activated by 5′ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Research. 2014; 42:7762–7775.
14. Kameshita I, Matsuda Z, Taniguchi T, Shizuta Y. Poly (ADP-Ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. The Journal of biological chemistry. 1984; 259:4770–4776.
15. Decker P, Isenberg D, Muller S. Inhibition of Caspase-3-mediated Poly(ADP-ribose) Polymerase (PARP) Apoptotic Cleavage by Human PARP Autoantibodies and Effect on Cells Undergoing Apoptosis. Journal of Biological Chemistry. 2000; 275:9043–9046.
16. Tao Z, Gao P, Hoffman DW, Liu HW. Domain C of human poly(ADP-ribose) polymerase-1 is important for enzyme activity and contains a novel zinc-ribbon motif. Biochemistry. 2008; 47:5804–5813.
17. Desmarais Y, Menard L, Lagueux J, Poirier GG. Enzymological properties of poly(ADP-ribose)polymerase: characterization of automodification sites and NADase activity. Biochimica et biophysica acta. 1991; 1078:179–186.
18. Altmeyer M, Messner S, Hassa PO, Fey M, Hottiger MO. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009; 37:3723–3738.
19. Masson M, Niedergang C, Schreiber V, Muller S, Menissier-de Murcia J, de Murcia G. XRCC1 Is Specifically Associated with Poly(ADP-Ribose) Polymerase and Negatively Regulates Its Activity following DNA Damage. Molecular and cellular biology. 1998; 18:3563–3571.
20. Langelier MF, Planck JL, Roy S, Pascal JM. Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: structural and functional insights into DNA-dependent PARP-1 activity. The Journal of biological chemistry. 2011; 286:10690–10701.
21. Wacker DA, Ruhl DD, Balagamwala EH, Hope KM, Zhang T, Kraus WL. The DNA Binding and Catalytic Domains of Poly(ADP-Ribose) Polymerase 1 Cooperate in the Regulation of Chromatin Structure and Transcription. Molecular and cellular biology. 2007; 27:7475–7485.
22. Sastry SS, Kun E. The interaction of adenosine diphosphoribosyl transferase (ADPRT) with a cruciform DNA. Biochemical and biophysical research communications. 1990; 167:842–847.
23. Gradwohl G, Mazen A, de Murcia G. Poly(ADP-ribose) polymerase forms loops with DNA. Biochemical and biophysical research communications. 1987; 148:913–919.
24. Lonskaya I, Potaman VN, Shlyakhtenko LS, Oussatcheva EA, Lyubchenko YL, Soldatenkov VA. Regulation of poly(ADP-ribose) polymerase-1 by DNA structure-specific binding. The Journal of biological chemistry. 2005; 280:17076–17083.
25. Tapodi A, Debreceni B, Hanto K, Bognar Z, Wittmann I, Gallyas F Jr, Varbiro G, Sumegi B. Pivotal role of Akt activation in mitochondrial protection and cell survival by poly(ADP-ribose)polymerase-1 inhibition in oxidative stress. The Journal of biological chemistry. 2005; 280:35767–35775.
26. Maffi SK, Rathinam ML, Cherian PP, Pate W, Hamby-Mason R, Schenker S, Henderson GI. Glutathione content as a potential mediator of the vulnerability of cultured fetal cortical neurons to ethanol-induced apoptosis. Journal of neuroscience research. 2008; 86:1064–1076.
27. Zhou J, Ng S, Huang Q, Wu YT, Li Z, Yao SQ, Shen HM. AMPK mediates a pro-survival autophagy downstream of PARP-1 activation in response to DNA alkylating agents. FEBS Lett. 2013; 587:170–177.
28. Duan Y, Gross RA, Sheu SS. Ca(2+)-dependent generation of mitochondrial reactive oxygen species serves as a signal for poly(ADP-ribose) polymerase-1 activation during glutamate excitotoxicity. The Journal of Physiology. 2007; 585:741–758.
29. Liaudet L, Pacher P, Mabley JG, Virag L, Soriano FG, Hasko G, Szabo C. Activation of poly(ADP-Ribose) polymerase-1 is a central mechanism of lipopolysaccharide-induced acute lung inflammation. American journal of respiratory and critical care medicine. 2002; 165:372–377.
30. Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, Brownlee M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. The Journal of clinical investigation. 2003; 112:1049–1057.
31. Chiu HJ, Fischman DA, Hammerling U. Vitamin A depletion causes oxidative stress, mitochondrial dysfunction, and PARP-1-dependent energy deprivation. FASEB journal. 2008; 22:3878–3887.
32. Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, Nielsen ML. Proteome-wide Identification of Poly(ADP-Ribosyl)ation Targets in Different Genotoxic Stress Responses. Molecular cell. 2013; 52:272–285.
33. Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992; 356:356–358.
34. Gagne JP, Isabelle M, Lo KS, Bourassa S, Hendzel MJ, Dawson VL, Dawson TM, Poirier GG. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. 2008; 36:6959–6976.
35. Satoh MS, Poirier GG, Lindahl T. Dual function for poly(ADP-ribose) synthesis in response to DNA strand breakage. Biochemistry. 1994; 33:7099–7106.
36. Pleschke JM, Kleczkowska HE, Strohm M, Althaus FR. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. The Journal of biological chemistry. 2000; 275:40974–40980.
37. Dantzer F, de la Rubia G, Ménissier-de Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Base Excision Repair Is Impaired in Mammalian Cells Lacking Poly(ADP-ribose) Polymerase-1. Biochemistry. 2000; 39:7559–7569.
38. El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003; 31:5526–5533.
39. Vodenicharov MD, Sallmann FR, Satoh MS, Poirier GG. Base excision repair is efficient in cells lacking poly(ADP-ribose) polymerase 1. Nucleic Acids Research. 2000; 28:3887–3896.
40. Allinson SL, Dianova, II, Dianov GL. Poly(ADP-ribose) polymerase in base excision repair: always engaged, but not essential for DNA damage processing. Acta biochimica Polonica. 2003; 50:169–179.
41. Ström CE, Johansson F, Uhlén M, Szigyarto CAK, Erixon K, Helleday T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011; 39:3166–3175.
42. Campalans A, Kortulewski T, Amouroux R, Menoni H, Vermeulen W, Radicella JP. Distinct spatiotemporal patterns and PARP dependence of XRCC1 recruitment to single-strand break and base excision repair. Nucleic Acids Research. 2013; 41:3115–29.
43. Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, Hensbergen P, Deelder A, de Groot A, Matsumoto S, Sugasawa K, Thoma N, Vermeulen W, et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. The Journal of cell biology. 2012; 199:235–249.
44. Liu Y, Kadyrov FA, Modrich P. PARP-1 enhances the mismatch-dependence of 5′-directed excision in human mismatch repair in vitro. DNA repair. 2011; 10:1145–1153.
45. Ruscetti T, Lehnert BE, Halbrook J, Le Trong H, Hoekstra MF, Chen DJ, Peterson SR. Stimulation of the DNA-dependent Protein Kinase by Poly(ADP-Ribose) Polymerase. Journal of Biological Chemistry. 1998; 273:14461–14467.
46. Gibbs-Seymour I, Fontana P, Rack JG, Ahel I. HPF1/C4orf27 Is a PARP-1-Interacting Protein that Regulates PARP-1 ADP-Ribosylation Activity. Molecular cell. 2016; 62:432–442.
47. Kang HC, Lee YI, Shin JH, Andrabi SA, Chi Z, Gagne JP, Lee Y, Ko HS, Lee BD, Poirier GG, Dawson VL, Dawson TM. Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:14103–14108.
48. Lin W, Amé JC, Aboul-Ela N, Jacobson EL, Jacobson MK. Isolation and Characterization of the cDNA Encoding Bovine Poly(ADP-ribose) Glycohydrolase. Journal of Biological Chemistry. 1997; 272:11895–11901.
49. Oka S, Kato J, Moss J. Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. The Journal of biological chemistry. 2006; 281:705–713.
50. de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, Walztinger C, Chambon P, de Murcia G. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proceedings of the National Academy of Sciences. 1997; 94:7303–7307.
51. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913–917.
52. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434:917–921.
53. Thomas HD, Calabrese CR, Batey MA, Canan S, Hostomsky Z, Kyle S, Maegley KA, Newell DR, Skalitzky D, Wang LZ, Webber SE, Curtin NJ. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Molecular cancer therapeutics. 2007; 6:945–956.
54. Bernges F, Zeller WJ. Combination effects of poly(ADP-ribose) polymerase inhibitors and DNA-damaging agents in ovarian tumor cell lines—with special reference to cisplatin. Journal of cancer research and clinical oncology. 1996; 122:665–670.
55. Guggenheim ER, Ondrus AE, Movassaghi M, Lippard SJ. Poly(ADP-ribose) polymerase-1 activity facilitates the dissociation of nuclear proteins from platinum-modified DNA. Bioorganic & medicinal chemistry. 2008; 16:10121–10128.
56. Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer research. 2012; 72:5588–5599.
57. Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, Durkacz BW, Hostomsky Z, Kumpf RA, Kyle S, Li J, Maegley K, Newell DR, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. Journal of the National Cancer Institute. 2004; 96:56–67.
58. Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD, Cox BF, DeWeese TL, Dillehay LE, Ferguson DC, Ghoreishi-Haack NS, Grimm DR, et al. ABT-888, an Orally Active Poly(ADP-Ribose) Polymerase Inhibitor that Potentiates DNA-Damaging Agents in Preclinical Tumor Models. Clinical Cancer Research. 2007; 13:2728–2737.
59. Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, Boulter R, Cranston A, O’Connor MJ, Martin NM, Borst P, Jonkers J. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proceedings of the National Academy of Sciences. 2008; 105:17079–17084.
60. Ali M, Telfer BA, McCrudden C, O’Rourke M, Thomas HD, Kamjoo M, Kyle S, Robson T, Shaw C, Hirst DG, Curtin NJ, Williams KJ. Vasoactivity of AG014699, a Clinically Active Small Molecule Inhibitor of Poly(ADP-ribose) Polymerase: a Contributory Factor to Chemopotentiation In vivo? Clinical Cancer Research. 2009; 15:6106–6112.
61. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. The New England journal of medicine. 2009; 361:123–134.
62. Khan OA, Gore M, Lorigan P, Stone J, Greystoke A, Burke W, Carmichael J, Watson AJ, McGown G, Thorncroft M, Margison GP, Califano R, Larkin J, et al. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. British journal of cancer. 2011; 104:750–755.
63. Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, Yerushalmi R, Macpherson E, Carmichael J, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. The Lancet Oncology. 2011; 12:852–861.
64. Mullard A. European regulators approve first PARP inhibitor. Nat Rev Drug Discov. 2014; 13:877–877.
65. Sukhanova MV, Abrakhi S, Joshi V, Pastre D, Kutuzov MM, Anarbaev RO, Curmi PA, Hamon L, Lavrik OI. Single molecule detection of PARP1 and PARP2 interaction with DNA strand breaks and their poly(ADP-ribosyl)ation using high-resolution AFM imaging. Nucleic Acids Research. 2016; 44:e60.
66. Tulin A, Spradling A. Chromatin Loosening by Poly(ADP)-Ribose Polymerase (PARP) at Drosophila Puff Loci. Science. 2003; 299:560–562.
67. Pinnola A, Naumova N, Shah M, Tulin AV. Nucleosomal core histones mediate dynamic regulation of poly(ADP-ribose) polymerase 1 protein binding to chromatin and induction of its enzymatic activity. The Journal of biological chemistry. 2007; 282:32511–32519.
68. Lapucci A, Pittelli M, Rapizzi E, Felici R, Moroni F, Chiarugi A. Poly(ADP-ribose) polymerase-1 is a nuclear epigenetic regulator of mitochondrial DNA repair and transcription. Molecular pharmacology. 2011; 79:932–940.
69. Huang D, Wang Y, Yang C, Liao Y, Huang K. Angiotensin II promotes poly(ADP-ribosyl)ation of c-Jun/c-Fos in cardiac fibroblasts. Journal of molecular and cellular cardiology. 2009; 46:25–32.
70. Berger NA, Sims JL, Catino DM, Berger SJ. Poly(ADP-ribose) polymerase mediates the suicide response to massive DNA damage: studies in normal and DNA-repair defective cells. Princess Takamatsu symposia. 1983; 13:219–226.
71. Cipriani G, Rapizzi E, Vannacci A, Rizzuto R, Moroni F, Chiarugi A. Nuclear poly(ADP-ribose) polymerase-1 rapidly triggers mitochondrial dysfunction. The Journal of biological chemistry. 2005; 280:17227–17234.
72. Formentini L, Macchiarulo A, Cipriani G, Camaioni E, Rapizzi E, Pellicciari R, Moroni F, Chiarugi A. Poly(ADP-ribose) catabolism triggers AMP-dependent mitochondrial energy failure. The Journal of biological chemistry. 2009; 284:17668–17676.
73. Buonvicino D, Formentini L, Cipriani G, Chiarugi A. Glucose Deprivation Converts Poly(ADP-ribose) Polymerase-1 Hyperactivation into a Transient Energy-producing Process. The Journal of biological chemistry. 2013; 288:36530–36537.
74. Fouquerel E, Goellner EM, Yu Z, Gagné JP, de Moura MB, Feinstein T, Wheeler D, Redpath P, Li J, Romero G, Migaud M, Van Houten B, Poirier GG, et al. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD(+) depletion. Cell reports. 2014; 8:1819–1831.
75. Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, Gagné JP, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111:10209–10214.
76. Miramar MD, Costantini P, Ravagnan L, Saraiva LM, Haouzi D, Brothers G, Penninger JM, Peleato ML, Kroemer G, Susin SA. NADH Oxidase Activity of Mitochondrial Apoptosis-inducing Factor. Journal of Biological Chemistry. 2001; 276:16391–16398.
77. Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103:18314–18319.
78. Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. The Journal of neuroscience. 2007; 27:9278–9293.
79. Vosler PS, Sun D, Wang S, Gao Y, Kintner DB, Signore AP, Cao G, Chen J. Calcium dysregulation induces apoptosis-inducing factor release: cross-talk between PARP-1- and calpain-signaling pathways. Experimental neurology. 2009; 218:213–220.
80. Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999; 397:441–446.
81. Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001; 410:549–554.
82. Wang Y, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA, Poirier GG, Dawson VL, Dawson TM. Poly (ADP-ribose) (PAR) Binding to Apoptosis-Inducing Factor Is Critical For PAR Polymerase-1-Dependent Cell Death (Parthanatos). Sci Signal. 4:ra20.
83. Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD(+) depletion is necessary and sufficient for PARP-1 – mediated neuronal death. The Journal of neuroscience. 2010; 30:2967–2978.
84. Takeyama N, Matsuo N, Tanaka T. Oxidative damage to mitochondria is mediated by the Ca(2+)-dependent inner-membrane permeability transition. The Biochemical journal. 1993; 294:719–725.
85. Von Sonntag C. The chemical basis of radiation biology. London; Philadelphia, PA: Taylor & Francis; 1989.
86. Szabados E, Literati-Nagy P, Farkas B, Sumegi B. BGP-15, a nicotinic amidoxime derivate protecting heart from ischemia reperfusion injury through modulation of poly(ADP-ribose) polymerase. Biochemical pharmacology. 2000; 59:937–945.
87. Zingarelli B, Salzman AL, Szabo C. Genetic disruption of poly (ADP-ribose) synthetase inhibits the expression of P-selectin and intercellular adhesion molecule-1 in myocardial ischemia/reperfusion injury. Circulation research. 1998; 83:85–94.
88. Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997; 3:1089–1095.
89. Szabo C, Lim LH, Cuzzocrea S, Getting SJ, Zingarelli B, Flower RJ, Salzman AL, Perretti M. Inhibition of poly (ADP-ribose) synthetase attenuates neutrophil recruitment and exerts antiinflammatory effects. The Journal of experimental medicine. 1997; 186:1041–1049.
90. Radons J, Heller B, Burkle A, Hartmann B, Rodriguez ML, Kroncke KD, Burkart V, Kolb H. Nitric oxide toxicity in islet cells involves poly(ADP-ribose) polymerase activation and concomitant NAD+ depletion. Biochemical and biophysical research communications. 1994; 199:1270–1277.
91. Masutani M, Suzuki H, Kamada N, Watanabe M, Ueda O, Nozaki T, Jishage K, Watanabe T, Sugimoto T, Nakagama H, Ochiya T, Sugimura T. Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proceedings of the National Academy of Sciences of the United States of America. 1999; 96:2301–2304.
92. LaPlaca MC, Raghupathi R, Verma A, Pieper AA, Saatman KE, Snyder SH, McIntosh TK. Temporal Patterns of Poly(ADP-Ribose) Polymerase Activation in the Cortex Following Experimental Brain Injury in the Rat. Journal of Neurochemistry. 1999; 73:205–213.
93. Halmosi R, Berente Z, Osz E, Toth K, Literati-Nagy P, Sumegi B. Effect of poly(ADP-ribose) polymerase inhibitors on the ischemia-reperfusion-induced oxidative cell damage and mitochondrial metabolism in Langendorff heart perfusion system. Molecular pharmacology. 2001; 59:1497–1505.
94. Veres B, Gallyas F Jr, Varbiro G, Berente Z, Osz E, Szekeres G, Szabo C, Sumegi B. Decrease of the inflammatory response and induction of the Akt/protein kinase B pathway by poly-(ADP-ribose) polymerase 1 inhibitor in endotoxin-induced septic shock. Biochemical pharmacology. 2003; 65:1373–1382.
95. Bijur GN, Jope RS. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J Neurochem. 2003; 87:1427–1435.
96. Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995; 81:727–736.
97. Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. The Journal of biological chemistry. 1998; 273:19929–19932.
98. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995; 378:785–789.
99. Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP Jr, Roth RA. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. The Journal of biological chemistry. 1999; 274:20281–20286.
100. Zhuo B, Li Y, Li Z, Qin H, Sun Q, Zhang F, Shen Y, Shi Y, Wang R. PI3K/Akt signaling mediated Hexokinase-2 expression inhibits cell apoptosis and promotes tumor growth in pediatric osteosarcoma. Biochemical and biophysical research communications. 2015; 464:401–406.
101. Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. The Journal of biological chemistry. 1997; 272:17269–17275.
102. Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, Terracciano L, Heim MH, Ruegg MA, Hall MN. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell metabolism. 2012; 15:725–738.
103. Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, Brugge JS. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. The Journal of cell biology. 2005; 171:1023–1034.
104. Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer research. 2000; 60:1541–1545.
105. Yang JY, Yeh HY, Lin K, Wang PH. Insulin stimulates Akt translocation to mitochondria: implications on dysregulation of mitochondrial oxidative phosphorylation in diabetic myocardium. Journal of molecular and cellular cardiology. 2009; 46:919–926.
106. Su CC, Yang JY, Leu HB, Chen Y, Wang PH. Mitochondrial Akt-regulated mitochondrial apoptosis signaling in cardiac muscle cells. American Journal of Physiology - Heart and Circulatory Physiology. 2012; 302:H716–723.
107. Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Molecular and cellular biology. 1999; 19:5800–5810.
108. Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes & development. 2001; 15:1406–1418.
109. Plas DR, Talapatra S, Edinger AL, Rathmell JC, Thompson CB. Akt and Bcl-xL promote growth factor-independent survival through distinct effects on mitochondrial physiology. The Journal of biological chemistry. 2001; 276:12041–12048.
110. Linden M, Gellerfors P, Nelson BD. Pore protein and the hexokinase-binding protein from the outer membrane of rat liver mitochondria are identical. FEBS letters. 1982; 141:189–192.
111. Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H. Glucose Phosphorylation and Mitochondrial Binding Are Required for the Protective Effects of Hexokinases I and II. Molecular and cellular biology. 2008; 28:1007–1017.
112. Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell death and differentiation. 2008; 15:521–529.
113. Geistrikh I, Visochek L, Klein R, Miller L, Mittelman L, Shainberg A, Cohen-Armon M. Ca2+-induced PARP-1 activation and ANF expression are coupled events in cardiomyocytes. Biochemical Journal. 2011; 438:337–347.
114. Homburg S, Visochek L, Moran N, Dantzer F, Priel E, Asculai E, Schwartz D, Rotter V, Dekel N, Cohen-Armon M. A fast signal-induced activation of Poly(ADP-ribose) polymerase: a novel downstream target of phospholipase c. The Journal of cell biology. 2000; 150:293–307.
115. Kong D, Xu L, Yu Y, Zhu W, Andrews DW, Yoon Y, Kuo TH. Regulation of Ca2+-induced permeability transition by Bcl-2 is antagonized by Drpl and hFis1. Molecular and cellular biochemistry. 2005; 272:187–199.
116. Neuspiel M, Zunino R, Gangaraju S, Rippstein P, McBride H. Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. The Journal of biological chemistry. 2005; 280:25060–25070.
117. Lee HJ, Jeong CH, Cha JH, Kim KW. PKC-delta inhibitors sustain self-renewal of mouse embryonic stem cells under hypoxia in vitro. Exp Mol Med. 2010; 42:294–301.
118. Ong SB, Hall AR, Dongworth RK, Kalkhoran S, Pyakurel A, Scorrano L, Hausenloy DJ. Akt protects the heart against ischaemia-reperfusion injury by modulating mitochondrial morphology. Thrombosis and haemostasis. 2015; 113:513–521.
119. Varanita T, Soriano ME, Romanello V, Zaglia T, Quintana-Cabrera R, Semenzato M, Menabò R, Costa V, Civiletto G, Pesce P, Viscomi C, Zeviani M, Di Lisa F, et al. The Opa1-Dependent Mitochondrial Cristae Remodeling Pathway Controls Atrophic, Apoptotic, and Ischemic Tissue Damage. Cell metabolism. 2015; 21:834–844.
120. Baker MJ, Lampe PA, Stojanovski D, Korwitz A, Anand R, Tatsuta T, Langer T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. The EMBO journal. 2014; 33:578–593.
121. Parra V, Verdejo HE, Iglewski M, Del Campo A, Troncoso R, Jones D, Zhu Y, Kuzmicic J, Pennanen C, Lopez-Crisosto C, Jana F, Ferreira J, Noguera E, et al. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFkappaB-Opa-1 signaling pathway. Diabetes. 2014; 63:75–88.
122. Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovascular research. 2009; 84:91–99.
123. Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR, Rabasa-Lhoret R, Wallberg-Henriksson H, Laville M, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. The Journal of biological chemistry. 2003; 278:17190–17197.
124. Walder K, Kerr-Bayles L, Civitarese A, Jowett J, Curran J, Elliott K, Trevaskis J, Bishara N, Zimmet P, Mandarino L, Ravussin E, Blangero J, Kissebah A, et al. The mitochondrial rhomboid protease PSARL is a new candidate gene for type 2 diabetes. Diabetologia. 2005; 48:459–468.
125. Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia. 2010; 53:1783–1794.
126. Caino MC, Ghosh JC, Chae YC, Vaira V, Rivadeneira DB, Faversani A, Rampini P, Kossenkov AV, Aird KM, Zhang R, Webster MR, Weeraratna AT, Bosari S, et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proceedings of the National Academy of Sciences of the United States of America. 2015; 112:8638–8643.
127. Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. Journal of cell science. 1993; 104:917–927.
128. Xu Y, Huang S, Liu ZG, Han J. Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. The Journal of biological chemistry. 2006; 281:8788–8795.
129. Mester L, Szabo A, Atlasz T, Szabadfi K, Reglodi D, Kiss P, Racz B, Tamas A, Gallyas F Jr, Sumegi B, Hocsak E, Gabriel R, Kovacs K. Protection against chronic hypoperfusion-induced retinal neurodegeneration by PARP inhibition via activation of PI-3-kinase Akt pathway and suppression of JNK and p38 MAP kinases. Neurotoxicity research. 2009; 16:68–76.
130. Radnai B, Antus C, Racz B, Engelmann P, Priber JK, Tucsek Z, Veres B, Turi Z, Lorand T, Sumegi B, Gallyas F. Protective effect of the poly(ADP-ribose) polymerase inhibitor PJ34 on mitochondrial depolarization-mediated cell death in hepatocellular carcinoma cells involves attenuation of c-Jun N-terminal kinase-2 and protein kinase B/Akt activation. Molecular Cancer. 2012; 11:34.
131. Racz B, Hanto K, Tapodi A, Solti I, Kalman N, Jakus P, Kovacs K, Debreceni B, Gallyas F Jr, Sumegi B. Regulation of MKP-1 expression and MAPK activation by PARP-1 in oxidative stress: a new mechanism for the cytoplasmic effect of PARP-1 activation. Free radical biology & medicine. 2010; 49:1978–1988.
132. Schroeter H, Boyd CS, Ahmed R, Spencer JP, Duncan RF, Rice-Evans C, Cadenas E. c-Jun N-terminal kinase (JNK)-mediated modulation of brain mitochondria function: new target proteins for JNK signalling in mitochondrion-dependent apoptosis. The Biochemical journal. 2003; 372:359–369.
133. Donovan N, Becker EB, Konishi Y, Bonni A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. The Journal of biological chemistry. 2002; 277:40944–40949.
134. Leboucher GP, Tsai YC, Yang M, Shaw KC, Zhou M, Veenstra TD, Glickman MH, Weissman AM. Stress-Induced Phosphorylation and Proteasomal Degradation of Mitofusin 2 Facilitates Mitochondrial Fragmentation and Apoptosis. Molecular cell. 2012; 47:547–557.
135. Senyilmaz D. Regulation of mitochondrial morphology and function by Stearoylation of TfR1. Nature. 2015; 525:124–128.
136. Brady NR, Hamacher-Brady A, Gottlieb RA. Proapoptotic BCL-2 family members and mitochondrial dysfunction during ischemia/reperfusion injury, a study employing cardiac HL-1 cells and GFP biosensors. Biochimica et biophysica acta. 2006; 1757:667–678.
137. Song ZF, Ji XP, Li XX, Wang SJ, Wang SH, Zhang Y. Inhibition of the activity of poly (ADP-ribose) polymerase reduces heart ischaemia/reperfusion injury via suppressing JNK-mediated AIF translocation. Journal of cellular and molecular medicine. 2008; 12:1220–1228.
138. Pirinen E, Canto C, Jo YS, Morato L, Zhang H, Menzies K, Williams EG, Mouchiroud L, Moullan N, Hagberg C, Li W, Timmers S, Imhof R, et al. Pharmacological Inhibition of Poly(ADP-Ribose) Polymerases Improves Fitness and Mitochondrial Function in Skeletal Muscle. Cell metabolism. 2014; 19:1034–1041.
139. Deres L, Bartha E, Palfi A, Eros K, Riba A, Lantos J, Kalai T, Hideg K, Sumegi B, Gallyas F, Toth K, Halmosi R. PARP-inhibitor treatment prevents hypertension induced cardiac remodeling by favorable modulation of heat shock proteins, Akt-1/GSK-3beta and several PKC isoforms. PloS one. 2014; 9:e102148.
140. Veto S. Inhibiting poly(ADP-ribose) polymerase: a potential therapy against oligodendrocyte death. Brain. 2010; 133:822–834.
141. Soriano FG, Liaudet L, Szabó É, Virág L, Mabley JG, Pacher P, Szabó C. Resistance to Acute Septic Peritonitis in Poly(ADP-ribose) Polymerase-1-Deficient Mice. Shock. 2002; 17:286–292.
142. Oliver FJ, Ménissier-de Murcia J, Nacci C, Decker P, Andriantsitohaina R, Muller S, de la Rubia G, Stoclet JC, de Murcia G. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. The EMBO journal. 1999; 18:4446–4454.
143. Hassa PO, Hottiger MO. A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biological chemistry. 1999; 380:953–959.
144. Oliver FJ, Menissier-de Murcia J, de Murcia G. Poly(ADP-ribose) polymerase in the cellular response to DNA damage, apoptosis, and disease. American Journal of Human Genetics. 1999; 64:1282–1288.
145. Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocrine reviews. 2010; 31:194–223.
146. Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. The Journal of biological chemistry. 2005; 280:43121–43130.
147. Mendoza-Alvarez H, Alvarez-Gonzalez R. Poly(ADP-ribose) polymerase is a catalytic dimer and the automodification reaction is intermolecular. Journal of Biological Chemistry. 1993; 268:22575–22580.
148. Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell metabolism. 2011; 13:461–468.
149. Kolthur-Seetharam U, Dantzer F, McBurney MW, de Murcia G, Sassone-Corsi P. Control of AIF-mediated cell death by the functional interplay of SIRT1 and PARP-1 in response to DNA damage. Cell Cycle. 2006; 5:873–877.
150. Bai P, Canto C, Brunyánszki A, Huber A, Szántó M, Cen Y, Yamamoto H, Houten Sander M, Kiss B, Oudart H, Gergely P, Menissier-de Murcia J, Schreiber V, et al. PARP-2 Regulates SIRT1 Expression and Whole-Body Energy Expenditure. Cell metabolism. 13:450–460.
151. Kiernan R, Bres V, Ng RW, Coudart MP, El Messaoudi S, Sardet C, Jin DY, Emiliani S, Benkirane M. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. The Journal of biological chemistry. 2003; 278:2758–2766.
152. Garcia S, Bodano A, Pablos JL, Gomez-Reino JJ, Conde C. Poly(ADP-ribose) polymerase inhibition reduces tumor necrosis factor-induced inflammatory response in rheumatoid synovial fibroblasts. Annals of the rheumatic diseases. 2008; 67:631–637.
153. Jijon HB, Churchill T, Malfair D, Wessler A, Jewell LD, Parsons HG, Madsen KL. Inhibition of poly(ADP-ribose) polymerase attenuates inflammation in a model of chronic colitis. American journal of physiology Gastrointestinal and liver physiology. 2000; 279:G641–651.
154. Altmeyer M, Barthel M, Eberhard M, Rehrauer H, Hardt WD, Hottiger MO. Absence of Poly(ADP-Ribose) Polymerase 1 Delays the Onset of Salmonella enterica Serovar Typhimurium-Induced Gut Inflammation. Infection and Immunity. 2010; 78:3420–3431.
155. Brunyánszki A, Hegedűs C, Szántó M, Erdélyi K, Kovács K, Schreiber V, Gergely S, Kiss B, Szabó É, Virág L, Bai P. Genetic Ablation of PARP-1 Protects Against Oxazolone-Induced Contact Hypersensitivity by Modulating Oxidative Stress. Journal of Investigative Dermatology. 2010; 130:2629–2637.
156. Szabo C, Biser A, Benko R, Bottinger E, Susztak K. Poly(ADP-ribose) polymerase inhibitors ameliorate nephropathy of type 2 diabetic Leprdb/db mice. Diabetes. 2006; 55:3004–3012.
157. Bardos G, Moricz K, Jaszlits L, Rabloczky G, Tory K, Racz I, Bernath S, Sumegi B, Farkas B, Literati-Nagy B, Literati-Nagy P. BGP-15, a hydroximic acid derivative, protects against cisplatin- or taxol-induced peripheral neuropathy in rats. Toxicology and applied pharmacology. 2003; 190:9–16.
158. Miwa M, Sugimura T, Inui N, Takayama S. Poly(adenosine diphosphate ribose) synthesis during the cell cycle of transformed hamster lung cells. Cancer research. 1973; 33:1306–1309.
159. Lodhi N, Kossenkov AV, Tulin AV. Bookmarking promoters in mitotic chromatin: poly(ADP-ribose)polymerase-1 as an epigenetic mark. Nucleic Acids Research. 2014; 42:7028–38.
160. Ye XQ, Li Q, Wang GH, Sun FF, Huang GJ, Bian XW, Yu SC, Qian GS. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. International journal of cancer. 2011; 129:820–831.
161. Szanto A, Hellebrand EE, Bognar Z, Tucsek Z, Szabo A, Gallyas F Jr, Sumegi B, Varbiro G. PARP-1 inhibition-induced activation of PI-3-kinase-Akt pathway promotes resistance to taxol. Biochemical pharmacology. 2009; 77:1348–1357.
162. Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, Holland EC. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell stem cell. 2009; 4:226–235.
163. Mogi M, Yang J, Lambert JF, Colvin GA, Shiojima I, Skurk C, Summer R, Fine A, Quesenberry PJ, Walsh K. Akt signaling regulates side population cell phenotype via Bcrp1 translocation. The Journal of biological chemistry. 2003; 278:39068–39075.
164. Takada T, Suzuki H, Gotoh Y, Sugiyama Y. Regulation of the cell surface expression of human BCRP/ABCG2 by the phosphorylation state of Akt in polarized cells. Drug metabolism and disposition. 2005; 33:905–909.
165. Wang H, Fan L, Wei J, Weng Y, Zhou L, Shi Y, Zhou W, Ma D, Wang C. Akt mediates metastasis-associated gene 1 (MTA1) regulating the expression of E-cadherin and promoting the invasiveness of prostate cancer cells. PloS one. 2012; 7:e46888.
166. Kang CD, Ahn BK, Jeong CS, Kim KW, Lee HJ, Yoo SD, Chung BS, Kim SH. Downregulation of JNK/SAPK activity is associated with the cross-resistance to P-glycoprotein-unrelated drugs in multidrug-resistant FM3A/M cells overexpressing P-glycoprotein. Experimental cell research. 2000; 256:300–307.
167. Hao C, Zhu PX, Yang X, Han ZP, Jiang JH, Zong C, Zhang XG, Liu WT, Zhao QD, Fan TT, Zhang L, Wei LX. Overexpression of SIRT1 promotes metastasis through epithelial-mesenchymal transition in hepatocellular carcinoma. BMC Cancer. 2014; 14.
168. Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, Todorova Kwan T, Morris R, Lauffer S, Nussenzweig A, Ramaswamy S, Benes CH, Haber DA, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes & development. 2017; 31:318–332.
169. Mathews MT, Berk BC. PARP-1 inhibition prevents oxidative and nitrosative stress-induced endothelial cell death via transactivation of the VEGF receptor 2. Arteriosclerosis, thrombosis, and vascular biology. 2008; 28:711–717.
170. Noren Hooten N, Kompaniez K, Barnes J, Lohani A, Evans MK. Poly(ADP-ribose) Polymerase 1 (PARP-1) Binds to 8-Oxoguanine-DNA Glycosylase (OGG1). The Journal of biological chemistry. 2011; 286:44679–44690.
171. Martin-Oliva D, Aguilar-Quesada R, O’Valle F, Muñoz-Gámez JA, Martínez-Romero R, García del Moral R, Ruiz de Almodóvar JM, Villuendas R, Piris MA, Oliver FJ. Inhibition of Poly(ADP-Ribose) Polymerase Modulates Tumor-Related Gene Expression, Including Hypoxia-Inducible Factor-1 Activation, during Skin Carcinogenesis. Cancer research. 2006; 66:5744–5756.
172. Ciccarone F, Valentini E, Zampieri M, Caiafa P. 5mC-hydroxylase activity is influenced by the PARylation of TET1 enzyme. Oncotarget. 2015; 6:24333–24347. doi: 10.18632/oncotarget.4476.
173. Malanga M, Pleschke JM, Kleczkowska HE, Althaus FR. Poly(ADP-ribose) Binds to Specific Domains of p53 and Alters Its DNA Binding Functions. Journal of Biological Chemistry. 1998; 273:11839–11843.
174. Kannan P, Yu Y, Wankhade S, Tainsky MA. PolyADP-ribose polymerase is a coactivator for AP-2-mediated transcriptional activation. Nucleic Acids Research. 1999; 27:866–874.
175. Cervellera MN, Sala A. Poly(ADP-ribose) Polymerase Is a B-MYB Coactivator. Journal of Biological Chemistry. 2000; 275:10692–10696.
176. Nie J, Sakamoto S, Song D, Qu Z, Ota K, Taniguchi T. Interaction of Oct-1 and automodification domain of poly(ADP-ribose) synthetase. FEBS letters. 1998; 424:27–32.
177. Butler AJ, Ordahl CP. Poly(ADP-ribose) polymerase binds with transcription enhancer factor 1 to MCAT1 elements to regulate muscle-specific transcription. Molecular and cellular biology. 1999; 19:296–306.
178. Oei SL, Shi Y. Transcription Factor Yin Yang 1 Stimulates Poly(ADP-Ribosyl)ation and DNA Repair. Biochemical and biophysical research communications. 2001; 284:450–454.
179. Martin KA, Cesaroni M, Denny MF, Lupey LN, Tempera I. Global Transcriptome Analysis Reveals That Poly(ADP-Ribose) Polymerase 1 Regulates Gene Expression through EZH2. Molecular and cellular biology. 2015; 35:3934–3944.
180. Zampieri M, Guastafierro T, Calabrese R, Ciccarone F, Bacalini MG, Reale A, Perilli M, Passananti C, Caiafa P. ADP-ribose polymers localized on Ctcf-Parp1-Dnmt1 complex prevent methylation of Ctcf target sites. The Biochemical journal. 2012; 441:645–652.
181. Reale A, Matteis GD, Galleazzi G, Zampieri M, Caiafa P. Modulation of DNMT1 activity by ADP-ribose polymers. Oncogene. 2005; 24:13–19.