
INTRODUCTION
Lung cancer is the leading cause of cancer-related mortality worldwide, and more than 1.5 million deaths is related with it every year [1]. The majority of patients present with locally advanced or metastatic disease. Approximately 85% of lung cancers are classified as non–small cell lung cancer (NSCLC), and include lung adenocarcinoma, squamous cell carcinoma (SCC), and large cell carcinoma (LCC) histologic subtypes. Over the past decade, major advances in the understanding of lung cancer, especially NSCLC, have been achieved [2]. NSCLC is defined as a group of different diseases, and as an oncogenic driver, epidermal growth factor receptor (EGFR) has been identified. Blockade of EGFR with specific tyrosine kinase inhibitors (TKIs) can generate dramatic tumor responses in NSCLC [3, 4].
EGFR is one of the four members of the HER family receptors, which compose of EGFR/HER1/erbB1, HER2/erbB2, HER3/erbB3, and HER4/erbB4. There are 11 species in the HER family of growth factors which can be broadly divided into those that specifically bind with EGFR (EGF, TGF-α, Amphiregulin (AR)), those that binding with EGFR and HER4 (BTC, HB-EGF, Epidermal regulators), and those that binding with HER3 and HER4 (Neuregulin). Although HER2 has no corresponding ligand, it usually binds to a ligand similar to the one that activates it, readily forming a dimer with other members of the HER family. Additionally, EGFR is a receptor of tyrosine kinase (RTK). It is consisted of a C-terminus intracellular region that possesses the kinase activity, and an N-terminus extracellular ligand-binding site, a hydrophobic transmembrane domain [5]. The EGFR signaling network plays a significant role in the epithelial tissues maintenance and growth, active EGFR signaling is frequently observed in lung cancer, and EGFR level is related with advanced stage of disease and bad prognosis [6]. As known, HER family receptor related malformation from primary lung tumors, NSCLC brain metastases have some strikingly differences [7]. Therefore, in the development of new drugs for cancer treatment, EGFR and its signaling components can be used as targets, such as chimeric monoclonal antibodies (panitumumab and cetuximab) [8] and TKIs (gefitinib, erlotinib, and afatinib) [9–11]. However, to date, cancer heterogeneity and the drug resistance greatly limit the usefulness of anti-EGFR agents [12].
EGFR gene is an oncogene-driven gene, tyrosine kinases (TK) active [13]. Meanwhile, discovered in 2004, EGFR mutation is the first molecular alteration in lung cancer that is shown to confer sensitivity to specific targeted therapies, namely TKIs [14–17]. EGFR-TKIs can inhibit EGFR autophosphorylation activation and its downstream signaling pathways through competitive binding with the EGFR binding region, preventing the binding of ATP and EGFR receptors. The EGFR signaling pathway is summarized in Figure 1.
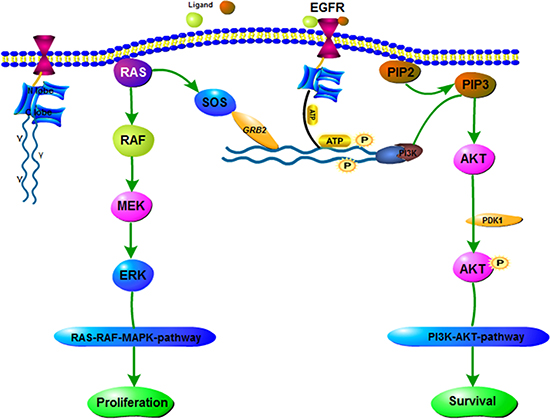
Figure 1: EGFR signaling pathway. EGFR is a receptor protein that spans the cell membrane. TKI consists of N lobe and C lobe. EGFR-TKI competes with ATP for inhibition of this site. If the growth factor (ligand) binds to the receptor, it forms an asymmetric dimer. A variety of proteins associated with the phosphorylation of tyrosine, the downstream protein is constantly activated, as shown in chart the RAS-RAF-MAPK pathway and PI3K-AKT pathway.
These molecular alterations could be influential in SCC and have important implications in lung cancer treatment [18]. Currently, none of the recurrent molecular alterations, which are commonly altered in lung squamous cell carcinoma, have proven to be as predictive for response to therapy as EGFR alterations in lung adenocarcinoma. Despite the establishment of tumor profiling for lung adenocarcinomas, its clinical benefits for other histologic subtypes of lung cancer, such as lung SCC and SCLC, remain unclear. Furthermore, a significant minority of patients with NSCLC have activating mutations in EGFR [19], patients typically develop resistance within 9 to 12 months. Moreover, anti-EGFR antibody therapy uses extensive in treatment cancer [20].
In summary, studies have been focused on the underpinning mechanisms the resistance for anti-EGFR agents, and EGFR mutations play a pivotal role in lung cancer.
EGFR AS A TARGET IN LUNG CANCER TREATMENT
Currently many studies have highlighted the relationship between EGFR and cancers. High expression of EGFR correlates with poor survival in cancer of the head and neck, as well as in cervical carcinoma and bladder [20–22]. In NSCLC, EGFR as a prognostic factor remains disputable. Some studies have verified that EGFR over-expression is predictive of a poor result in NSCLC [23–27], whereas others don’t found such situation [27–29].
In the progression of NSCLC, EGFR, regarded as the cancer driver gene [30, 31], demonstrates the effects of numerous oncogenic, including stimulating DNA synthesis, cell cycle, cell proliferation, cell metastasis and invasion [32, 33], and has been proposed as an attractive and promising target for anti-cancer treatment [34]. The expression of high level of EGFR, the pathway can also be up-regulated by coexpression of receptor ligands (such as transforming growth factor-α (TGF-α) or EGF). Moreover, the duration of the EGFR signaling pathways are stimulated by the positive feedback loop formed by EGFR, a ligand-releasing protease and the RAS-MAPK signaling pathway (Figure 1). As the signaling response can be prolonged in a cell that is efficient in recapturing the endogenous ligand, in spite of the levels of EGFR expressed may be low [35]. Furthermore, a research in breast cancer verified that co-expression of TGF-α and EGFR had a more remarkable effect on survival, comparing to the co-expression of HER-2 and EGFR [36]. In lung cancer, one study has reported that synchronous expression of EGFR and HER-2 is predictive of increased recurrence risk. Moreover, the success therapy with programmed death ligand 1 (PD-L1) blockade in lung cancer suggests that immune escape mechanisms can be conducive to lung tumor etiopathogenesis [37].
ACQUIRED RESISTANCE OF EGFR TKIS
In lung cancer, especially in NSCLC, a variety of EGFR mutations exist which are closely related to tumor development. The discovery of activating mutations in EGFR and the eventual approval of TKIs are the milestones in the history of NSCLC treatment.
First-generation EGFR TKIs
Gefitinib and Erlotinib have greatly improved the progression-free survival over standard chemotherapy for EGFR-positive NSCLC. Yet, due to the emergence of resistance, disease progression eventually occurs in almost all patients. Quinazoline has the core skeleton of gefitinib and erlotinib combined with EGFR reversibility. However, almost all patients who received gefitinib/erlotinib treatment eventually and inevitably acquired resistance, with a median progression-free survival (mPFS) of approximately 9 to 11 months [38]. The most common mechanism of acquired resistance is point mutation of exon 20 to T790M, with an incidence of approximately 50% to 60% [39] (Figure 2). The threonine (T) at site 790 of EGFR is located at the ATP bound pocket of the tail end, and mutations in the kinase receptor-binding region form a huge chain methionine(M) residue, resulting in steric hindrance, which affects EGFR TKIs from binding to the receptor. Other mechanisms of resistance include MET gene amplification [40], small cell transformation [41], epithelial mesenchymal transition (EMT) [42], and so on.
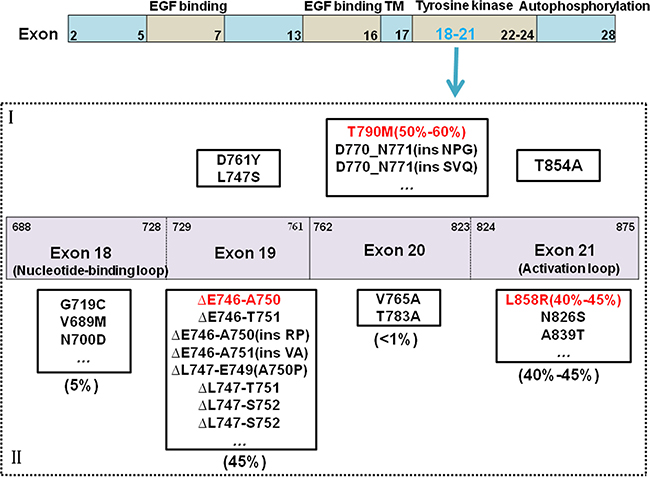
Figure 2: EGFR mutations and drug-resistant mechanism. As common mutant sites, the mutations of exon18-21 in EGFR are discovered. It’s including common mutations and rare mutations. Common mutations are involved deletion mutations in 45 percent of 19 exon, and point mutations of L858R in 40–45 percent of 21 exon. Others are rare mutations. The reason to raise drug-resistant is that it arises new mutations, the most important mutations is T790M in 50 percent. I stands for Mutations associated with drug resistant, II stands for Mutations associated with drug sensitivity.
Second-generation EGFR TKIs
The second-generation EGFR TKIs were designed, in order to get around the problem of gefitinib and erlotinib resistance, and mainly included BIBW 2992 (afatinib), PF00299804 (dacomitinib), HKI-272 (neratinib), CI-1033(canertinib). The second-generation EGFR TKIs have greater potential to overcome or delay withstand to the first-generation EGFR TKIs [43], and have improvements in target, binding form, and efficacy. Sufficient evidences have not yet been accumulated to determine whether or not they can overcome the resistance to first-generation EGFR TKIs entirely. Recent discoveries based on these principles continue to inspire the next generation of innovative clinical trials in this diseases.
Third-generation EGFR TKIs
To date, acquired resistance to EGFR-TKI is an unavoidable process and usually appears after 10–12 months of therapy. New EGFR-TKIs with specific capability bind to T790M mutated receptor have been developed and successfully tested in patients with acquired resistance [44, 45]. Moreover, the emerging third-generation EGFR TKIs have demonstrated high tolerability, through tested the higher ability to spare EGFR wild-type counterpart. The most famous of the third generation TKI is AZD9291 (osimertinib). It can response highly rate to drug-resistance in T790M mutations. Some patients showed resistance to this drug, and the major mutation site is C797S on the EGFR gene by the discovery of genome sequencing. The reason to drug resistance is including C-MET amplification, small cell lung cancer transformation, and downstream genes (KRAS or BRAF) activation. So patients can be controlled using a combination of the first and three generation EGFR-TKIs. With these evidences, AZD9291 (osimertinib), HM61713 (olmutinib), CO-1686 (rociletinib) and others (ASP8273, EGF816) are object of several clinical trials and AZD9291 has already obtained FDA and EMA approval for the therapy of EGFR mutant T790M-positive NSCLC [46].
In the future, when EGFR-TKI drug resistance occurs, genetic testing could be used to select the treatment method corresponding to the resistance mechanism.
PRECISION MEDICINE OF EGFR IN LUNG CANCER
Precision medicine in the treatment of lung cancer has dramatically impacted diagnostic pathology, and precision medicine provides a better comprehension of both the mechanism of the disease at the molecular level.
The discovery of EGFR mutations and ALK-rearrangements were the first molecular alterations in lung adenocarcinoma that confer sensitivity to TKIs in 2004 and 2007 (Figure 3), heralding the initiation of the era of precision medicine for lung [15, 47]. Among EGFR mutations, EGFRT790M mutation was a clear target for drug development to address the important precision medicine need, which identified as a mechanism of resistance to TKIs (Figure 3). The remarkable responses to TKIs observed in patients as well as the discoveries made studying these molecular subsets of lung cancer, served as catalysts for further exploration of the lung cancer genome, leading to the incorporation of molecular testing in routine clinical practice. Clinical trials have revealed that treatment of advanced EGFR lung cancers is superior to chemotherapy, using the appropriate TKIs [19, 48]. Conversely, it has also been shown that patients with the mutant EGFR lung cancers rarely respond to EGFR TKIs and are more likely to benefit from chemotherapy, underlining the importance of matching tumor genotype to therapy [48]. Here, we show the timeline of the history of EGFR development in recent years (Figure 3). At present and future efforts to find new types of precision medicine for lung cancers is necessary to improve outcomes for patients with lung cancer, as well as biomarker-driven clinical trials. In lung cancer, the impact of precision medicine has resulted in considerable changes as well as challenges in diagnostic pathology [49].
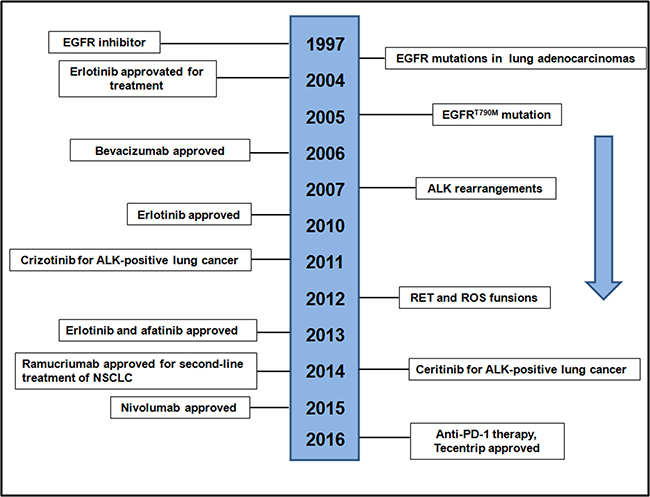
Figure 3: Timeline of EGFR-related drug development.
NONCODING RNA AND EGFR
MiRNAs in EGFR-targeted therapies for lung cancer
MicroRNAs (miRNAs) are a class of small noncoding RNAs that act as key post-transcriptional regulators of gene expression. They can functionally impact cell fate determination through the regulation of critical protein expression, thus playing a pivotal role in the diverse processes of human cancer, acting as either tumor oncogenes or suppressors [50–53]. Recently, increasing numbers of miRNAs have been correlated with the drug resistance of lung cancer cells to anti-EGFR agents, indicating that miRNAs may serve as novel targets or promising predictive biomarkers for anti-EGFR therapy. Moreover, miRNA-based therapy has been suggested to be a rational and potentially effective approach for the therapeutic targeting of EGFR [54]. Recently, a number of miRNAs, such as miR-200a, miR-27a/27b, miR-133a, and miR-134 have been verified to directly target EGFR [54–58] (Table 1). These EGFR-miRNAs regulation network studies demonstrated that miRNA-based therapy could possibly be utilized to target EGFR, except for TKIs and classical mAbs for EGFR-targeted therapies [54].
Table 1: miRNAs that target EGFR signaling pathway involved in cancer
miRNAs |
Carcinomas |
Biological effect |
Regulation |
Citations |
|---|---|---|---|---|
miR-200 |
anaplastic thyroid cancer/bladder cancer |
Regulate EMT and reverse resistance of EGFR therapy |
Down |
|
miR-23b/27b |
bladder cancer |
Regulate EGFR and suppress cancer |
Down |
[55] |
miR-27a |
renal cell carcinoma |
Suppress human RCC cell proliferation and induce cell apoptosis |
Down |
[56] |
miR-133a |
NSCLC |
Suppresses multiple oncogenic membrane receptors and cell invasion |
Down |
[57] |
miR-134 |
NSCLC |
Inhibit proliferation |
Down |
[58] |
miR-7 |
various cancer cells |
Inhibit EGFR-PI3K-AKT signaling and reverse radio resistance |
Down |
[67] |
miR-34a |
solid cancer |
Regulate Axl receptor tyrosine kinase by targeting SIRT1 and MEK1 |
Down |
[68] |
miR-145 |
lung cancer |
Negatively regulate EGFR expression |
Down |
[69] |
miR-146a |
NSCLC |
Inhibit EGFR in NSCLC cancer cells |
Down |
|
miR-146b-5p |
glioblastoma |
Suppress EGFR expression |
Down |
[72] |
miR-206 |
squamous lung cancer |
Suppress EGFR signaling |
Down |
[73] |
miR-135a-1 |
prostate cancer |
Inhibit cell growth and migration |
Down |
[74] |
miR-133a |
NSCLC |
Suppress EGFR signaling |
Down |
[57] |
miR-133b |
NSCLC |
Suppress EGFR pathway signaling and enhance susceptibility to EGFR-TKI |
Down |
[75] |
miR-1203,1237,541,542-5p |
human lung cancer |
Downregulate EGFR |
Down |
|
miR-199a-3p |
prostate cancer |
Suppress the expansion and tumor |
Down |
[77] |
miR-2861 |
cervical cancer |
Inhibit tumor growth |
Down |
[78] |
miR-25 |
lung cancer |
Upregulate EGFR |
Up |
[79] |
miR-24 |
Activates EGFR signaling |
Up |
[80] |
|
miR-21 |
glioblastoma |
Regulate the EGFR/AKT pathway in a PTEN independent manner |
Up |
[81] |
Our lab has confirmed that miR-34a, miR-181a-5p, miR-32, and miR-486-5p play vital roles in the progression of NSCLC [59–61]. Our recent study demonstrated that miR-34a can suppress NSCLC by directly targeting EGFR in vitro and in vivo (paper in under-decision).These findings demonstrate that altered miRNA expression may be related to the oncogenesis of lung cancer. Some studies have verified that EGFR mutations can be regulated by miRNA in cancer therapies. At present study, small RNA possesses the best potential as a diagnostic biomarkers and therapeutic drug for cancer, which is the most mature miRNA. For instance, because of its pivotal role in lung cancer, liver cancer, breast cancer, and among others, miR-34a is considered to be the most likely of the miRNAs to become a diagnostic marker and target of drugs [62–64] .
In conclusion, the emerging role of miRNAs as regulators could not only active EGFR signaling, but also the lung cancer cells resistance to anti-EGFR therapy. Additionally, miRNAs could also be employed as novel therapeutic targets to circumvent the resistance of lung cancer cells to EGFR inhibitors, and as biomarkers for response to anti-EGFR agents.
LncRNAs in EGFR- TKIs for lung cancer
Long non-coding RNAs (lncRNAs) are non-coding RNAs with a length > 200 nt. It has been revealed that lncRNAs are involved in a number of biological processes, such as chromatin modification, gene regulation, transcription activation and interference, and cellular processes, including cell apoptosis, migration, tumor invasion, metastasis, and drug resistance [82–84]. They play vital roles in the tumor incidence and development.
Currently study shown that lncRNAs can connect to transcription sites and regulate both the expression of alleles and a long fragment, whereas coding genes and miRNAs have no such functions [85]. This suggests that lncRNAs may be better epigenetic regulators in gene expression regulation. Research has shown that some lncRNAs, including lncRNA UCA1, H19, BC200, and BC087858, were increased in gefitinib-resistant human lung cancer cells, as determined by lncRNA microarray analysis [86]. Studies have also demonstrated that lncRNA UCA1 may stimulate non-T790M acquired resistance for EGFR-TKIs by activating the AKT/mTOR pathway and EMT [87]. Moreover, over-expression of lncRNA BC087858 could act via a novel mechanism by which acquired resistance for EGFR-TKIs can develop in EGFR-mutant NSCLC patients without T790M mutation [88]. Another study showed that stimulation of the PI3K/AKT and MEK/ERK pathways, as well as EMT, could be implicated in the resistance to EGFR-TKIs [89, 90]. Herein, we illustrate that lncRNAs are involved in the EGFR signaling pathway, as displayed in Figure 4.
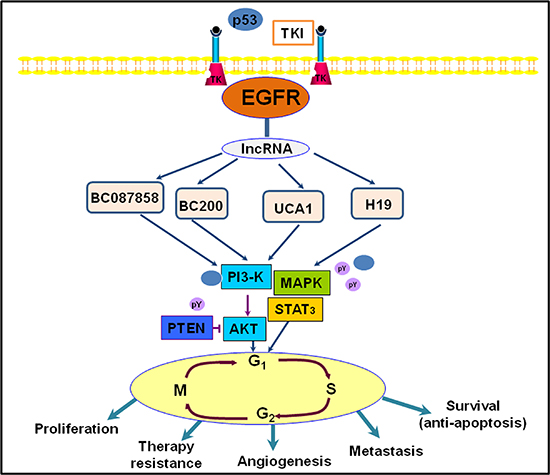
Figure 4: An illustration representing long noncoding RNAs (lncRNAs) and they involved in EGFR signaling pathway in lung cancer.
In conclusion, further exploration of the function and mechanism of lncRNAs will reveal their critical role in the process of lung cancer generation, and their significance as a diagnostic tool as well as in the treatment of cancer.
tRNA-derived RNA fragments and EGFR
A close connection has been established between cancers and a variety of small noncoding RNAs, such as miRNAs, piRNAs, and circRNAs, but not including tRNA-derived RNA fragments (tRFs). tRFs, the class of small RNAs, are noncoding-stranded RNAs 14–35 nt in length, which always derive from the 5′ end or 3′ end of tRNA in the particular environment [91]. The length and the generation of tRFs are very similar to those of miRNAs. Through the action of anticodon-cleaving enzymes, some tRNA cleavages can be generated from mature tRNAs. The biological function of tRFs is still unclear. Some studies have shown that, in biological processes, tRFs have the capacity to regulate some cellular processes, including translational efficiency under stress conditions [92], oncogenic transformation [93], and mitochondrial-mediated apoptosis [94]. EGFR and transferrin receptors (TFR) are known to be involved in cell growth and to be expressed in normal human epidermis. Currently, about EGFR and TRF expression in human cancer is few.
However, their biological function has drawn the attention of many research efforts. Some studies have shown that, in biological processes, the function of 5’ tRFs might extend much farther than that of 3’ tRFs [95, 96]. In addition, it has been found that tRFs suppress breast cancer progression via YBX1 displacement [97]. Furthermore, some tRFs can active cellular functions through argonaute engagement, such as cell proliferation and RNA silencing [98–100]. TRF expression is not associated with cellular proliferation, in embryonic and fetal epidermis, whereas EGFR appears to be correlated with proliferation and undifferentiated cells [101].
PERSPECTIVES AND CHALLENGE IN EGFR-RELATED STUDY
The evolution of modern medical technology has enabled advancements in research to reveal the mechanism of the development of a variety of incurable disease. In the case of cancer, the emerging view is that cancer is a “genetic disease”. With next generation sequencing technology and promotion of the human genome project, the treatment of cancer has been gradually moving towards the era of precision therapy. Under this condition, new therapies take advantage of RNA and other powerful features targeting genes that play a critical role in tumorigenesis.
The importance of miRNA regulatory network’s role in tumors has been revealed more and more. Meanwhile, we should combine molecular mechanisms to overcome the emergence of resistance. As described in this review, several molecular pathways underlying the mechanisms of this disease have been elaborated in part, among them the EGFR pathway is one of the well-known signal cascades that play a pivotal role in oncogenesis. In lung cancer, dysregulation of EGFR signaling is frequently found. Strategies to effectively inhibit the EGFR signaling pathway have been mounted for developing anticancer therapeutic agents. However, on account of the development of drug-resistance, most anti-EGFR-targeted agents are unable to repress cancer progression. Therefore, studies of the mechanisms underpinning the resistance toward anti-EGFR agents may afford important findings for the use of anti-EGFR therapies in lung cancer treatment.
Finally, when determining the applicability of EGFR-TKI in practice, clinical benefit should be carefully analyzed based on clinical background and the prediction of the presence or absence of EGFR mutations. Furthermore, immune-based cancer prevention can also influence premalignant biology. It has been shown that cancer vaccines reprogram the immune response to prevent, reject, and detect premalignant cells, which might be applicable in EGFR therapy [102]. Going forward, in lung cancer, EGFR may be the rising star in the era of precision medicine.
ACKNOWLEDGMENTS
We acknowledge Dr Deng Pan (Dana-Farber/Harvard Cancer Center) for him critical comments and discussions. This work was supported by and National Natural Science Foundation of China (91543123) and (81572122).
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87–108.
2. Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nature reviews Cancer. 2014; 14:535–546.
3. Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, Lorimer I, Zhang T, Liu N, Daneshmand M, Marrano P, da Cunha Santos G, Lagarde A, et al. Erlotinib in lung cancer - molecular and clinical predictors of outcome. The New England journal of medicine. 2005; 353:133–144.
4. Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, Thomas M, O’Byrne KJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. The New England journal of medicine. 2013; 368:2385–2394.
5. Yoshida T, Zhang G, Haura EB. Targeting epidermal growth factor receptor: central signaling kinase in lung cancer. Biochemical pharmacology. 2010; 80:613–623.
6. Janne PA, Engelman JA, Johnson BE. Epidermal growth factor receptor mutations in non-small-cell lung cancer: implications for treatment and tumor biology. Journal of clinical oncology. 2005; 23:3227–3234.
7. Sun M, Behrens C, Feng L, Ozburn N, Tang X, Yin G, Komaki R, Varella-Garcia M, Hong WK, Aldape KD, Wistuba II. HER family receptor abnormalities in lung cancer brain metastases and corresponding primary tumors. Clinical cancer research. 2009; 15:4829–4837.
8. Wang Y, Deng G, Liu X, Cho WC. Monoclonal antibodies in lung cancer. Expert opinion on biological therapy. 2013; 13:209–226.
9. Perez R, Crombet T, de Leon J, Moreno E. A view on EGFR-targeted therapies from the oncogene-addiction perspective. Frontiers in pharmacology. 2013; 4:53.
10. Remon J, Moran T, Majem M, Reguart N, Dalmau E, Marquez-Medina D, Lianes P. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: a new era begins. Cancer treatment reviews. 2014; 40:93–101.
11. Yu HA, Riely GJ, Lovly CM. Therapeutic strategies utilized in the setting of acquired resistance to EGFR tyrosine kinase inhibitors. Clinical cancer research. 2014; 20:5898–5907.
12. Lin Y, Wang X, Jin H. EGFR-TKI resistance in NSCLC patients: mechanisms and strategies. American journal of cancer research. 2014; 4:411–435.
13. Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer cell. 2007; 11:217–227.
14. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007; 131:1190–1203.
15. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. The New England journal of medicine. 2004; 350:2129–2139.
16. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004; 304:1497–1500.
17. Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101:13306–13311.
18. Gandara DR, Hammerman PS, Sos ML, Lara PN Jr, Hirsch FR. Squamous cell lung cancer: from tumor genomics to cancer therapeutics. Clinical cancer research. 2015; 21:2236–2243.
19. Riely GJ, Yu HA. EGFR: The Paradigm of an Oncogene-Driven Lung Cancer. Clinical cancer research. 2015; 21:2221–2226.
20. Herbst RS, Kim ES, Harari PM. IMC-C225, an anti-epidermal growth factor receptor monoclonal antibody, for treatment of head and neck cancer. Expert opinion on biological therapy. 2001; 1:719–732.
21. Colquhoun AJ, Mellon JK. Epidermal growth factor receptor and bladder cancer. Postgraduate medical journal. 2002; 78:584–589.
22. Kim YB, Kim GE, Cho NH, Pyo HR, Shim SJ, Chang SK, Park HC, Suh CO, Park TK, Kim BS. Overexpression of cyclooxygenase-2 is associated with a poor prognosis in patients with squamous cell carcinoma of the uterine cervix treated with radiation and concurrent chemotherapy. Cancer. 2002; 95:531–539.
23. Ohsaki Y, Tanno S, Fujita Y, Toyoshima E, Fujiuchi S, Nishigaki Y, Ishida S, Nagase A, Miyokawa N, Hirata S, Kikuchi K. Epidermal growth factor receptor expression correlates with poor prognosis in non-small cell lung cancer patients with p53 overexpression. Oncology reports. 2000; 7:603–607.
24. Pastorino U, Andreola S, Tagliabue E, Pezzella F, Incarbone M, Sozzi G, Buyse M, Menard S, Pierotti M, Rilke F. Immunocytochemical markers in stage I lung cancer: relevance to prognosis. Journal of clinical oncology. 1997; 15:2858–2865.
25. Selvaggi G, Novello S, Torri V, Leonardo E, De Giuli P, Borasio P, Mossetti C, Ardissone F, Lausi P, Scagliotti GV. Epidermal growth factor receptor overexpression correlates with a poor prognosis in completely resected non-small-cell lung cancer. Annals of oncology. 2004; 15:28–32.
26. Harpole DH Jr, Richards WG, Herndon JE 2nd, Sugarbaker DJ. Angiogenesis and molecular biologic substaging in patients with stage I non-small cell lung cancer. The Annals of thoracic surgery. 1996; 61:1470–1476.
27. Pfeiffer P, Clausen PP, Andersen K, Rose C. Lack of prognostic significance of epidermal growth factor receptor and the oncoprotein p185HER-2 in patients with systemically untreated non-small-cell lung cancer: an immunohistochemical study on cryosections. British journal of cancer. 1996; 74:86–91.
28. Rusch V, Baselga J, Cordon-Cardo C, Orazem J, Zaman M, Hoda S, McIntosh J, Kurie J, Dmitrovsky E. Differential expression of the epidermal growth factor receptor and its ligands in primary non-small cell lung cancers and adjacent benign lung. Cancer research. 1993; 53:2379–2385.
29. Cox G, Jones JL, O’Byrne KJ. Matrix metalloproteinase 9 and the epidermal growth factor signal pathway in operable non-small cell lung cancer. Clinical cancer research. 2000; 6:2349–2355.
30. Dias F, Teixeira AL, Santos JI, Gomes M, Nogueira A, Assis J, Medeiros R. Renal cell carcinoma development and miRNAs: a possible link to the EGFR pathway. Pharmacogenomics. 2013; 14:1793–1803.
31. Stumm G, Eberwein S, Rostock-Wolf S, Stein H, Pomer S, Schlegel J, Waldherr R. Concomitant overexpression of the EGFR and erbB-2 genes in renal cell carcinoma (RCC) is correlated with dedifferentiation and metastasis. International journal of cancer. 1996; 69:17–22.
32. Lui VW, Grandis JR. EGFR-mediated cell cycle regulation. Anticancer research. 2002; 22:1–11.
33. Shelton JG, Steelman LS, Abrams SL, White ER, Akula SM, Franklin RA, Bertrand FE, McMahon M, McCubrey JA. Conditional EGFR promotes cell cycle progression and prevention of apoptosis in the absence of autocrine cytokines. Cell cycle. 2005; 4:822–830.
34. Kari C, Chan TO, Rocha de Quadros M, Rodeck U. Targeting the epidermal growth factor receptor in cancer: apoptosis takes center stage. Cancer research. 2003; 63:1–5.
35. Shvartsman SY, Hagan MP, Yacoub A, Dent P, Wiley HS, Lauffenburger DA. Autocrine loops with positive feedback enable context-dependent cell signaling. American journal of physiology Cell physiology. 2002; 282:C545–559.
36. Umekita Y, Ohi Y, Sagara Y, Yoshida H. Co-expression of epidermal growth factor receptor and transforming growth factor-alpha predicts worse prognosis in breast-cancer patients. International journal of cancer. 2000; 89:484–487.
37. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, Wilkerson MD, Fecci PE, Butaney M, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer discovery. 2013; 3:1355–1363.
38. Paz-Ares L, Soulieres D, Melezinek I, Moecks J, Keil L, Mok T, Rosell R, Klughammer B. Clinical outcomes in non-small-cell lung cancer patients with EGFR mutations: pooled analysis. Journal of cellular and molecular medicine. 2010; 14:51–69.
39. Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009; 28:S24–31.
40. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316:1039–1043.
41. Popat S, Wotherspoon A, Nutting CM, Gonzalez D, Nicholson AG, O’Brien M. Transformation to “high grade” neuroendocrine carcinoma as an acquired drug resistance mechanism in EGFR-mutant lung adenocarcinoma. Lung cancer. 2013; 80:1–4.
42. Suda K, Tomizawa K, Fujii M, Murakami H, Osada H, Maehara Y, Yatabe Y, Sekido Y, Mitsudomi T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. Journal of thoracic oncology. 2011; 6:1152–1161.
43. Metro G, Cappuzzo F. Emerging drugs for small-cell lung cancer. Expert opinion on emerging drugs. 2009; 14:591–606.
44. Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. The New England journal of medicine. 2015; 372:1689–1699.
45. Kim ES. Olmutinib: First Global Approval. Drugs. 2016; 76:1153–1157.
46. Minari R, Bordi P, Tiseo M. Third-generation epidermal growth factor receptor-tyrosine kinase inhibitors in T790M-positive non-small cell lung cancer: review on emerged mechanisms of resistance. Translational lung cancer research. 2016; 5:695–708.
47. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007; 448:561–566.
48. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. The New England journal of medicine. 2009; 361:947–957.
49. Politi K, Herbst RS. Lung cancer in the era of precision medicine. Clinical cancer research. 2015; 21:2213–2220.
50. Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nature reviews Molecular cell biology. 2010; 11:252–263.
51. Hoshino I, Matsubara H. MicroRNAs in cancer diagnosis and therapy: from bench to bedside. Surgery today. 2013; 43:467–478.
52. Wang F, Chan LW, Law HK, Cho WC, Tang P, Yu J, Shyu CR, Wong SC, Yip SP, Yung BY. Exploring microRNA-mediated alteration of EGFR signaling pathway in non-small cell lung cancer using an mRNA:miRNA regression model supported by target prediction databases. Genomics. 2014; 104:504–511.
53. Gomez GG, Wykosky J, Zanca C, Furnari FB, Cavenee WK. Therapeutic resistance in cancer: microRNA regulation of EGFR signaling networks. Cancer biology & medicine. 2013; 10:192–205.
54. Zhen Q, Liu J, Gao L, Liu J, Wang R, Chu W, Zhang Y, Tan G, Zhao X, Lv B. MicroRNA-200a Targets EGFR and c-Met to Inhibit Migration, Invasion, and Gefitinib Resistance in Non-Small Cell Lung Cancer. Cytogenetic and genome research. 2015; 146:1–8.
55. Chiyomaru T, Seki N, Inoguchi S, Ishihara T, Mataki H, Matsushita R, Goto Y, Nishikawa R, Tatarano S, Itesako T, Nakagawa M, Enokida H. Dual regulation of receptor tyrosine kinase genes EGFR and c-Met by the tumor-suppressive microRNA-23b/27b cluster in bladder cancer. International journal of oncology. 2015; 46:487–496.
56. Li Y, Li J, Sun X, Chen J, Sun X, Zheng J, Chen R. MicroRNA-27a functions as a tumor suppressor in renal cell carcinoma by targeting epidermal growth factor receptor. Oncology letters. 2016; 11:4217–4223.
57. Wang LK, Hsiao TH, Hong TM, Chen HY, Kao SH, Wang WL, Yu SL, Lin CW, Yang PC. MicroRNA-133a suppresses multiple oncogenic membrane receptors and cell invasion in non-small cell lung carcinoma. PloS one. 2014; 9:e96765.
58. Qin Q, Wei F, Zhang J, Wang X, Li B. miR-134 inhibits non-small cell lung cancer growth by targeting the epidermal growth factor receptor. Journal of cellular and molecular medicine. 2016; 20:1974–83.
59. Ma ZL, Hou PP, Li YL, Wang DT, Yuan TW, Wei JL, Zhao BT, Lou JT, Zhao XT, Jin Y, Jin YX. MicroRNA-34a inhibits the proliferation and promotes the apoptosis of non-small cell lung cancer H1299 cell line by targeting TGFbetaR2. Tumour biology. 2015; 36:2481–2490.
60. Ma Z, Qiu X, Wang D, Li Y, Zhang B, Yuan T, Wei J, Zhao B, Zhao X, Lou J, Jin Y, Jin Y. MiR-181a-5p inhibits cell proliferation and migration by targeting Kras in non-small cell lung cancer A549 cells. Acta biochimica et biophysica Sinica. 2015; 47:630–638.
61. Shao Y, Shen YQ, Li YL, Liang C, Zhang BJ, Lu SD, He YY, Wang P, Sun QL, Jin YX, Ma ZL. Direct repression of the oncogene CDK4 by the tumor suppressor miR-486–5 p in non-small cell lung cancer. Oncotarget. 2016; 7:34011–21. doi: 10.18632/oncotarget.8514.
62. Del Vescovo V, Denti MA. microRNA, Lung Cancer. Advances in experimental medicine and biology. 2015; 889:153–177.
63. Yu T, Liu L, Li J, Yan M, Lin H, Liu Y, Chu D, Tu H, Gu A, Yao M. MiRNA-10a is upregulated in NSCLC and may promote cancer by targeting PTEN. Oncotarget. 2015; 6:30239–30250. doi: 10.18632/oncotarget.4972.
64. Yang Y, Liu L, Zhang Y, Guan H, Wu J, Zhu X, Yuan J, Li M. MiR-503 targets PI3K p85 and IKK-beta and suppresses progression of non-small cell lung cancer. International journal of cancer. 2014; 135:1531–1542.
65. Adam L, Zhong M, Choi W, Qi W, Nicoloso M, Arora A, Calin G, Wang H, Siefker-Radtke A, McConkey D, Bar-Eli M, Dinney C. miR-200 expression regulates epithelial-to-mesenchymal transition in bladder cancer cells and reverses resistance to epidermal growth factor receptor therapy. Clinical cancer research. 2009; 15:5060–5072.
66. Zhang Z, Liu ZB, Ren WM, Ye XG, Zhang YY. The miR-200 family regulates the epithelial-mesenchymal transition induced by EGF/EGFR in anaplastic thyroid cancer cells. International journal of molecular medicine. 2012; 30:856–862.
67. Webster RJ, Giles KM, Price KJ, Zhang PM, Mattick JS, Leedman PJ. Regulation of epidermal growth factor receptor signaling in human cancer cells by microRNA-7. The Journal of biological chemistry. 2009; 284:5731–5741.
68. Mudduluru G, Ceppi P, Kumarswamy R, Scagliotti GV, Papotti M, Allgayer H. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene. 2011; 30:2888–2899.
69. Cho WC, Chow AS, Au JS. MiR-145 inhibits cell proliferation of human lung adenocarcinoma by targeting EGFR, NUDT1. RNA biology. 2011; 8:125–131.
70. Chen G, Umelo IA, Lv S, Teugels E, Fostier K, Kronenberger P, Dewaele A, Sadones J, Geers C, De Greve J. miR-146a inhibits cell growth, cell migration and induces apoptosis in non-small cell lung cancer cells. PloS one. 2013; 8:e60317.
71. Xu B, Wang N, Wang X, Tong N, Shao N, Tao J, Li P, Niu X, Feng N, Zhang L, Hua L, Wang Z, Chen M. MiR-146a suppresses tumor growth and progression by targeting EGFR pathway and in a p-ERK-dependent manner in castration-resistant prostate cancer. The Prostate. 2012; 72:1171–1178.
72. Katakowski M, Zheng X, Jiang F, Rogers T, Szalad A, Chopp M. MiR-146b-5p suppresses EGFR expression and reduces in vitro migration and invasion of glioma. Cancer investigation. 2010; 28:1024–1030.
73. Mataki H, Seki N, Chiyomaru T, Enokida H, Goto Y, Kumamoto T, Machida K, Mizuno K, Nakagawa M, Inoue H. Tumor-suppressive microRNA-206 as a dual inhibitor of MET, EGFR oncogenic signaling in lung squamous cell carcinoma. International journal of oncology. 2015; 46:1039–1050.
74. Xu B, Tao T, Wang Y, Fang F, Huang Y, Chen S, Zhu W, Chen M. hsa-miR-135a-1 inhibits prostate cancer cell growth and migration by targeting EGFR. Tumour biology. 2016.
75. Liu L, Shao X, Gao W, Zhang Z, Liu P, Wang R, Huang P, Yin Y, Shu Y. MicroRNA-133b inhibits the growth of non-small-cell lung cancer by targeting the epidermal growth factor receptor. The FEBS journal. 2012; 279:3800–3812.
76. Yamaguchi G, Takanashi M, Tanaka M, Fujita K, Ohira T, Kuroda M, Ikeda N. Isolation of miRNAs that target EGFR mRNA in human lung cancer. Biochemical and biophysical research communications. 2012; 420:411–416.
77. Liu R, Liu C, Zhang D, Liu B, Chen X, Rycaj K, Jeter C, Calhoun-Davis T, Li Y, Yang T, Wang J, Tang DG. miR-199a-3p targets stemness-related and mitogenic signaling pathways to suppress the expansion and tumorigenic capabilities of prostate cancer stem cells. Oncotarget. 2016; 7:56628–56642. doi: 10.18632/oncotarget.10652.
78. Xu J, Wan X, Chen X, Fang Y, Cheng X, Xie X, Lu W. miR-2861 acts as a tumor suppressor via targeting EGFR/AKT2/CCND1 pathway in cervical cancer induced by human papillomavirus virus 16 E6. Scientific reports. 2016; 6:28968.
79. Dacic S, Kelly L, Shuai Y, Nikiforova MN. miRNA expression profiling of lung adenocarcinomas: correlation with mutational status. Modern pathology. 2010; 23:1577–1582.
80. Du WW, Fang L, Li M, Yang X, Liang Y, Peng C, Qian W, O’Malley YQ, Askeland RW, Sugg SL, Qian J, Lin J, Jiang Z, et al. MicroRNA miR-24 enhances tumor invasion and metastasis by targeting PTPN9 and PTPRF to promote EGF signaling. Journal of cell science. 2013; 126:1440–1453.
81. Zhou X, Ren Y, Moore L, Mei M, You Y, Xu P, Wang B, Wang G, Jia Z, Pu P, Zhang W, Kang C. Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of PTEN status. Laboratory investigation. 2010; 90:144–155.
82. Chen J, Wang R, Zhang K, Chen LB. Long non-coding RNAs in non-small cell lung cancer as biomarkers and therapeutic targets. Journal of cellular and molecular medicine. 2014; 18:2425–2436.
83. Esteller M. Non-coding RNAs in human disease. Nature reviews Genetics. 2011; 12:861–874.
84. Cheetham SW, Gruhl F, Mattick JS, Dinger ME. Long noncoding RNAs and the genetics of cancer. British journal of cancer. 2013; 108:2419–2425.
85. Li T, Mo X, Fu L, Xiao B, Guo J. Molecular mechanisms of long noncoding RNAs on gastric cancer. Oncotarget. 2016; 7:8601–8612. doi: 10.18632/oncotarget.6926.
86. Cheng N, Li X, Zhao C, Ren S, Chen X, Cai W, Zhao M, Zhang Y, Li J, Wang Q, Zhou C. Microarray expression profile of long non-coding RNAs in EGFR-TKIs resistance of human non-small cell lung cancer. Oncology reports. 2015; 33:833–839.
87. Cheng N, Cai W, Ren S, Li X, Wang Q, Pan H, Zhao M, Li J, Zhang Y, Zhao C, Chen X, Fei K, Zhou C, et al. Long non-coding RNA UCA1 induces non-T790M acquired resistance to EGFR-TKIs by activating the AKT/mTOR pathway in EGFR-mutant non-small cell lung cancer. Oncotarget. 2015; 6:23582–23593. doi: 10.18632/oncotarget.4361.
88. Pan H, Jiang T, Cheng N, Wang Q, Ren S, Li X, Zhao C, Zhang L, Cai W, Zhou C. Long non-coding RNA BC087858 induces non-T790M mutation acquired resistance to EGFR-TKIs by activating PI3K/AKT and MEK/ERK pathways and EMT in non-small-cell lung cancer. Oncotarget. 2016; 7:49948–49960. doi: 10.18632/oncotarget.10521.
89. Hampton KK, Craven RJ. Pathways driving the endocytosis of mutant and wild-type EGFR in cancer. Oncoscience. 2014; 1:504–512. doi: 10.18632/oncoscience.67.
90. Rolfo C, Giovannetti E, Hong DS, Bivona T, Raez LE, Bronte G, Buffoni L, Reguart N, Santos ES, Germonpre P, Taron M, Passiglia F, Van Meerbeeck JP, et al. Novel therapeutic strategies for patients with NSCLC that do not respond to treatment with EGFR inhibitors. Cancer treatment reviews. 2014; 40:990–1004.
91. Lee YS, Shibata Y, Malhotra A, Dutta A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes & development. 2009; 23:2639–2649.
92. Ivanov P, Emara MM, Villen J, Gygi SP, Anderson P. Angiogenin-induced tRNA fragments inhibit translation initiation. Molecular cell. 2011; 43:613–623.
93. Marshall L, Kenneth NS, White RJ. Elevated tRNA(iMet) synthesis can drive cell proliferation and oncogenic transformation. Cell. 2008; 133:78–89.
94. Mei Y, Yong J, Liu H, Shi Y, Meinkoth J, Dreyfuss G, Yang X. tRNA binds to cytochrome c and inhibits caspase activation. Molecular cell. 2010; 37:668–678.
95. Dhahbi JM, Spindler SR, Atamna H, Yamakawa A, Boffelli D, Mote P, Martin DI. 5’ tRNA halves are present as abundant complexes in serum, concentrated in blood cells, and modulated by aging and calorie restriction. BMC genomics. 2013; 14:298.
96. Andersen KL, Collins K. Several RNase T2 enzymes function in induced tRNA and rRNA turnover in the ciliate Tetrahymena. Molecular biology of the cell. 2012; 23:36–44.
97. Goodarzi H, Liu X, Nguyen HC, Zhang S, Fish L, Tavazoie SF. Endogenous tRNA-Derived Fragments Suppress Breast Cancer Progression via YBX1 Displacement. Cell. 2015; 161:790–8 02.
98. Gebetsberger J, Polacek N. Slicing tRNAs to boost functional ncRNA diversity. RNA biology. 2013; 10:1798–1806.
99. Haussecker D, Huang Y, Lau A, Parameswaran P, Fire AZ, Kay MA. Human tRNA-derived small RNAs in the global regulation of RNA silencing. Rna. 2010; 16:673–695.
100. Garcia-Silva MR, Cabrera-Cabrera F, Guida MC, Cayota A. Hints of tRNA-Derived Small RNAs Role in RNA Silencing Mechanisms. Genes. 2012; 3:603–614.
101. Zambruno G, Girolomoni G, Manca V, Segre A, Giannetti A. Epidermal growth factor and transferrin receptor expression in human embryonic and fetal epidermal cells. Archives of dermatological research. 1990; 282:544–548.
102. Spira A, Disis ML, Schiller JT, Vilar E, Rebbeck TR, Bejar R, Ideker T, Arts J, Yurgelun MB, Mesirov JP, Rao A, Garber J, Jaffee EM, et al. Leveraging premalignant biology for immune-based cancer prevention. Proceedings of the National Academy of Sciences of the United States of America. 2016; 113:10750–8.