
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer worldwide and the second most common cause of cancer-related mortality. It has been well established that colorectal tumorigenesis is a multistep process with an accumulation of multiple, successive genetic alterations, including chromosomal abnormalities, gene mutations, and/or epigenetic changes transforming normal colonic epithelium to colorectal carcinoma [1]. APC, TP53, RAS, RAF, and PIK3CA gene mutations are the most commonly noted aberrations in metastatic CRC (mCRC). While the prognostic and predictive implications of a few such aberrations, including those of RAF, RAS, and deficient mismatch repair (MMR), are well established in CRC and now routinely assessed as part of clinical care [2–4], the clinical implications of other genetic aberrations in CRC are unclear in spite of extensive work over the past few decades. Intense research efforts are ongoing to identify reliable, novel biomarkers to help clinicians make personalized treatment decisions in CRC.
A potential avenue to this end involves ubiquitin-mediated proteolysis, which regulates the degradation of many proteins involved in the control of cell growth and differentiation. F-box proteins, the substrate-recognition subunit of SKP1–Cullin1–F-box protein ubiquitin E3 ligase complexes, have been well characterized and shown to play important roles in degradation of proteins regulating cell cycle progression. So far, more than 70 putative F-box proteins have been identified in the human genome; however, the function and the substrates of most F-box proteins remain elusive [5].
FBXW7 is a tumor suppressor gene on human chromosome 4q that encodes the substrate recognition components of SKP1–Cullin1–F-box protein ubiquitin E3 ligase complexes [6]. These specific E3 ligase complexes negatively regulate the intracellular abundance of an expanding list of key oncogenic proteins such as cyclin E [7], c-JUN [8, 9], c-MYC [10, 11], MCL1 (myeloid cell leukemia 1) [12, 13], NOTCH [14–17], AURKA (aurora kinase A) [18, 19], KLF5 (Krüppel-like factor 5) [20], mTOR [21], and TGIF1 [22]. Therefore, the loss of FBXW7 function results in accumulation of its substrates, which leads to oncogenesis and progression of multiple cancers including CRC [23, 24]. A study of over 500 primary tumors of diverse tissue origins suggested that FBXW7 mutations occurred in approximately 6% of all evaluated tumors. Of these, the most commonly affected tumors were cholangiocarcinoma (35%), (T-cell acute lymphocytic leukemia, 31%), endometrial cancer (9%), and gastric cancer (6%) [24]. FBXW7 has also consistently been identified as one of the most commonly mutated genes in CRC [25], observed in 6% to 10% of all cases [24–27].
FBXW7 is structurally composed of several conserved protein-protein interaction domains, including the 40–amino acid F-box, which recruits the SKP1–Cullin1–F-box complex; eight WD40 repeats that bind to substrates; and the D domain located just before the F-box, which facilitates dimerization of FBXW7. WD40 repeats 3 and 4 contain three highly conserved arginine residues that play a key role in binding to phosphothreonine residues on substrates, while residues within the other WD40 repeats contribute incrementally [28–31]. The FBXW7 mutational range is rather atypical; over 70% are missense point mutations affecting amino acids within substrate-binding sites, and two of the key arginine residues described above (Arg465 and Arg479) are mutational hotspots [5]. The rest are mostly nonsense mutations leading to premature termination of translation of FBXW7, while the loss of an entire allele is very rarely noted [24]. Tumor-derived FBXW7 alleles show a very strong predisposition for missense mutations over nonsense mutations. In fact, some tumor types such as T-cell acute lymphoblastic leukemia have only FBXW7 missense mutations. This marked skew toward full-length FBXW7 mutations with impaired substrate binding, instead of prematurely terminating mutants (due to nonsense mutations), is thought to be due to the former group’s ability to act as more potent dominant negatives negating the impact of the wild-type protein in FBXW7 dimers [5].
However, to date, the prognostic significance of FBXW7 missense mutations in CRC remains to be elucidated. In the current study, we extensively evaluated the clinicopathologic and molecular characteristics of FBXW7 missense mutations in mCRC and the associated survival outcomes.
RESULTS
A total of 855 mCRC patients were included in the Assessment of Targeted Therapies Against Colorectal Cancer program at The University of Texas MD Anderson Cancer Center between February 13, 2009, and November 18, 2015. Of these, we identified 571 patients for the current study with data available on FBXW7 status. The median age of the cohort was 55 years (range 20-82 years), and the ratio of males to females was 1.27. The majority of primary tumors were left-sided colon tumors (243 patients, 42.6%), followed by right-sided colon tumors (195 patients, 34.2%), and the rest were rectal tumors (128 patients, 22.4%). Patient and tumor characteristics are shown in (Table 1).
Table 1: Clinicopathological characteristics of study population, n (%)
Variable |
Value |
% |
|---|---|---|
No. of patients |
571 |
100 |
Median age (yr, range) |
55, 20-82 |
|
Age |
||
<50 years |
197 |
34.5 |
≥50 years |
374 |
65.5 |
Sex |
||
Female |
251 |
44 |
Male |
320 |
56 |
Race/ethnicity |
||
Asian |
31 |
5.4 |
Black |
50 |
8.8 |
Hispanic |
54 |
9.5 |
White |
432 |
75.7 |
No data |
4 |
0.7 |
Primary tumor site |
||
Right sided |
195 |
34.2 |
Left sided |
243 |
42.6 |
Rectum |
128 |
22.4 |
No data |
5 |
0.9 |
Metastases type |
||
Synchronous |
368 |
64.4 |
Metachronous |
203 |
35.6 |
Metastatic site |
||
Liver only |
51 |
8.9 |
Not Limited to Liver |
520 |
91.1 |
Differentiated |
||
Well |
1 |
0.2 |
Mod |
423 |
74.1 |
Poorly |
141 |
24.7 |
No data |
6 |
1 |
KRAS |
||
wt |
284 |
49.7 |
mt |
286 |
50.1 |
Variant |
1 |
0.2 |
NRAS |
||
wt |
545 |
95.4 |
mt |
26 |
4.6 |
BRAF |
||
wt |
524 |
91.8 |
mt |
47 |
8.2 |
PIK3CA |
||
wt |
477 |
83.5 |
mt |
90 |
15.8 |
Variant |
4 |
0.7 |
MMR status |
||
Proficient |
395 |
69.2 |
Deficient |
19 |
3.3 |
No data |
157 |
27.5 |
FBXW7 |
||
wt |
527 |
92.3 |
mt |
43 |
7.5 |
Variant |
1 |
0.2 |
wt: wild type, mt: mutation
Frequency of KRAS, NRAS, BRAF, PIK3CA, and FBXW7 mutations and MSI-H
DNA was extracted from 363 primary CRC tissues and 208 metastatic tissues. One hundred and ninety four cases were tested with the 46-gene panel while 377 cases were tested with the 50-gene panel. KRAS, NRAS, BRAF, and PIK3CA mutations were present in 50.1%, 4.6%, 8.2%, and 16.5%, respectively, of the 571 patients in our cohort. FBXW7 mutations were identified in 43 patients (7.5%) (Table 1). Of these, 37 patients had missense mutations, while four had nonsense mutations, one had insertion, and one had deletion. R465C in exon 9 affecting Arg465 was the most common FBXW7 missense mutation (18.6%). R465H (affecting Arg465) and R505C (affecting Arg505) were the second most common FBXW7 missense mutations (16.3% each). No difference in mutation frequency in FBXW7 was noted either between primary CRC and metastatic tissues (8.8% vs 5.3%, respectively; P=0.12) (Supplementary Table 1) or between the two gene panels (7.2% vs 7.7%, respectively; p=0.39) (Supplementary Table 2). The frequencies and types of FBXW7 mutations are shown in (Figure 1 and Supplementary Table 3). MSI-H was found in 3.3% of patients tested (19/414).
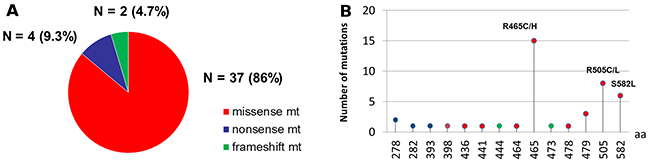
Figure 1: Frequency and spectrum of FBXW7 mutations. (A) FBXW7 mutations were identified in 43 patients. Of these, 37 patients had missense mutations, while four had nonsense mutations, and two had frameshift mutations. (B) R465C/H were the most common FBXW7 missense mutation (n = 15). R505C/L were the second most common FBXW7 missense mutations (n = 8) followed by S582L missense mutations (n = 6).
Associations between FBXW7 mutations and clinicopathologic factors
The clinicopathologic variables compared with FBXW7 status included age, sex, race/ethnicity, site of the primary tumor, histologic grade, and KRAS, NRAS, BRAF, PIK3CA, and microsatellite instability status. Among these factors, only PIK3CA mutations were significantly associated with FBXW7 mutations (p=0.012). A subset analysis limited to FBXW7 missense mutations showed a similar association between the missense mutations and PIK3CA mutations (p=0.022) (Table 2). However, after excluding PIK3CA variant mutations (n = 4), there was only a trend towards co-occurrence of PIK3CA and FBXW7 mutations (P=0.10).
Table 2: FBXW7 status and associated clinicopathological factors
Variable |
FBXW7 status |
|||||||
|---|---|---|---|---|---|---|---|---|
wt |
All FBXW7 mutations |
FBXW7 missense mutations |
||||||
N |
% |
N |
% |
P value |
N |
% |
P value |
|
Age (N=570) |
||||||||
<50 years |
183 |
34.7 |
14 |
32.6 |
0.77 |
13 |
35.1 |
0.96 |
≥50 years |
344 |
65.3 |
29 |
67.4 |
24 |
64.9 |
||
Sex (N=570) |
||||||||
Female |
233 |
44.2 |
18 |
41.9 |
0.77 |
14 |
37.8 |
0.45 |
Male |
294 |
55.8 |
25 |
58.1 |
23 |
62.2 |
||
Race/ethnicity (N=566) |
||||||||
Asian |
29 |
5.5 |
2 |
4.7 |
0.99 |
1 |
2.7 |
0.89 |
Black |
46 |
8.8 |
4 |
9.3 |
3 |
8.1 |
||
Hispanic |
50 |
9.6 |
4 |
9.3 |
4 |
10.8 |
||
White |
398 |
76.1 |
33 |
76.7 |
29 |
78.4 |
||
Primary tumor site (N=565) |
||||||||
Rt.sided |
182 |
34.9 |
12 |
27.9 |
0.42 |
8 |
21.6 |
0.17 |
Lt.sided |
225 |
43.1 |
18 |
41.9 |
17 |
45.9 |
||
Rectum |
115 |
22 |
13 |
30.2 |
12 |
32.4 |
||
Metastases type |
||||||||
Synchronous |
341 |
64.7 |
26 |
60.5 |
0.58 |
21 |
56.8 |
0.33 |
Metachronous |
186 |
35.3 |
17 |
39.5 |
16 |
43.2 |
||
Metastatic site |
||||||||
Liver only |
46 |
8.7 |
5 |
11.6 |
0.52 |
4 |
10.8 |
0.67 |
Not Limited to Liver |
481 |
91.3 |
38 |
88.4 |
33 |
89.2 |
||
Differentiated (N=564) |
||||||||
Well to moderately |
392 |
45.2 |
32 |
74.4 |
0.91 |
27 |
73 |
0.76 |
Poorly |
129 |
24.8 |
11 |
25.6 |
10 |
27 |
||
KRAS (N=569) |
||||||||
wt |
265 |
50.4 |
19 |
44.2 |
0.44 |
15 |
40.5 |
0.25 |
mt |
261 |
49.6 |
24 |
55.8 |
22 |
59.5 |
||
NRAS (N=570) |
||||||||
wt |
503 |
95.4 |
41 |
95.3 |
1 |
35 |
94.6 |
0.69 |
mt |
24 |
4.6 |
2 |
4.7 |
2 |
5.4 |
||
BRAF (N-570) |
||||||||
wt |
485 |
92 |
38 |
88.4 |
0.39 |
33 |
89.2 |
0.53 |
mt |
42 |
8 |
5 |
11.6 |
4 |
10.8 |
||
PIK3CA (N=570) |
||||||||
wt |
446 |
84.6 |
30 |
69.8 |
0.012 |
26 |
70.3 |
0.022 |
mt |
81 |
15.4 |
13 |
30.2 |
12 |
29.7 |
||
MMR status (N=414) |
||||||||
Proficient |
365 |
96.1 |
30 |
88.2 |
0.06 |
26 |
89.7 |
0.13 |
Deficient |
15 |
3.9 |
4 |
11.8 |
3 |
10.3 |
||
wt: wild type, mt: mutation
Survival analysis
The median follow-up time was 30.4 months. At the time of data cut-off (March 1, 2016), there were 266 patients alive and 305 patients who had died.
Univariate analysis of OS was performed using previously established prognostic factors: the aforementioned clinicopathologic variables as well as FBXW7 mutations. Factors that were associated with statistically significantly worse OS in this analysis included an age of <50 years (p=0.016), right-sided tumor origin (p<0.001), poor differentiation (p<0.001), KRAS mutations (p<0.001), BRAF mutations (p=0.004), and PIK3CA mutations (p=0.007). Patients with FBXW7 mutations had significantly worse OS (median OS 31.3 mo, 95% confidence interval [CI] 18.7-43.9 mo) than patients with wild-type FBXW7 (median OS 46.6 mo, 95% CI 43.0-50.1 mo; p=0.002 (Table 3, Figure 2A). Patients with FBXW7 missense mutations had significantly worse OS (median OS 28.7 mo, 95% CI 17.8-39.6 mo) than patients with other FBXW7 mutations (median OS 43.0 mo, 95% CI not reached (Table 3, Figure 2B). We also looked at FBXW7arg and other missense mutations but found no significant associated prognostic effects (Supplementary Figure 1).
Table 3: Survival analysis
Variables |
N |
Univariate analysis |
Multivariate analysis |
||||
|---|---|---|---|---|---|---|---|
Median survival (mo) |
95%CI |
P value |
HR |
95%CI |
P value |
||
Age |
|||||||
<50 years |
197 |
40.96 |
34.88-47.05 |
0.016 |
1.42 |
1.10-1.83 |
0.006 |
≥50 years |
373 |
47.57 |
43.58-51.56 |
Ref |
|||
Sex |
|||||||
Female |
251 |
45.47 |
41.19-49.75 |
0.842 |
|||
Male |
320 |
45.70 |
39.61-51.78 |
||||
Site |
|||||||
Rt.sided |
195 |
38.20 |
30.33-46.07 |
<0.001 |
1.46 |
1.03-2.05 |
0.031 |
Lt.sided |
243 |
50.27 |
44.92-55.62 |
0.98 |
0.71-1.34 |
0.891 |
|
Rectum |
128 |
48.33 |
40.44-56.22 |
Ref |
|||
Differentiated |
|||||||
Well to moderately |
424 |
48.26 |
44.34-52.19 |
<0.001 |
Ref |
||
Poorly |
141 |
36.49 |
28.61-44.38 |
1.52 |
1.18-1.96 |
0.001 |
|
KRAS |
|||||||
wt |
284 |
50.47 |
43.62-57.31 |
<0.001 |
Ref |
1.13-1.91 |
0.004 |
mt |
286 |
41.03 |
35.41-46.65 |
1.47 |
|||
NRAS |
|||||||
wt |
545 |
45.67 |
42.28-49.06 |
0.163 |
|||
mt |
26 |
37.18 |
29.37-45.00 |
||||
BRAF |
|||||||
wt |
524 |
46.55 |
42.72-50.39 |
0.004 |
Ref |
||
mt |
47 |
37.71 |
16.55-58.87 |
1.72 |
1.11-2.67 |
0.015 |
|
PIK3CA |
|||||||
wt |
477 |
47.24 |
43.55-50.94 |
0.007 |
Ref |
||
mt |
94 |
40.73 |
31.54-49.93 |
1.12 |
0.83-1.52 |
0.451 |
|
MMR status |
|||||||
Proficient |
395 |
45.67 |
40.82-50.51 |
0.073 |
Ref |
||
Deficient |
19 |
35.93 |
14.76-57.10 |
1.53 |
0.72-3.24 |
0.270 |
|
Unknown |
157 |
45.70 |
39.95-51.45 |
1.13 |
0.88-1.45 |
0.352 |
|
FBXW7 |
|||||||
wt |
527 |
46.55 |
43.00-50.11 |
0.003 |
Ref |
||
Missense mt |
37 |
28.67 |
17.76-39.58 |
2.0 |
1.27-3.16 |
0.003 |
|
Other mt |
6 |
42.97 |
Not reached |
0.98 |
0.24-4.08 |
0.980 |
|
wt: wild type, mt: mutation, Ref: reference
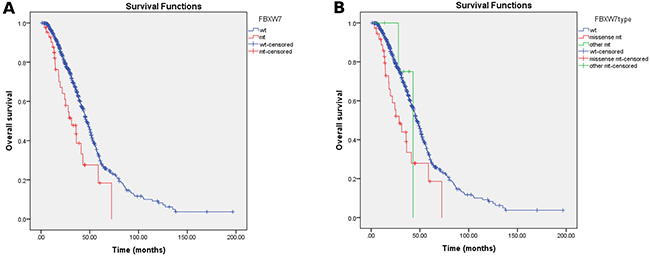
Figure 2: Kaplan-Meier survival curves according to FBXW7 status. (A) Patients with FBXW7 mutations had significantly worse OS (median OS 31.3 mo, 95% confidence interval [CI] 18.7-43.9 mo) than patients with wild-type FBXW7 (median OS 46.6 mo, 95% CI 43.0-50.1 mo; p = 0.002). (B) Patients with FBXW7 missense mutations had significantly worse OS (median OS 28.7 mo, 95% CI 17.8-39.6 mo) than patients with other FBXW7 mutations "(median OS 43.0 mo, 95% CI not reached; p = 0.003).
Multivariate Cox proportional hazards regression analysis of OS was performed using the factors mentioned above. In this analysis, FBXW7 missense mutations emerged as the strongest negative prognostic factor for OS (hazard ratio [HR] 2, 95% CI 1.3-3.2; p=0.003). Other factors associated with worse OS were an age of <50 years (HR 1.4, 95% CI 1.1-1.8; p=0.006), right-sided tumor origin (HR 1.5, 95% CI 1.0-2.1; p=0.031), poor differentiation (HR 1.5, 95% CI 1.2-2.0; p=0.001), KRAS mutations (HR 1.5, 95% CI 1.1-1.9; p=0.004), and BRAF mutations (HR 1.7, 95% CI 1.1-2.7; p=0.015) (Table 3, Figure 3).
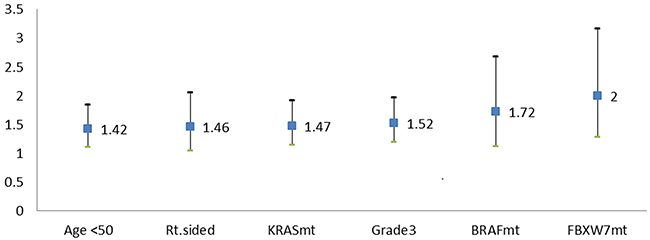
Figure 3: Negative prognostic factors for OS rate (HR with 95% CI). Factors associated with worse OS were an age of <50 years (HR 1.42), right-sided tumor origin (HR 1.46), KRAS mutations (HR 1.47), poor differentiation (HR 1.52), BRAF mutations (HR 1.72), and FBXW7 mutations (HR=2).
DISCUSSION
In this large cohort of mCRC patients, those who harbored FBXW7 mutations had significantly worse survival. We showed for the first time that FBXW7 missense mutations portend a worse prognosis in this population than do other type of mutations
In CRC, the frequency of FBXW7 mutations has been shown to vary between 6% and 10% and is consistently one of the most frequently mutated genes [24–27]. In our study, the frequency of FBXW7 mutations was 7.5%, which is consistent with prior studies. In a study by Akhoondi et al. [24] using 1556 samples from a wide range of cancers, including 523 CRC samples, the most frequently noted mutations were single-nucleotide changes, commonly missense mutations, of which nearly half (43%) occurred at two mutation hotspots positioned at codons Agr465 (29%) and Arg479 (14%). Other mutation hotspots detected in that study were at Ser582(4%), Arg278 (4%), Arg224 (3%), and Arg393 (3%). Jardim et al. [32] reported similar results in which the most common mutations were in two hot spots at codons Arg465 and Arg479.
In our study, most of the detected FBXW7 mutations were missense (37/43, 86%). FBXW7 mutations were most commonly found in codon Arg465 (15 cases, 34.9%) and were observed in codon Arg479in only three cases (7%). Interestingly, our study had a slightly higher frequency of FBXW7 mutations in codon Arg505 (eight cases, 18.6%) than previously described [24, 32]. S582L was noted in six cases (14%) and was the third most common FBXW7 mutation in our study. Chang et al. [33] reported a similar frequency of FBXW7 mutations (7.5%, 114/1519) in CRC patients but noted that Ser582 (S582L) was the most frequent type (19.3%), ahead of Arg465 (R465H, 16.6%), Arg505 (R505C, 14.9%), and Arg479 (R479Q, 14.9%). These variations are slight and within the realm of inter-study statistical variations. Furthermore, data from the Cancer Genome Atlas Network [34] project confirm that missense mutations affecting the hotspot arginine residues are the most common types in CRC, validating our findings.
FBXW7 missense mutations, especially those that are FBXW7Arg mutations, have a much more profound impact on substrate binding activity than other FBXW7 gene aberrations because these residues make up a critical substrate-binding interface, and thus disrupt substrate binding [29, 30]. Most of the remaining FBXW7 mutations are nonsense codons that lead to premature termination of FBXW7 translation that largely leads to production of non-functional alleles that may or may not dimerize. In contrast, full-length missense mutations result in alleles with dominant negative activity. Multiple mechanisms have been proposed for the more profound effects of missense mutations, including dominant negative effects on wild-type FBXW7 in dimers by retaining partial activity and stably binding to substrates, preventing their accessibility to wild-type FBXW7 monomers. Overall, these data suggest that missense mutations in a single allele of FBXW7 impair the activity of FBXW7 more profoundly than either allelic loss or stop codons. These data were confirmed in a CRC mouse model with FBXW7Arg/+ that showed increased tumorigenesis compared with FBXW7+/-mice. In this model, downstream FBXW7 substrates were elevated in FBXW7Arg/+ but not in FBXW7+/- tumors [35].
In our study, multivariate analysis demonstrated that FBXW7 missense mutations were independently associated with poor OS in mCRC and in fact were the strongest negative prognostic factor (HR 2.0) (Figure 3). In particular, FBXW7 missense mutations had a stronger prognostic association with OS than all FBXW7 mt or non-missense mutations. This finding confirms our hypothesis that FBXW7 mutation type has prognostic implications. Given the small numbers in the current study, these results will need to be confirmed in larger datasets.
In contrast to our study, several other CRC studies evaluating the prognostic effects of FBXW7 mutations have shown varying and even conflicting results from ours. In a study of 1,519 patients with CRC at all stages, Chang et al. [33] showed that FBXW7 mutations did not impact prognosis. In fact, in that study, subgroup analyses of FBXW7 mutations showed that R465H, R465C, and R479Q were associated with better 5-year OS rates than other FBXW7 mutation types were (76.9% vs 56.0%; p=0.012). Similarly, in the VICTOR trial report, FBXW7 mutations were not associated with disease-free survival in stage II/III CRC [36]. Finally, data from The Cancer Genome Atlas suggest that FBXW7 mutations were not associated with overall survival. How do we explain these disparate findings? One obvious explanation would be the difference in patient populations. All these datasets included predominantly early-stage disease, while our study was limited to patients with metastatic disease. The differential prognostic effect of a biomarker across advancing stages of CRC is a well-established concept. For instance, BRAF gene mutations are a known strong negative prognostic maker for stage IV CRC [2, 37] but not stage II-III CRC [38]. Similarly, MSI-H has positive prognostic effects in early-stage CRC but little if any in stage IV CRC [4]. Secondly, these studies looked at different end points, ranging from disease-free survival to OS. Finally, as demonstrated in multiple studies [29, 30, 35] missense mutations have much more profound implications than other mutations do, which was not specifically addressed in the prior studies. Future studies should be designed to specifically evaluate the role of missense mutations, especially in the metastatic setting.
Our study demonstrated the co-occurrence of FBXW7 mutations with PIK3CA mutations (p=0.012). However, after excluding PIK3CA variants in codon I391M which could be potential germline polymorphisms [39] (4 cases), there was only a tendency towards co-occurrence of these two mutations (P=0.10). The TCGA dataset [34] from 212 sequenced CRC cases suggests that FBXW7 mutations co-occur with BRAF mutations (p=0.03) and also have a tendency towards co-occurrence with PIK3CA mutations (p=0.25). It should be noted that TCGA dataset did not include PIK3CA I391M variants in their analysis. Alternatively, this difference might also be related to the difference in study populations with our study including only advanced stage of tumor while TCGA dataset included all stages with only 29/212 (13.7%) being stage IV. The correlation between FBXW7 mutations with other gene mutations across different stages needs to be validated in future studies. Lupini et al. reported that FBXW7 mutations were associated with resistance to anti-epidermal growth factor receptor antibodies (anti-EGFRab). The ability to predict response to anti-EGFRab in KRAS WT patients was significantly improved by adding FBXW7 to a NRAS-BRAF-PIK3CA mutation panel (P=0.016 and 0.045 respectively). In this study, the prevalence of FBXW7 mt was 17.8% (5/28 cases) and 2.7% (1/37 cases) in the non-responder and responder groups, respectively (P=0.08). [40]. However, this is a small dataset and requires further larger studies to confirm this effect.
An obvious direction of future work based on the current findings would be FBXW7-directed therapy. Directly, targeting FBXW7 mutations may be challenging given the disparate spectrum of changes noted and as yet unclear implications of the various aberrations. An easier and more direct approach might be targeting the downstream oncogenic substrates of FBXW7. Since FBXW7 has multiple substrates, it would be important to identify the most important ones in mCRC. Aydin et al. [41] reported that FBXW7 mutational inactivation represents a mechanism for NOTCH1 activation in melanoma and that anti-NOTCH treatment strategies showed promise in reduction of tumor growth in a xenograft model, making the NOTCH pathway a compelling target for therapeutic intervention. Similar findings have been noted in T-cell acute lymphocytic leukemia as well [42]. Intriguingly, a study by Sancho et al. in APCmin/+mouse models lacking FBXW7 activity in the intestine showed more aggressive adenomatous polyposis coli–mediated tumorigenesis and progression. These tumors were noted to have an abundance of Notch pathway substrates. These findings were confirmed in a small set of human CRC samples (five tumor and six control samples) that confirmed increased Notch activity, suggesting that a failure to antagonize Notch activity is an evolutionarily conserved mechanism of loss of function of FBXW7 [43]. Whether the Notch pathway is truly activated in CRC patients with FBXW7 mutations and whether the pathway would serve as a predictive biomarker for Notch pathway inhibitors will need to be evaluated.
Our study had several limitations. Firstly, it has the inherent drawbacks of a retrospective study. Secondly, our analysis of FBXW7 function was limited to only hotspot FBXW7 gene mutations, and therefore it is likely we missed other mechanisms leading to the loss of function of this protein, including other gene aberrations (such as truncation or deletion) or post-translational modifications. However, to the best of our knowledge, this is the first study to report the clinical characteristics and outcomes associated with FBXW7 missense mutations in mCRC and show the strong negative prognostic effects of such mutations.
In summary, this is the first and largest clinical dataset of FBXW7 missense mutations which showed negative prognostic impact on survival in mCRC. Since FBXW7 was found more frequently in CRC and since most FBXW7 substrates are oncoproteins, further studies are required to identify downstream pathways underlying this worse prognosis and potential therapeutic targets.
MATERIALS AND METHODS
In this single-institution, retrospective study, medical records of patients with mCRC who were enrolled in the Assessment of Targeted Therapies Against Colorectal Cancer program at The University of Texas MD Anderson Cancer Center between February 13, 2009, and November 18, 2015, were reviewed, and only patients who had next-generation sequencing data available were included in the study. The study protocol was approved by the MD Anderson Cancer Center institutional review board. All patients provided written informed consent for sequencing of their tumors according to institutional guidelines. The primary objective of this study was to determine the prognostic effect of FBXW7 missense mutations on overall survival (OS); the secondary objectives were to examine associations between FBXW7 mutations and various clinicopathologic characteristics and to evaluate the prognostic effect of all FBXW7 mutations, irrespective of type, on OS.
Clinical characteristics
Demographic information including age, sex, race, primary tumor site, date of diagnosis with stage IV disease, date of last follow-up, and date of death were collected from the review of the medical records. Right-sided colon cancer was defined as cancer in the region from the cecum to the splenic flexure, while left-sided colon cancer was defined as cancer in the region from the descending colon through the sigmoid colon, and the rectum was considered a separate site. Staging was done per the American Joint Committee on Cancer/Union for International Cancer Control TMN staging system (version 7, 2010) [44]. OS was defined as the interval between the date of diagnosis of metastatic disease and the date of death from any cause; patients alive at the time of analysis were censored at their last known follow-up date.
Molecular characterization
DNA was extracted from paraffin-embedded formalin-fixed tumor tissue. Samples were evaluated by using a next-generation sequencing platform with 46- or 50-gene panels for the detection of frequently reported point mutations in human malignancies in a Clinical Laboratory Improvement Amendments–certified molecular diagnostics laboratory which determined the effective lower limit of detection (analytical sensitivity) for single nucleotide variations to be in the range of 5% (one mutant allele in the background of nineteen wild type alleles) to 10% (one mutant allele in the background of nine wild type alleles). Details of codons and exons tested in KRAS, NRAS, BRAF, PIK3CA, and FBXW7 are shown in (Supplementary Table 4).
Determination of MMR status by microsatellite instability testing
MMR status was determined by immunohisto-chemical analysis of MMR protein expression or by polymerase chain reaction analysis of microsatellite instability. Deficient MMR was defined as the presence of high-level microsatellite instability on polymerase chain reaction and/or as the loss of MMR protein expression on immunohistochemical analysis. Proficient MMR was defined as the presence of microsatellite stability or low-level microsatellite instability on polymerase chain reaction and/or as the presence of normal MMR protein expression on immunohistochemical analysis.
Immunohistochemical analysis of MMR expression
Immunoperoxidase stains were performed on sections from formalin-fixed paraffin-embedded tissues with antibodies for the DNA mismatch repair enzymes MLH1, MSH2, MSH6, and PMS2. Staining for MMR proteins was performed using the following primary antibodies: mouse anti-human MLH1 (clone G168-728; Cell Marque Corporation, Rocklin, CA), 1:300 dilution; mouse anti-human MSH2 (clone FE11; Calbiochem/Oncogene Research Products, Cambridge, MA), 1:100 dilution; mouse anti-human MSH6 (clone 44; BD Biosciences, San Jose, CA), 1:300 dilution; and mouse anti-human PMS2 (clone A16-4; BD Biosciences), 1:125 dilution. Loss of MMR protein was defined as the absence of nuclear staining of tumor cells in the presence of positive nuclear staining in normal epithelial cells and lymphocytes. Interpretation of immunohistochemistry for mismatch repair status is shown in (Supplementary Table 5).
Polymerase chain reaction analysis for microsatellite instability testing
DNA extracted from microdissected paraffin-embedded tumor sections and non-neoplastic tissues was analyzed by a polymerase chain reaction–based method followed by capillary electrophoretic detection. A panel of seven microsatellite markers (BAT25, BAT26, BAT40, D2S123, D5S346, D17S250, and TGFBR2) was evaluated to detect changes in the number of microsatellite repeats in tumor tissue compared with normal tissue. Tumors were classified as MSI-H (> 40% of markers altered), MSI-L (< 40% of markers altered) and MSS (no marker altered).
Statistical analysis
Patient characteristics were summarized with descriptive statistics. The relationships between clinicopathologic variables and FBXW7 status were assessed using Pearson’s χ2 or Fisher exact test as appropriate. The associations of patient and molecular characteristics with OS were assessed by Kaplan-Meier estimation, log-rank test, and Cox proportional hazards regression models. Median follow-up time was calculated using the reverse Kaplan-Meier method. All tests were two-sided, and p<0.05 was considered statistically significant. Calculations were carried out using SPSS version 23.0 software (IBM Corporation, Armonk, NY).
Abbreviations
CRC: colorectal cancer; mCRC: metastatic colorectal cancer; OS: overall survival; MMR: mismatch repair; MCL1: myeloid cell leukemia 1; AURKA: Aurora kinase A; KLF5: Krüppel-like factor 5; MSI: Microsattelite instability; anti-EGFRab: anti-epidermal growth factor receptor antibodies
Author contributions
KK performed the research, data collection, acquired and analyzed the data, and drafted the manuscript. SM participated in data collection. VKM, MJO, DRF, BK, KPSR, IS, KE, DM, SRH, and SK participated in revised the manuscript for important intellectual content. AD designed the study, supervised the study, critically revised the manuscript, and approved the final version for publication. All authors read, reviewed, and approved the final manuscript.
ACKNOWLEDGMENTS
The University of Texas MD Anderson Cancer Center is supported in part by the National Institutes of Health through Cancer Center Support Grant P30CA016672.
CONFLICTS OF INTEREST
The authors declare that no conflicts of interest exist.
FUNDING
The University of Texas MD Anderson Cancer Center is supported by the NCI Core Center Grant 5P30CA016672-39
REFERENCES
1. Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988; 319: 525-532.
2. Therkildsen C, Bergmann TK, Henrichsen-Schnack T, Ladelund S, Nilbert M. The predictive value of kras, nras, braf, pik3ca and pten for anti-egfr treatment in metastatic colorectal cancer: a systematic review and meta-analysis. Acta Oncol. 2014; 53: 852-864.
3. Rowland A, Dias MM, Wiese MD, Kichenadasse G, McKinnon RA, Karapetis CS, Sorich MJ. Meta-analysis of braf mutation as a predictive biomarker of benefit from anti-egfr monoclonal antibody therapy for ras wild-type metastatic colorectal cancer. Br J Cancer. 2015; 112: 1888-1894.
4. Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005; 23: 609-618.
5. Welcker M, Clurman BE. Fbw7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008; 8: 83-93.
6. Spruck CH, Strohmaier H, Sangfelt O, Müller HM, Hubalek M, Müller-Holzner E, Marth C, Widschwendter M, Reed SI. Hcdc4 gene mutations in endometrial cancer. Cancer Res. 2002; 62: 4535-4539.
7. Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-dependent ubiquitination of cyclin e by the scffbw7 ubiquitin ligase. Science. 2001; 294: 173-177.
8. Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase scffbw7 antagonizes apoptotic jnk signaling. Science. 2004; 303: 1374-1378.
9. Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG. The v-jun point mutation allows c-jun to escape gsk3-dependent recognition and destruction by the fbw7 ubiquitin ligase. Cancer Cell. 2005; 8: 25-33.
10. Welcker M, Orian A, Jin J, Grim JE, Grim JA, Harper JW, Eisenman RN, Clurman BE. The fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-myc protein degradation. Proc Natl Acad Sci U S A. 2004; 101: 9085-9090.
11. Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-myc is mediated by the f-box protein fbw7. EMBO J. 2004; 23: 2116-2125.
12. Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, Xiao Y, Christie AL, Aster J, et al. Scf(fbw7) regulates cellular apoptosis by targeting mcl1 for ubiquitylation and destruction. Nature. 2011; 471: 104-109.
13. Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, Belmont LD, Kaminker JS, O’Rourke KM, et al. Sensitivity to antitubulin chemotherapeutics is regulated by mcl1 and fbw7. Nature. 2011; 471: 110-114.
14. Snyder JL, Kearns CA, Appel B. Fbxw7 regulates notch to control specification of neural precursors for oligodendrocyte fate. Neural Dev. 2012; 7: 15.
15. Fryer CJ, White JB, Jones KA. Mastermind recruits cycc:Cdk8 to phosphorylate the notch icd and coordinate activation with turnover. Mol Cell. 2004; 16: 509-520.
16. Tetzlaff MT, Yu W, Li M, Zhang P, Finegold M, Mahon K, Harper JW, Schwartz RJ, Elledge SJ. Defective cardiovascular development and elevated cyclin e and notch proteins in mice lacking the fbw7 f-box protein. Proc Natl Acad Sci U S A. 2004; 101: 3338-3345.
17. Tsunematsu R, Nakayama K, Oike Y, Nishiyama M, Ishida N, Hatakeyama S, Bessho Y, Kageyama R, Suda T, Nakayama KI. Mouse fbw7/sel-10/cdc4 is required for notch degradation during vascular development. J Biol Chem. 2004; 279: 9417-9423.
18. Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. Fbxw7/cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004; 432: 775-779.
19. Kwon YW, Kim IJ, Wu D, Lu J, Stock WA, Liu Y, Huang Y, Kang HC, DelRosario R, Jen KY, Perez-Losada J, Wei G, Balmain A, et al. Pten regulates aurora-a and cooperates with fbxw7 in modulating radiation-induced tumor development. Mol Cancer Res. 2012; 10: 834-844.
20. Wang R, Wang Y, Liu N, Ren C, Jiang C, Zhang K, Yu S, Chen Y, Tang H, Deng Q, Fu C, Li R, Liu M, et al. Fbw7 regulates endothelial functions by targeting klf2 for ubiquitination and degradation. Cell Res. 2013; 23: 803-819.
21. Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, Balmain A. Fbxw7 targets mtor for degradation and cooperates with pten in tumor suppression. Science. 2008; 321: 1499-1502.
22. Bengoechea-Alonso MT, Ericsson J. Tumor suppressor fbxw7 regulates tgfβ signaling by targeting tgif1 for degradation. Oncogene. 2010; 29: 5322-5328.
23. Cao J, Ge MH, Ling ZQ. Fbxw7 tumor suppressor: a vital regulator contributes to human tumorigenesis. Medicine (Baltimore). 2016; 95: e2496.
24. Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Dofou D, Marth C, Mueller-Holzner E, Corcoran M, et al. Fbxw7/hcdc4 is a general tumor suppressor in human cancer. Cancer Res. 2007; 67: 9006-9012.
25. Malapelle U, Pisapia P, Sgariglia R, Vigliar E, Biglietto M, Carlomagno C, Giuffrè G, Bellevicine C, Troncone G. Less frequently mutated genes in colorectal cancer: evidences from next-generation sequencing of 653 routine cases. J Clin Pathol. 2016.
26. Kemp Z, Rowan A, Chambers W, Wortham N, Halford S, Sieber O, Mortensen N, von Herbay A, Gunther T, Ilyas M, Tomlinson I. Cdc4 mutations occur in a subset of colorectal cancers but are not predicted to cause loss of function and are not associated with chromosomal instability. Cancer Res. 2005; 65: 11361-11366.
27. Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C. Inactivation of hcdc4 can cause chromosomal instability. Nature. 2004; 428: 77-81.
28. Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. Skp1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the f-box. Cell. 1996; 86: 263-274.
29. Orlicky S, Tang X, Willems A, Tyers M, Sicheri F. Structural basis for phosphodependent substrate selection and orientation by the scfcdc4 ubiquitin ligase. Cell. 2003; 112: 243-256.
30. Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a fbw7-skp1-cyclin e complex: multisite-phosphorylated substrate recognition by scf ubiquitin ligases. Mol Cell. 2007; 26: 131-143.
31. Welcker M, Clurman BE. Fbw7/hcdc4 dimerization regulates its substrate interactions. Cell Div. 2007; 2: 7.
32. Jardim DL, Wheler JJ, Hess K, Tsimberidou AM, Zinner R, Janku F, Subbiah V, Naing A, Piha-Paul SA, Westin SN, Roy-Chowdhuri S, Meric-Bernstam F, Hong DS. Fbxw7 mutations in patients with advanced cancers: Clinical and molecular characteristics and outcomes with mtor inhibitors. PLoS One. 2014; 9: e89388.
33. Chang CC, Lin HH, Lin JK, Lin CC, Lan YT, Wang HS, Yang SH, Chen WS, Lin TC, Jiang JK, Chang SC. Fbxw7 mutation analysis and its correlation with clinicopathological features and prognosis in colorectal cancer patients. Int J Biol Markers. 2015; 30: e88-95.
34. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487: 330-337.
35. Davis H, Lewis A, Behrens A, Tomlinson I. Investigation of the atypical fbxw7 mutation spectrum in human tumours by conditional expression of a heterozygous propellor tip missense allele in the mouse intestines. Gut. 2014; 63: 792-799.
36. Mouradov D, Domingo E, Gibbs P, Jorissen RN, Li S, Soo PY, Lipton L, Desai J, Danielsen HE, Oukrif D, Novelli M, Yau C, Holmes CC, et al. Survival in stage ii/iii colorectal cancer is independently predicted by chromosomal and microsatellite instability, but not by specific driver mutations. Am J Gastroenterol. 2013; 108: 1785-1793.
37. Maughan TS, Adams RA, Smith CG, Meade AM, Seymour MT, Wilson RH, Idziaszczyk S, Harris R, Fisher D, Kenny SL, Kay E, Mitchell JK, Madi A, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 mrc coin trial. Lancet. 2011; 377: 2103-2114.
38. Shen Y, Han X, Wang J, Wang S, Yang H, Lu SH, Shi Y. Prognostic impact of mutation profiling in patients with stage ii and iii colon cancer. Sci Rep. 2016; 6: 24310.
39. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, et al. Cosmic: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015; 43: D805-811.
40. Lupini L, Bassi C, Mlcochova J, Musa G, Russo M, Vychytilova-Faltejskova P, Svoboda M, Sabbioni S, Nemecek R, Slaby O, Negrini M. Prediction of response to anti-egfr antibody-based therapies by multigene sequencing in colorectal cancer patients. BMC Cancer. 2015; 15: 808.
41. Aydin IT, Melamed RD, Adams SJ, Castillo-Martin M, Demir A, Bryk D, Brunner G, Cordon-Cardo C, Osman I, Rabadan R, Celebi JT. Fbxw7 mutations in melanoma and a new therapeutic paradigm. J Natl Cancer Inst. 2014; 106: dju107.
42. O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, Draetta G, Sears R, Clurman BE, et al. Fbw7 mutations in leukemic cells mediate notch pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007; 204: 1813-1824.
43. Sancho R, Jandke A, Davis H, Diefenbacher ME, Tomlinson I, Behrens A. F-box and wd repeat domain-containing 7 regulates intestinal cell lineage commitment and is a haploinsufficient tumor suppressor. Gastroenterology. 2010; 139: 929-941.
44. Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010; 17: 1471-1474.