
INTRODUCTION
Recurrent and metastatic sarcomas are a rare and heterogeneous group of diseases. With well over 70 subtypes, the exact diagnosis alone can be challenging to make [1]. When sarcomas progress beyond efficacious local control, the standard practice with few notable exceptions is to treat with cytotoxic agents until progression or intolerance [2]. Unfortunately, these cytotoxic agents yield overall response rates of around 25% [3, 4]. Over the last three decades recurrent translocations have been found that drive the development of certain sarcomas and are now used as an adjunctive diagnostic tool [5, 6]. Unfortunately, in clinical practice most sarcoma therapies are not yet targeting these unique and simple fusions. While the transcription factor fusions pose an enormous drug development challenge, the kinase fusions are potentially targetable with current technology. A notable exception is non-fusion genomic alterations in kinases, with gastrointestinal stromal tumors serving as the paradigm of druggable c-Kit alterations by imatinib [7, 8]. A separate sub-group of sarcomas have complex cytogenetic changes hallmarked by genomic instability and are not characterized by discrete gene fusions [9].
The promise of personalized medicine has become a realization for many malignancies. Pairing genomic alterations and targeted therapy has transformed diseases like lung cancer, leukemia, and breast cancer [10-12]. Outside of gastrointestinal stromal tumors, inflammatory myofibroblastic tumors, and PECOMAs [13], targeted therapies in sarcomas have not seen such breakthroughs. Perhaps it is the staggering heterogeneity of the disease, relative rarity, or difficulty making a definitive diagnosis outside tertiary care centers that makes it challenging [14-20]. To compound the problem, some of the sarcomas are a group of biologically complex and resistant diseases and, outside of surgically curable local disease, portend an exceptionally poor prognosis when metastatic [21, 22]. A large portion of these patients will be referred for clinical trials partly because of their young age, preserved performance status, or scarcity of treatment options [23, 24]. To date much has been published about potentially targetable alterations, but clinical translation in sarcoma has been minimal [25-31]. We undertook a systematic analysis of potentially druggable alterations in sarcomas based on comprehensive genomic profiling (CGP) performed in the course of clinical care and evaluated clinical response in patients receiving molecularly matched therapies. Here we present 102 sarcoma patients that were referred to the Investigational Therapeutics Department which is the phase 1 clinical trials program at MD Anderson Cancer Center.
RESULTS
All patients in this study had advanced or metastatic relapsed or refractory sarcoma or did not have any other standard care therapies available when they presented for clinical trials. There were 48 men (47%) and 54 women (53%) in this patient cohort with a median age at diagnosis of 45.5 years (range 8-76). There were 36 patients (35%) who had primary site biopsied and sent for next generation sequencing (NGS) and 66 (65%) who had a metastatic site sent for NGS. Of the 102 patients, 38 were metastatic at time of diagnosis. The most common histologies seen in our study was leiomyosarcoma (8 uterine and 11 non-uterine, 18.6%), dedifferentiated liposarcoma (11%), osteosarcoma (11%), well-differentiated liposarcoma (7%), carcinosarcoma (6%), and rhabdomyosarcoma (6%) (Table 1). All tumors were reviewed and histology confirmed by MD Anderson pathology department. The most frequently seen genomic alterations are summarized in (Figure 1A). Ninety-five out of 102 patients (93%) had at least one genomic alteration identified with a mean of six alterations per patient. The vast majority (49%) of all alterations were amino acid substitutions. Amplifications were the second most common alteration (31%). The remaining alterations were split as described in (Figure 1B). The most commonly altered genes were TP53 (31.4%), CDK4 (23.5%), MDM2 (21.6%), RB1 (18.6%), and CDKN2A/B (13.7%) (Figure 1A).
Table 1: Patient characteristics
Patient Characteristics |
||
|---|---|---|
Median age at Dx |
45.5 |
8-76 years |
#Men |
48 |
47.06% |
#Women |
54 |
52.94% |
Race |
||
Caucasian |
78 |
76.47% |
AA |
10 |
9.80% |
Hispanic |
13 |
12.75% |
Asian |
1 |
0.98% |
Histology |
||
LEIOMYOSARCOMA |
19 |
18.63% |
DEDIFFERENTIATED LIPOSARCOMA |
11 |
10.78% |
OSTEOSARCOMA |
11 |
10.78% |
WELL DIFFERENTIATED LIPOSARCOMA |
7 |
6.86% |
CARCINOSARCOMA |
6 |
5.88% |
RHABDOMYOSARCOMA (NOS) |
6 |
5.88% |
GASTROINTESTINAL STROMAL TUMOR |
5 |
4.90% |
SPINDLE CELL SARCOMA |
5 |
4.90% |
SYNOVIAL SARCOMA |
4 |
3.92% |
ALVEOLAR SOFT PART SARCOMA |
3 |
2.94% |
CHONDROSARCOMA |
4 |
3.92% |
CHORDOMA |
3 |
2.94% |
CLEAR CELL SARCOMA |
3 |
2.94% |
EWING SARCOMA |
3 |
2.94% |
UNCLASSIFIED |
3 |
2.94% |
ALVEOLAR RHABDOMYOSARCOMA |
2 |
1.96% |
FIBROSARCOMA |
2 |
1.96% |
BRAIN GLIOSARCOMA |
1 |
0.98% |
DESMOPLASTIC SMALL ROUND CELL TUMOR |
1 |
0.98% |
PLEOMORPHIC SARCOMA |
1 |
0.98% |
MALIGNANT PERIPHERAL NERVE SHEATH |
1 |
0.98% |
MYXOIDLIPOSARCOMA |
1 |
0.98% |
102 |
100.00% |
|
Metastasis at diagnosis |
38 |
|
Metastasis at biopsy |
86 |
|
Biopsy site |
||
Primary |
36 |
35% |
Metastasis |
66 |
65% |
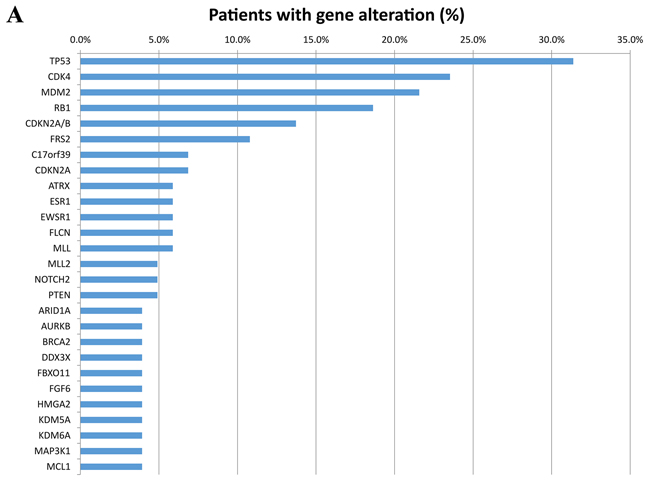
Figure 1A: Frequency of the most common genes altered by percentage of 102 patients with diverse sarcomas. Only alterations seen in at least 4% of patients are included. Different alteration in the same gene are listed under the same gene name.
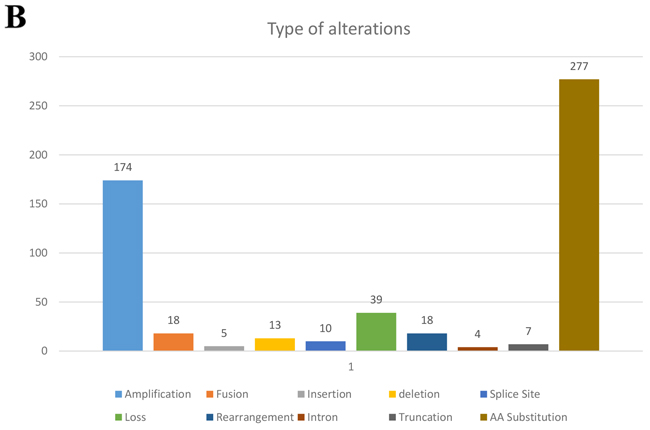
Figure 1B: Types of gene alterations seen as a percentage of 102 patients with diverse sarcomas.
Two of the most common alterations (MDM2 and CDK4 amplifications) are both actionable. MDM2 was altered in 22 patients and all were amplifications. All 22 of these patients also had CDK4 amplifications. One additional patient had a CDK4 amplification without MDM2 amplification. Ten of these co-amplified CDK4/MDM2 cases were dedifferentiated liposarcomas, seven were well-differentiated liposarcomas, two rhabdomyosarcomas (one pleomorphic and one nos), one osteosarcoma, one ewing sarcoma, and one unclassified soft tissue sarcoma. Four of the well-differentiated liposarcoma patients were treated with an investigational MDM2 inhibitor and all achieved at least stable disease, some showing a very durable response.
Other notable mutations include three FRS2 and FGF co-amplifications seen in a rhabdomyosarcoma, osteosarcoma, and dedifferentiated liposarcoma. The rhabdomyosarcomas were FOXO1 fusion-negative, hinting at a higher number of mutations [32]. We also identified previously reported fusions of SSX with SS18 in synovial sarcoma, as well as HMGA2 in liposarcoma. Three of the leiomyosarcomas had a mutation in the Lynch syndrome gene MSH2 (two uterine and one non-uterine). A complete list of all identified mutations can be seen in (Table 2).
Table 2: All identified mutations from the NGS panel
ABL1 |
CCT6B |
FAM123B |
IGF1R |
MSH6 |
RANBP2 |
WHSC1 |
ACTB |
CD274 |
FAM46C |
IL7R |
MTOR |
RB1 |
WT1 |
AKT1 |
CD36 |
FANCA |
INPP4B |
MYC |
RELN |
YY1AP1 |
AKT2 |
CD70 |
FANCD2 |
INPP5D |
MYCL1 |
RICTOR |
ZNF703 |
AKT3 |
CDK12 |
FANCE |
IRF2 |
MYO18A |
ROS1 |
|
ALK |
CDK4 |
FAS |
IRS2 |
MYST3 |
RUNX1 |
|
APC |
CDKN2A |
FAT1 |
JAK1 |
NF1 |
RUNX1T1 |
|
APH1A |
CDKN2A/B |
FBXO11 |
JAK2 |
NF2 |
SETD2 |
|
AR |
CEBPA |
FBXW7 |
JAK3 |
NFKBIA |
SMARCA1 |
|
ARID1A |
CHD2 |
FDF23 |
JUN |
NKX2-1 |
SMARCA4 |
|
ARID1B |
CHEK2 |
FGF10 |
KDM5A |
NOD1 |
SMARCB1 |
|
ASXL1 |
CIC |
FGF14 |
KDM5C |
NOTCH1 |
SMC1A |
|
ATM |
CIITA |
FGF23 |
KDM6A |
NOTCH2 |
SOCS2 |
|
ATR |
CPS1 |
FGF6 |
KDR |
NRAS |
SPOP |
|
ATRX |
CREBBP |
FGFR1 |
KEAP1 |
nsT |
SPTA1 |
|
AURKA |
CSF1R |
FGFR2 |
KIT |
NTRK1 |
SSX |
|
AURKB |
CTNNB1 |
FLCN |
KRAS |
NTRK3 |
SSX2 |
|
BARD1 |
CUX1 |
FLT4 |
LRP1B |
PAG1 |
STAG2 |
|
BCL11B |
DAXX |
FLYWCH1 |
LYN |
PAK3 |
STAT5B |
|
BCL2A1 |
DDIT3 |
FOXO3 |
MAFB |
PALB2 |
STAT6 |
|
BCL2L2 |
DDR2 |
FRS2 |
MALT1 |
PASK |
STK11 |
|
BCOR |
DDX3X |
gement |
MAP2K2 |
PAX5 |
SUFU |
|
BCORL1 |
DNM2 |
GNA12 |
MAP2K4 |
PC |
SYK |
|
BIRC3 |
DNMT3A |
GNAS |
MAP3K1 |
PCLO |
TCL1A |
|
BLM |
DOT1L |
GPR124 |
MAP3K14 |
PDCD1LG2 |
TET2 |
|
BRAF |
DTX1 |
GRIN2A |
MCL1 |
PDGFRA |
TGFBR2 |
|
BRCA1 |
EBF1 |
HDAC4 |
MDM2 |
PDGFRB |
TLL2 |
|
BRCA2 |
EGFR |
HGF |
MDM4 |
PIK3CA |
TNFAIP3 |
|
BRD4 |
EMSY |
HIST1H1C |
MED12 |
PIK3R1 |
TNFRSF17 |
|
BTG1 |
EP300 |
HIST1H1D |
MET |
PIM1 |
TOP1 |
|
C17orf39 |
EPHA5 |
HIST1H2AC |
MIB1 |
PRDM1 |
TOP2A |
|
CARD11 |
EPHA7 |
HIST1H2AG |
MKI67 |
PRKDC |
TP53 |
|
CBFB |
EPHB1 |
HLGGSSCSTC |
MLL |
PTCH1 |
TSC1 |
|
CBL |
ERBB4 |
HMGA2 |
MLL2 |
PTEN |
TSC2 |
|
CCND1 |
ERG |
HSP90AA1 |
MLL3 |
PTPN11 |
TSHR |
|
CCND2 |
ESR1 |
ICK |
MPL |
PTPRO |
TYK2 |
|
CCND3 |
EWSR1 |
IDH1 |
MSH2 |
RAD21 |
VHL |
|
CCNE1 |
EWSR1-NFATC2 |
IDH2 |
MSH3 |
RAD50 |
WDR90 |
Of the 102 patients in our cohort, forty (39%) had either no reported mutation (7%) or no actionable mutation (32%). The remaining 62 (61%) patients all had a potentially actionable alteration. Fourteen (14%) patients had an alteration that could be targeted with an approved drug in sarcoma (on-label). This was either an off-target effect of pazopanib or imatinib and included five patients with PDGFR (1 GIST), four with FGFR, three with KIT (2 GIST), and two with KDR gene aberrations.
Forty-six (45%) patients had an alteration that could be targeted with a drug approved in another disease (off-label). Sixty-one (60%) patients had an alteration that could potentially be targeted by a drug currently available in clinical trials and, barring particular exclusion criteria, all of them could have been enrolled on a matching trial. Fifty-eight (57%) had an alteration for which a drug currently in pre-clinical development could be used (Figure 2).
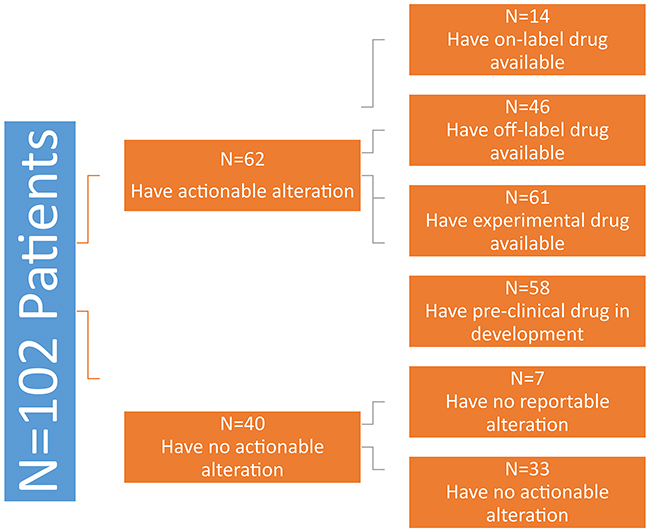
Figure 2: Number of sarcoma patients with actionable mutations divided by drug availability. Patients had overlap between approved, off-label, and experimental drug options.
A subtype analysis of all sarcoma types revealed that the probability of having an actionable mutation was related to histology (Figure 3). Notable sarcoma subtypes include dedifferentiated liposarcoma (100%), well-differentiated liposarcoma (100%), and carcinosarcoma (83%) all of which had an exceptional number of patients with actionable mutations. As noted above, dedifferentiated/well-differentiated liposarcomas had a preponderance of MDM2 and CDK4 mutations. Carcinosarcomas had targetable mutations in AKT2 and harbored a resistance mutation in ESR1 (Table 3).
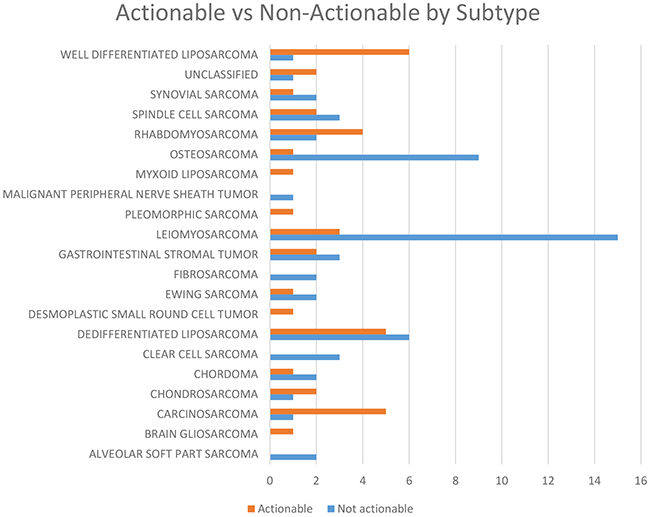
Figure 3: Number of patients with actionable as compared to non-actionable distributed by sarcoma subtype.
Table 3: Actionable alteration by sarcoma subtype
Histology (patients) |
No reportable alteration, n (%) |
Patients had alteration(s), but none actionable, n (%) |
Patients with approved drug(s) in the disease available, n (%) (on-label) |
Patients with approved drug(s) in another disease available, n (%) (off-label) |
Patients with experimental treatment options (clinical trials), n (%) |
Patients with pre-clinical treatment options, n(%) |
|---|---|---|---|---|---|---|
LEIOMYOSARCOMA |
1 |
10 |
3 |
6 |
8 |
7 |
DEDIFFERENTIATED LIPOSARCOMA |
0 |
0 |
11 |
10 |
10 |
|
OSTEOSARCOMA |
1 |
6 |
1 |
2 |
4 |
4 |
WELL DIFFERENTIATED LIPOSARCOMA |
0 |
1 |
7 |
7 |
7 |
|
CARCINOSARCOMA |
0 |
3 |
5 |
6 |
6 |
|
RHABDOMYOSARCOMA |
1 |
1 |
2 |
5 |
5 |
|
GASTROINTESTINAL STROMAL TUMOR |
1 |
3 |
3 |
4 |
4 |
|
SPINDLE CELL SARCOMA |
1 |
1 |
2 |
4 |
3 |
|
SYNOVIAL SARCOMA |
1 |
2 |
0 |
1 |
1 |
1 |
ALVEOLAR SOFT PART SARCOMA |
1 |
1 |
0 |
1 |
1 |
1 |
CHONDROSARCOMA |
1 |
1 |
0 |
0 |
2 |
1 |
CHORDOMA |
1 |
0 |
1 |
2 |
2 |
|
CLEAR CELL SARCOMA |
3 |
0 |
0 |
0 |
0 |
|
EWING SARCOMA |
2 |
0 |
1 |
1 |
1 |
|
UNCLASSIFIED |
1 |
0 |
1 |
2 |
2 |
|
ALVEOLAR RHABDOMYOSARCOMA |
2 |
0 |
0 |
0 |
0 |
0 |
FIBROSARCOMA |
2 |
0 |
0 |
0 |
0 |
|
BRAIN GLIOSARCOMA |
0 |
0 |
1 |
1 |
1 |
|
DESMOPLASTIC SMALL ROUND CELL TUMOR |
0 |
0 |
1 |
1 |
1 |
|
PLEOMORPHIC SARCOMA |
0 |
1 |
1 |
1 |
1 |
|
MALIGNANT PERIPHERAL NERVE SHEATH TUMOR |
1 |
0 |
0 |
0 |
0 |
|
MYXOID LIPOSARCOMA |
0 |
0 |
0 |
1 |
1 |
|
7 |
33 |
14 |
46 |
61 |
58 |
|
6.86% |
32.35% |
13.73% |
45.10% |
59.80% |
56.86% |
Clinical response was highly variable. Forty-three patients (42%) only received tumor sequencing, but did not participate in a clinical trial. The remaining fifty-nine patients (58%) chose to participate in a clinical trial. Of these, sixteen (16%) received therapy directed by molecular profile. Of these sixteen patients, eight (50%) had at least stable disease (Table 4).
Table 4: Results of sixteen sarcoma patients treated with targeted therapy based on NGS results
Patients treated with targeted therapy based on NGS result |
||||
|---|---|---|---|---|
Histology |
Gene |
Mutation |
Treatment and Best response |
Comments and Referenes |
BRAIN GLIOSARCOMA |
BRAF |
V600E |
vemurafenib --> PR |
86% decrease, duration of Response 16 months [44] |
CARCINOSARCOMA |
ESR1 |
A569T |
anastrozole plus everolimus --> PD |
IHC for PTEN was positive. ER was 3+ per IHC. ESR1 is a resistance mutation |
DEDIFFERENTIATED LIPOSARCOMA |
ROS1 |
amplification |
ceritinib --> SD |
Best response SD x5 months [20] |
DEDIFFERENTIATED LIPOSARCOMA |
MDM2 |
amplification |
MDM2 inhibitor --> PR |
Best response PR x3 cycles |
GASTROINTESTINAL STROMAL TUMOR |
KIT, |
amplification |
Imatinib - PD sutent -PD, regorafenib-PD, |
Best response PR, progressed after 22 cycles. Initially dx as wt kit and pdgfr, FM later showed akt, kit, mdm4, MCL1 amplification |
LEIOMYOSARCOMA |
ROS1 |
D1538V |
pazopanib and crizotinib --> SD |
SD x 6 months |
LEIOMYOSARCOMA |
PTEN |
Loss |
PI3K Inhibitor --> PD |
Deceased after 3 days on study |
LEIOMYOSARCOMA |
ROS1 |
D1538V |
pazopanib and crizotinib --> PD |
Patient deceased prior to restaging scans |
PLEOMORPHIC SARCOMA |
ALK |
MEMO1-ALK fusion |
ceritinib --> PD |
Progressed after 4 cycles [20] |
MYXOID LIPOSARCOMA |
AKT1 |
E17K |
AKT inhibitor --> SD |
Stopped after 1 cycle due to ggt elevation |
OSTEOSARCOMA |
PDGFRA |
amplification |
Sorafenib, Avastin, and Torisel --> PD |
PD after 1 cycle [29] |
SPINDLE CELL SARCOMA |
BRAF |
KIAA1549-BRAF fusion |
Sorafenib, Avastin, and Torisel --> SD |
Best response 28% reduction per RECIST. Also PTEN Loss. SD for 11 cycles, until death [31]. |
WELL DIFFERENTIATED LIPOSARCOMA |
MDM2 |
amplification |
MDM-2 --> SD |
Best response SD x8 cycles |
WELL DIFFERENTIATED LIPOSARCOMA |
MDM2 |
amplification |
MDM2 inhibitor --> CR |
On since 2008, has had several resections during this period. Now NED again |
WELL DIFFERENTIATED LIPOSARCOMA |
MDM2 |
amplification |
MDM2/MDMX inhibitor --> SD |
Stopped after 2 cycles due to side effects |
WELL DIFFERENTIATED LIPOSARCOMA |
MDM2 |
amplification |
MDM2 inhibitor --> SD |
SD x23 months, stopped due to patient preference |
All patients were treated on clinical trial. Eight patients had clinical benefit as defined by at least stable disease.
DISCUSSION
Overall survival continues to be poor in metastatic sarcoma as a group. With small numbers and large diversity of subtypes, even the prospect of initiating and accruing a study in this population is daunting. Given the success of targeted therapy in other diseases, we sought to discover if CGP could aid in diagnosis and treatment of sarcomas as a whole. Using CGP we discovered that 61% of our patients had a potentially actionable mutation which could be targeted with either an off-label or an investigational therapeutic available in a clinical trial. A relative minority of samples had alterations targetable by on-label drugs (Pazopanib or imatinib). This is almost certainly driven by the paucity of approved (targeted) therapies in sarcoma. There was a skew toward certain histologic subtypes that harbor more potentially actionable mutations. Well-differentiated and dedifferentiated liposarcomas as well as carcinosarcomas stand out. Almost every patient who was a candidate for an off-label drug also had a drug available in a clinical trial. This speaks to the large variety of compounds available in trials, and the need to get sarcoma patients enrolled early and often.
One of the most frequent mutations seen in our cohort was MDM2 amplification. This was exclusively seen co-existing with a CDK4 amplification. This alteration has been reported most commonly in dedifferentiated/well-differentiated liposarcoma [33] and rarely in osteosarcoma [34]. In addition to the aforementioned subtypes our study detected this co-amplification in rhabdomyosarcoma, Ewing sarcoma (EWSR1 fusion positive), and an unclassified sarcoma. We hypothesize that this co-amplified duo may be more prevalent in other subtypes than previously thought. MDM2 and CDK4 have previously been proposed as tantalizing personalized targets in liposarcoma and indeed clinical trials are underway [35]. However, our small dataset suggests that such trials should be opened to all sarcoma subtypes and based on CGP rather than histology due to the occurrence of previously unreported mutations in the subtypes mentioned above.
Similar to the MDM2 and CDK4 co-amplification, we found FGF and FRS2 to be co-amplified. This has previously been observed in dedifferentiated liposarcoma and even found to be co-expressed with MDM2 and CDK4. This co-amplification is not surprising since FRS2 is the receptor substrate for FGF [36]. Despite this known overexpression in liposarcoma, to our knowledge this has never been reported in rhabdomyosarcoma or osteosarcoma. Given the relatively well-studied pathway, this FGF and FRS2 pathway is a salient potential target. Within the last few months, Ponatinib has been reported as a very potent inhibitor of this pathway in endometrial cancer [37]. Potentially, this serves as a novel therapeutic target in FRS2 and FGF co-amplified sarcomas.
While MDM2 and FRS2 are enticing for targeted therapy, our finding of MSH2 in leiomyosarcomas presents a potential for immunotherapy. MSH2 is an integral component of the mismatch repair machinery and causes microsatellite instability, creating a target for PD-1 blockade [38]. Successful treatment has been reported with PD-1 inhibitors in MSI-high colon cancer resulting in long-term disease control where chemotherapy had not been effective. MSH mutations have been reported in sarcomas previously, especially in uterine sarcomas [39]. While none of our three leiomyosarcoma patients received immunotherapy, this would have been a potentially useful therapy and opens up the possibility of a basket trial with all-comer MSI-high tumors treated with anti PD-1 drugs.
Previous studies have assessed genomic biomarker actionability [40, 41]. These studies included larger numbers of patients and reported high frequencies of clinically actionable genomic markers. However, we believe this is the first study to look specifically at sarcomas. We report significantly fewer actionable mutations (61%) than previous studies of other cancers (>90%) and this may be related to the fusion proteins-associated sarcomas which comprise approximately 30% of all sarcomas. Furthermore, this may suggest that many sarcomas are driven by copy number alterations rather than somatic mutations. It was encouraging to see that in our center genomic testing is being used to drive clinical decision in some patients. It was even more encouraging to see that almost half of those patients (47%) derived clinical benefit from mutational analysis based on at least stable disease as per RECIST.
MDM2 is a negative regulator of the tumor suppressor gene P53 and is a powerful oncogene. MDM2 and CDK4 (12q13-15 amplification) are co-amplified in well differentiated liposarcoma. The response to current therapies is poor. As in (Table 2) several patients with MDM2 aberration benefitted clinically from MDM2 inhibitors in early phase clinical trials. Clinical trials are underway in liposarcoma using MDM2 inhibitors either singly or in combination with CDK4 inhibitors. The efficacy of these agents as a group are to be determined soon.
Our study confirms several previously described overexpressed pathways in sarcomas such as MDM2-CKD4 and FRS2-FGF. Importantly it demonstrates that these are not unique to the previously described sarcomas, and indeed are present in other subtypes. This underscores the importance of NGS in all sarcoma patients to find these potentially actionable mutations. Additionally, it highlights the need for basket trials in sarcoma that are targeted to mutations and pathways rather than histologic subtypes. With properly designed trials, these could even be accepted for drug registration or expanded indications.
Limitations abound in a retrospective observational study such as ours. While we consider our census size to be adequate, there was a wide variety of subtypes. Many of these subtypes included a single individual making any kind of conclusion impossible. This is an unfortunate consequence of sarcoma heterogeneity. However, this created a distinct advantage in showing that certain pathways are deranged in diverse subtypes. Our definition of an actionable mutation is based on aggregation of myriad studies. The true clinical utility of any given drug to target a particular mutation is not known until a prospective trial is done. However, our observational study was able to demonstrate at least anecdotal evidence of clinical benefit from targeted therapy.
In conclusion, based on our findings we believe that future studies in sarcomas should be guided by NGS and actionable alterations rather than histologic subtypes. Sarcomas are lacking in development of targeted therapy, but we demonstrate that there are myriad targets with novel therapeutic potential. We believe that personalization will shape future therapy in oncology. A rare and heterogeneous neoplasm like sarcoma would especially benefit from such a personalized approach.
MATERIALS AND METHODS
The electronic medical records of 102 diverse sarcoma patients were reviewed and history, laboratory and clinical findings were abstracted. These patients were referred to the Investigational Therapeutics Department at MD Anderson Cancer Center (MDACC). All pathology had previously been reviewed and confirmed by an MDACC pathologist with experience in bone and soft-tissue sarcomas. Therapies differed based on clinical trial opportunities at date of visit. All patients had a commercially available comprehensive genomic panel from Foundation Medicine (FoundationOne, http://www.foundationone.com). Profiling could have been performed as part of prior care. Otherwise, genomic profiling was performed upon phase 1 clinic presentation.
Patient attributes noted from the chart included age, sex, race, tumor histology, and whether the biopsy was from primary tumor or metastasis. Additional data recorded include type of investigational therapy and start date, as well as best overall response and duration of response based on Response Evaluation Criteria in Solid Tumors (RECIST V1.1). Date of death or last follow-up were also noted.
Each of the represented clinical trials in this review were independently approved by the MD Anderson institutional review board (IRB) and patients provided written consent to be treated with the corresponding investigational therapy. This retrospective review was also approved by the MD Anderson IRB.
NGS was performed by Foundation Medicine (FoundationOne, http://www.foundationone.com), a clinical grade CLIA-approved NGS test analyzing 236 or 315 cancer-related genes in at least 50ng of DNA from routine formalin-fixed and paraffin-embedded (FFPE) clinical specimens [42].
Actionable gene alteration was defined as any gene alteration that is either directly targeted or a pathway component of a directly targeted gene by an approved or investigational drug [43] (Table 5).
Table 5: FDA-approved drugs that target genes with published evidence
Gene |
Drugs |
|---|---|
ABL1 |
Bosutinib, Dasatinib, Imatinib, Sorafenib, Vandetanib |
ALK |
Alectinib, Ceritinib, Crizotinib |
AR |
Bicalutamide, Enzalutamide, Flutamide |
BRAF |
Dabrafenib, Regorafenib, Sorafenib |
CDK4 |
Palbociclib |
CSF1R |
Sunitinib |
DDR2 |
Dasatinib |
DNMT3A |
Azacitidine |
EGFR |
Afatinib, Cetuximab, Erlotinib, Gefitinib, Lapatinib, Osimertinib, Panitumumab, Vandetanib |
FGFR1 |
Lenvatinib, Pazopanib, Regorafenib, Sorafenib, Sunitinib |
FGFR2 |
Lenvatinib, Pazopanib, Regorafenib, Sorafenib, Sunitinib |
FLT4 |
Axitinib, Cabozantinib, Lenvatinib, Pazopanib, Sorafenib, Sunitinib, Vandatenib |
JAK1 |
Ruxolitinib |
JAK2 |
Ruxolitinib |
JAK3 |
Ruxolitinib, Tofacitinib |
KDR |
Axitinib, Cabozantinib, Lenvatinib, Pazopanib, Ramucirumab, Regorafenib, Sorafenib, Sunitinib, Vandetanib |
KIT |
Axitinib, Cabozantinib, Dasatinib, Imatinib, Lenvatinib, Pazopanib, Regorafenib, Sorafenib, Sunitinib |
MAP2K2 |
Trametinib |
MET |
Cabozantinib, Crizotinib |
MPL |
Eltrombopag Olamine, Romiplostim |
MTOR |
Everolimus, Sirolimus, Temsirolimus |
NTRK1 |
Crizotinib, Regorafenib |
PDGFRA |
Axitinib, Dasatinib, Imatinib, Lenvatinib, Pazopanib, Regorafenib, Sorafenib, Sunitinib |
PDGFRB |
Axitinib, Cabozantinib, Dasatinib, Imatinib, Lenvatinib, Pazopanib, Regorafenib, Sorafenib, Sunitinib |
ROS1 |
Ceritinib, Crizotinib |
Electronic medical records were reviewed for above mentioned demographic and diagnostic data. The respective molecular diagnostic reports were reviewed for alterations with a potentially actionable mutation either on-label, off-label, or in clinical trials. If patients received treatment with an investigational therapeutic, this was recorded along with the response.
Abbreviations
PD = progressive disease; PR = partial response; SD = stable disease; CR = complete response; ER = estrogen receptor; IHC = immunohistochemistry. Dx = diagnosed; WT = wild type; FM = foundation medicine.
Author contributions
Dr Groisberg and Dr Subbiah contributed equally to the work, had full access to all of the data in the study, and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Groisberg, Subbiah.
Acquisition, analysis, or interpretation of data: All authors.
Drafting of the manuscript: Groisberg, Subbiah.
Critical revision of the manuscript for important intellectual content: All authors.
Obtained funding: Subbiah.
Study supervision: Subbiah.
CONFLICTS OF INTEREST
Roman Groisberg – none.
David S Hong – Novartis, Genentech, Eisai, Astra-Zenica, Pfizer, MiRNA.
Filip Janku – Novartis.
Sarina Piha-Paul – funding: Merck, Puma, Samumed, Curis, Cerulean, Incyte, AbbVie, FivePrime, GlaxoSmithKline, Helix, Biomarin, Bayer, XuanZhu.
Vinod Ravi – none.
Robert Benjamin – none.
Shreyas Kumar Patel – consulting: J&J, CytRx, EMD Serono. funding: J&J, Eisai, Morphotek.
Neeta Somaiah – none.
Anthony Conley – EMD Serono, Novartis, Nektar.
Siraj Ali, Jeff Ross, Phil Stephens, Vincent Miller- Are Employees of Foundation Medicine and own stock in the company.
Shiraj Sen – none.
Cynthia Herzog – J&J, Amgen, Sanofi, Roche.
Vijaykumar Holla – none.
Funda Meric-Bernstam – Novartis, Genentech.
Vivek Subbiah – Novartis, Bayer, GSK, Nanocarrier, Vegenics, Northwest Biotherapeutics, Berghealth, Incyte, Fujifilm, Pharmamar, D3, Pfizer, Amgen, and Abbvie.
GRANT SUPPORT
The University of Texas MD Anderson Cancer Center is supported by the National Institutes of Health Cancer Center Support Grant CA016672. VS acknowledges the Shannon Wilkes Sarcoma Research funds. This work was supported in part by Cancer Prevention Research Institute of Texas Grant RP110584 and National Center for Advancing Translational Sciences Grant UL1 TR000371 (Center for Clinical and Translational Sciences).
Role of the Funder/Sponsor
The funding sources had no input into the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
REFERENCES
1. World Health Organization. Pathology and genetics of tumours of soft tissue and bone [the WHO classification of tumours of soft tissue and bone presented in this book reflects the views of a working group that convened for an editorial and consensus conference in Lyon, France, April 24–28, 2002]. (Lyon: IARC Press).
2. Linch M, Miah AB, Thway K, Judson IR, Benson C. Systemic treatment of soft-tissue sarcoma—gold standard and novel therapies. Nat Rev Clin Oncol. 2014; 11: 187-202.
3. Judson I, Verweij J, Gelderblom H, Hartmann JT, Schöffski P, Blay JY, Kerst JM, Sufliarsky J, Whelan J, Hohenberger P, Krarup-Hansen A, Alcindor T, Marreaud S, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol. 2014; 15: 415-423.
4. Santoro A, Tursz T, Mouridsen H, Verweij J, Steward W, Somers R, Buesa J, Casali P, Spooner D, Rankin E. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol. 1995; 13: 1537-1545.
5. Aurias A, Rimbaut C, Buffe D, Zucker JM, Mazabraud A. Translocation involving chromosome 22 in Ewing’s sarcoma. A cytogenetic study of four fresh tumors. Cancer Genet Cytogenet. 1984; 12: 21-25.
6. Khan J, Wei JS, Ringnér M, Saal LH, Ladanyi M, Westermann F, Berthold F, Schwab M, Antonescu CR, Peterson C, Meltzer PS. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001; 7: 673-679.
7. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998; 279: 577-580.
8. Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, Raymond AK, Bramwell VH, Baker LH, Maki RG, Tanaka M, Hecht JR, Heinrich MC, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008; 26: 626-632.
9. Helman LJ, Meltzer P. Mechanisms of sarcoma development. Nat Rev Cancer. 2003; 3: 685-694.
10. Larsen JE, Cascone T, Gerber DE, Heymach JV, Minna JD. Targeted therapies for lung cancer: clinical experience and novel agents. Cancer J. 2011; 17: 512-527.
11. Shafer D, Grant S. Update on rational targeted therapy in AML. Blood Rev. 2016; 30:275–283.
12. Rimawi MF, Schiff R, Osborne CK. Targeting HER2 for the treatment of breast cancer. Annu Rev Med. 2015; 66: 111-128.
13. Dickson MA, Schwartz GK, Antonescu CR, Kwiatkowski DJ, Malinowska IA. Extrarenal perivascular epithelioid cell tumors (PEComas) respond to mTOR inhibition: clinical and molecular correlates. Int J Cancer. 2013; 132: 1711-1717.
14. Italiano A, Di Mauro I, Rapp J, Pierron G, Auger N, Alberti L, Chibon F, Escande F, Voegeli AC, Ghnassia JP, Keslair F, Laé M, Ranchère-Vince D, et al. Clinical effect of molecular methods in sarcoma diagnosis (GENSARC): a prospective, multicentre, observational study. Lancet Oncol. 2016.
15. Subbiah V, Anderson P. Targeted therapy of Ewing’s sarcoma. Sarcoma. 2011; 2011:686985.
16. Subbiah V, Anderson P, Lazar AJ, Burdett E, Raymond K, Ludwig JA. Ewing’s sarcoma: standard and experimental treatment options. Curr Treat Options Oncol. 2009; 10: 126-140.
17. Livingston JA, Hess KR, Naing A, Hong DS, Patel S, Benjamin RS, Ludwig JA, Conley A, Herzog CE, Anderson P, Meric-Bernstam F, Kurzrock R, Subbiah V. Validation of prognostic scoring and assessment of clinical benefit for patients with bone sarcomas enrolled in phase I clinical trials. Oncotarget. 2016; 7:64421–64430. doi: 10.18632/oncotarget.10910.
18. Egas-Bejar D, Anderson PM, Agarwal R, Corrales-Medina F, Devarajan E, Huh WW, Brown RE, Subbiah V. Theranostic profiling for actionable aberrations in advanced high risk osteosarcoma with aggressive biology reveals high molecular diversity: the human fingerprint hypothesis. Oncoscience. 2014; 1:167-179. doi: 10.18632/oncoscience.21.
19. Subbiah V. Prospects and pitfalls of personalizing therapies for sarcomas: from children, adolescents, and young adults to the elderly. Curr Oncol Rep. 2014; 16: 401.
20. Subbiah V, Hess KR, Khawaja MR, Wagner MJ, Tang C, Naing A, Fu S, Janku F, Piha-Paul S, Tsimberidou AM, Herzog CE, Ludwig JA, Patel S, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016; 6: 35448.
21. Vlenterie M, Litière S, Rizzo E, Marréaud S, Judson I, Gelderblom H, Le Cesne A, Wardelmann E, Messiou C, Gronchi A, van der Graaf WTA. Outcome of chemotherapy in advanced synovial sarcoma patients: review of 15 clinical trials from the European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group; setting a new landmark for studies in this entity. Eur J Cancer. 2016; 58: 62-72.
22. Rosa F, Fiorillo C, Tortorelli AP, Sánchez AM, Costamagna G, Doglietto GB, Alfieri S. Surgical management of retroperitoneal soft tissue sarcomas: role of curative resection. Am Surg. 2016; 82: 128-133.
23. Lange SE, Liu J, Adkins DR, Powell MA, Van Tine BA, Mutch DG. Improved clinical trial enrollments for uterine leiomyosarcoma patients after gynecologic oncology partnership with a sarcoma center. Gynecol Oncol. 2016; 140: 307-312.
24. Subbiah V, Kurzrock R. Phase 1 clinical trials for sarcomas: the cutting edge. Curr Opin Oncol. 2011; 23: 352-360.
25. Radford JA, Cowan RA, Flanagan M, Dunn G, Crowther D, Johnson RJ, Eddleston B. The significance of residual mediastinal abnormality on the chest radiograph following treatment for Hodgkin’s disease. J Clin Oncol. 1988; 6: 940-946.
26. Meric-Bernstam F, Brusco L, Shaw K, Horombe C, Kopetz S, Davies MA, Routbort M, Piha-Paul SA, Janku F, Ueno N, Hong D, De Groot J, Ravi V, et al. Feasibility of Large-Scale Genomic Testing to Facilitate Enrollment Onto Genomically Matched Clinical Trials. J Clin Oncol. 2015; 33: 2753-2762.
27. Wheler JJ, Janku F, Naing A, Li Y, Stephen B, Zinner R, Subbiah V, Fu S, Karp D, Falchook GS, Tsimberidou AM, Piha-Paul S, Anderson R, et al. Cancer therapy directed by comprehensive genomic profiling: a single center study. Cancer Res. 2016; 76: 3690-3701.
28. Jour G, Scarborough JD, Jones RL, Loggers E, Pollack SM, Pritchard CC, Hoch BL. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Hum Pathol. 2014; 45: 1563-1571.
29. Subbiah V, Wagner MJ, McGuire MF, Sarwari NM, Devarajan E, Lewis VO, Westin S, Kato S, Brown RE, Anderson P. Personalized comprehensive molecular profiling of high risk osteosarcoma: implications and limitations for precision medicine. Oncotarget. 2015; 6: 40642-40654. doi: 10.18632/oncotarget.5841
30. Subbiah V, McMahon C, Patel S, Zinner R, Silva EG, Elvin JA, Subbiah IM, Ohaji C, Ganeshan DM, Anand D, Levenback CF, Berry J, Brennan T, et al. STUMP un“stumped”: anti-tumor response to anaplastic lymphoma kinase (ALK) inhibitor based targeted therapy in uterine inflammatory myofibroblastic tumor with myxoid features harboring DCTN1-ALK fusion. J Hematol Oncol. 2015; 8: 66.
31. Subbiah V, Westin SN, Wang K, Araujo D, Wang WL, Miller VA, Ross JS, Stephens PJ, Palmer GA, Ali SM. Targeted therapy by combined inhibition of the RAF and mTOR kinases in malignant spindle cell neoplasm harboring the KIAA1549-BRAF fusion protein. J Hematol Oncol. 2014; 7: 8.
32. Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, Tolman C, Hurd L, Liao H, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014; 4: 216-231.
33. Thway K, Jones RL, Noujaim J, Zaidi S, Miah AB, Fisher C. Dedifferentiated liposarcoma: updates on morphology, genetics, and therapeutic strategies. Adv Anat Pathol. 2016; 23: 30-40.
34. Righi A, Gambarotti M, Benini S, Gamberi G, Cocchi S, Picci P, Bertoni F. MDM2 and CDK4 expression in periosteal osteosarcoma. Hum Pathol. 2015; 46: 549-553.
35. Lencioni R, Llovet JM. Modified RECIST (mRECIST) assessment for hepatocellular carcinoma. Semin Liver Dis. 2010; 30: 52-60.
36. Zhang K, Chu K, Wu X, Gao H, Wang J, Yuan YC, Loera S, Ho K, Wang Y, Chow W, Un F, Chu P, Yen Y. Amplification of FRS2 and activation of FGFR/FRS2 signaling pathway in high-grade liposarcoma. Cancer Res. 2013; 73: 1298-1307.
37. Kim DH, Kwak Y, Kim ND, Sim T. Antitumor effects and molecular mechanisms of ponatinib on endometrial cancer cells harboring activating FGFR2 mutations. Cancer Biol Ther. 2016; 17: 65-78.
38. Dudley JC, Lin MT, Le DT, Eshleman JR. Microsatellite instability as a biomarker for PD-1 blockade. Clin Cancer Res. 2016; 22: 813-820.
39. Hoang LN, Ali RH, Lau S, Gilks CB, Lee CH. Immunohistochemical survey of mismatch repair protein expression in uterine sarcomas and carcinosarcomas. Int J Gynecol Pathol. 2014; 33: 483-491.
40. Schwaederle M, Daniels GA, Piccioni DE, Fanta PT, Schwab RB, Shimabukuro KA, Parker BA, Kurzrock R. On the road to precision cancer medicine: analysis of genomic biomarker actionability in 439 patients. Mol Cancer Ther. 2015; 14: 1488-1494.
41. Ross JS, Wang K, Khaira D, Ali SM, Fisher HA, Mian B, Nazeer T, Elvin JA, Palma N, Yelensky R, Lipson D, Miller VA, Stephens PJ, et al. Comprehensive genomic profiling of 295 cases of clinically advanced urothelial carcinoma of the urinary bladder reveals a high frequency of clinically relevant genomic alterations. Cancer. 2016; 122: 702-711.
42. Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, Sun J, Juhn F, Brennan K, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013; 31: 1023-1031.
43. Meric-Bernstam F, Johnson A, Holla V, Bailey AM, Brusco L, Chen K, Routbort M, Patel KP, Zeng J, Kopetz S, Davies MA, Piha-Paul SA, Hong DS, et al. A decision support framework for genomically informed investigational cancer therapy. J Natl Cancer Inst. 2015; 107.
44. Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A, Gervais R, Elez-Fernandez ME, Italiano A, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015; 373: 726-736.