
INTRODUCTION
Oral cancer has a disfiguring, disabling, and painful prognosis [1, 2]. It can profoundly affect essential daily functions, such as food consumption, speech, sight, and hearing; these insults are compounded by distortions in facial appearance. Despite improvements in surgery and chemotherapy, the prognosis of advanced oral cancer remains poor.
Abnormal epigenetic changes including DNA methylation and histone modifications are hallmarks of cancer in humans [3]. Histone lysine-specific demethylase 1 (LSD1, also known as KDM1A) was the first identified FAD-dependent histone demethylase; it participates in gene regulation as a nuclear homolog of amine oxidases, specifically in demethylation of monomethylated and dimethylated histone 3 (H3) at lysine 4 (H3K4me 1/2) and of H3 at lysine 9 (H3K9me 1/2) [4, 5] and in demethylation of other nonhistone substrates, such as p53 and DNA (cytosine-5-)-methyltransferase 1 in some biological situations [6]. LSD1-overexpressing tongue squamous cell carcinoma is associated with tumor size, pathological grade, and reduced overall survival [7]. LSD1 has a major role in cancer development and progression and has therapeutic potential [8–10]; therefore, optimizing drugs for the treatment of this oral cancer type is crucial.
Melatonin (N-acetyl-5-methoxytryptamine), a natural compound present in animals, plants, and microbes [11], is effective against various cancers [12–14]; however, its effects against oral cancer are unclear. In addition, melatonin has anxiolytic, analgesic, and antidepressant effects, and it increases sleep efficiency [15, 16]. We suspected the clinical benefit may improve if melatonin is used for cancer therapy. Moreover, melatonin can induce H3 acetylation [17, 18]. In addition, inhibition of LSD1 activity or suppression of LSD1 expression enhances H3 acetylation [19]. Thus, melatonin may possess strong LSD1-associated anticancer characteristics.
A lack of appropriate preclinical models is mainly impeding the progress of oncological drug research. Patient-derived tumor xenograft (PDTX) models can offer several advantages compared with standard cell-line xenograft models in preclinical trials of novel anticancer drugs because PDTXs have the molecular, genetic, and histological heterogeneity typical of the tumors of origin propagated through serial passaging in mice [20, 21]. We recently established oral cancer PDTX models. Here, we examined the LSD1 status in oral cancer by using human tumor tissue arrays. We evaluated the anti-oral cancer effects of melatonin through suppression of LSD1 expression in our PDTX models in vivo and in vitro.
RESULTS
LSD1 is a potential novel diagnostic marker and therapeutic target in OSCC
LSD1 has an oncogenic role in various malignancies [22]. To assess the status of LSD1 in oral cancers, we evaluated the expression of LSD1 by using oral cancer tissue arrays (n = 78; Figure 1a, Supplementary Table 1, 2) containing different oral cancer grades as well as normal mucosal tissues and the percentage of stained (positive) cells was calculated as described previously [23]. LSD1 expression was significantly higher in oral cancer tissue than in normal oral mucosa (P< 0.0001; Figure 2a); it was positively correlated with survival rate and disease free survival rate (Figure 1b). We further analyzed LSD1 mRNA levels in OSCC tissues from eight patients and their eight matched normal mucosal tissues. Among these, five (62.5%) patient samples displayed higher LSD1 RNA levels in the OSCC tissues than in the adjacent normal mucosal tissues (P = 0.0434; Figure 2b). We also used a bioinformatics databank (NCBI Gene Expression Omnibus profiles) to assess the expression of LSD1 in tongue cancer and observed that LSD1 RNA levels were higher in OSCC tissues than in adjacent normal mucosal tissues (P < 0.0001; Figure 2c). Western blot and Q-PCR analyses of oral cancer cell lines displayed higher LSD1 expression in most of the tested oral cancer cell lines (Figure 2d, 2e). These results suggest that LSD1 is highly expressed in OSCC, demonstrating its potential as a novel diagnostic marker and therapeutic target.
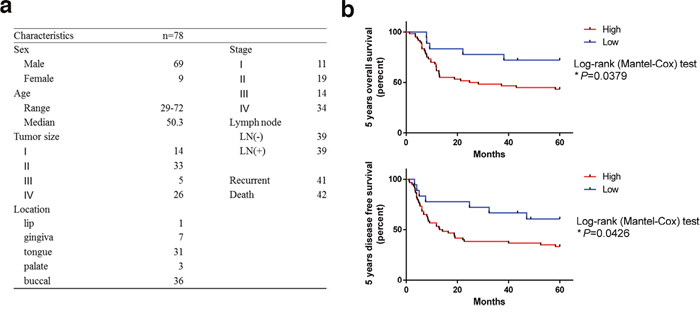
Figure 1: Overexpression of LSD1 is linked to poor outcome in oral cancer. (a) Clinicopathological data of the 78 oral cancer patients used in the study. (b) The Kaplan-Meier curve compares the 5 years overall survival and 5 years disease free survival of cancer with high or low level LSD1 protein products. Definition of LSD1 high is samples that have IHC scores equate or above 4.
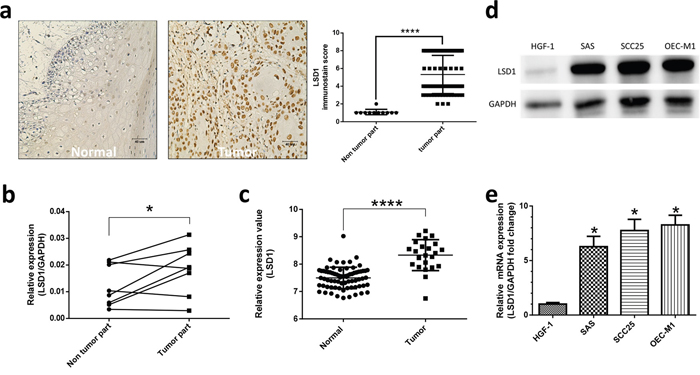
Figure 2: High expression of LSD1 existed in oral cancer. (a) Positive nuclear immunostaining of LSD1 in normal mucosal and oral cancer tissues. (b) Q-PCR results from oral cancer tissues from eight patients and their eight matched normal mucosal tissues. (c) LSD1 mRNA expression in human tongue cancer. Data were obtained from NCBI Gene Expression Omnibus profiles (http://www.ncbi.nlm.nih.gov/geoprofiles; Reporter: GDS4562). LSD1 protein and mRNA levels in four oral cancer cell lines were determined through western blot analysis (d) and Q-PCR (e), respectively. Normal human gingival fibroblast HGF-1 cells were used as the negative control. Data are presented as the mean ± SD. *P < 0.05 (Student t test).
Histological and molecular characterization of oral cancer PDTX models
We successfully established oral cancer PDTX models; histological examinations employing H&E sections from the generation 4 xenografts of both 127R and 134 PDTX models (Figure 3c, 3e) revealed poorly differentiated OSCC, which was consistent with the original clinical cancer tissues (Figure 3b, 3d). Next, LSD1 expression was evaluated through immunohistochemical analysis: both clinical samples and PDTXs of 127R and 134 models displayed LSD1 overexpression (Figure 3g–3j).
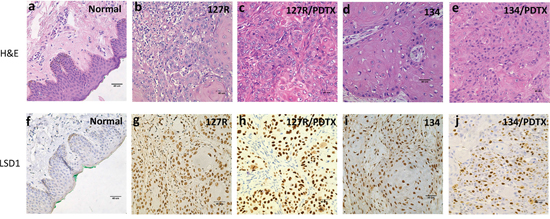
Figure 3: Histological and molecular characterization of the oral cancer PDTX models. H&E staining of normal oral mucosa (a), 127R clinical sample (b), 127R PDTX model (c), 134 clinical sample (d), and 134 PDTX sample (e). LSD1-specific immunohistochemical staining of normal oral sample (f), 127R clinical sample (g), 127R PDTX model (h), 134 clinical sample (i), and 134 PDTX sample (j). Immunodetectable proteins are stained brown; nuclei are counterstained blue. Original magnification, 400×.
Melatonin inhibited tumor growth in oral cancer PDTX models
Melatonin is effective against various cancers, but its action mechanism against oral cancer is unclear. In this study, we further examined the effects of melatonin on growth in the oral cancer PDTX (preclinical) models. We observed that melatonin significantly inhibits tumor growth compared with the vehicle (PBS) control both in 127R and 134 oral cancer PDTX models (Figure 4a), where 5-FU was the positive control. No apparent toxicity or weight loss in the mice was observed after melatonin administration during the experimental period (Figure 4b).
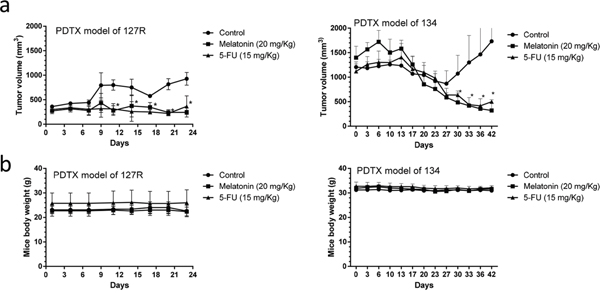
Figure 4: Melatonin inhibited tumor growth in LSD1-overexpressing oral cancer PDTX models. (a) Changes in tumor volume in 127R and 134 oral cancer PDTX models, which were treated for 24 and 42 days, respectively, with melatonin (20 mg/kg daily i.p.), 5-FU (15 mg/kg), and PBS (a vehicle control). Diameters were measured twice a week for 24 or 42 days by using Vernier calipers, and the tumor volume was calculated as 1/2 × L × W2, where W and L are the shortest and longest diameters, respectively. Tumor volumes were compared with those of controls. All data are expressed as mean ± SD. *P < 0.05 (Student t test). (b) No significant change was observed in mice body weight compared with that of the vehicle control.
Melatonin repressed LSD1 expression in oral cancer PDTX models
Abnormal activation of LSD1 signaling contributes to tumor survival, growth, and metastasis. In IHC analysis, we verified the LSD1 and Ki-67 expression in oral cancer PDTXs and observed that this expression decreases after melatonin administration in clinical tumor tissue-bearing mice compared with that in controls (Figure 5a, 5b). We observed that LSD1 expression significantly decreased in the melatonin-treated groups in both 127R and 134 PDTX models (Figure 5a, 5b). The expressions of Ki-67 (as proliferation maker) also highly decreased in the melatonin-treated group (Figure 5a, 5b).
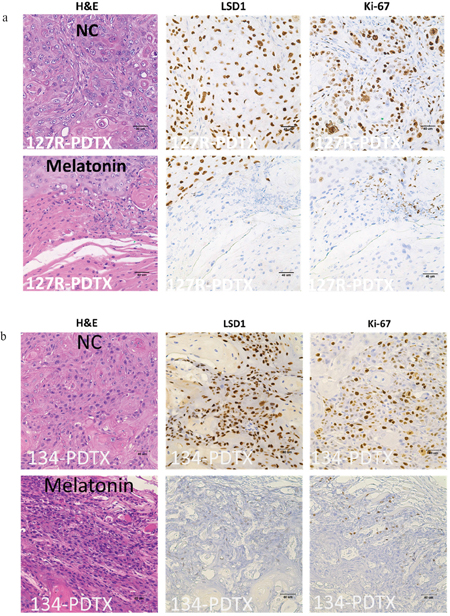
Figure 5: Effects of melatonin administration on the expressions of LSD1 and Ki-67 in oral cancer PDTX models. H&E staining and IHC were performed after administration of melatonin or PBS (a vehicle control). Both 127R (a) and 134 (b) oral cancer PDTX models stained positive for LSD1 and Ki-67. Immunodetectable proteins are stained brown; nuclei are counterstained blue. Original magnification, 400×.
Melatonin decreased tumor growth and LSD1 expression in oral cancer xenograft model
To verify the therapeutic effect of melatonin inhibition on the growth of oral cancer cells in vivo, we applied a mouse-based subcutaneous oral cancer xenograft model. NOD/SCID mice were subcutaneously implanted with 2 × 106 SCC25 cells in their flank regions. Three days after injecting tumor cells, the mice were administered melatonin or PBS through i.p. injection. Tumor volumes were measured two times weekly, and mice were euthanized 42 days after treatment. Compared with PBS treatment, melatonin treatment significantly reduced the tumor growth and tumor weight (Figure 6a). No toxicities were observed in the treatment group, as determined through behavioral changes; mouse body weight did not differ significantly between the control and melatonin-treated groups (Figure 6a). LSD1 expression was significantly lower in the melatonin-treated group than in the controls (Figure 6b). These results suggested that melatonin-treated reduction in xenograft tumor growth involves reduced LSD1 expression and oral cancer cell growth.
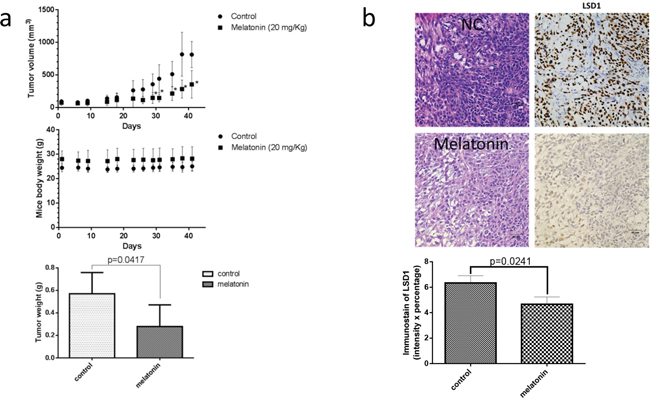
Figure 6: Melatonin inhibited oral cancer growth in SCC25 xenograft. (a) SCC25 cells were subcutaneously xenografted into NOD/SCID mice (n = 5 per group). The tumor size was analyzed 42 days after administration of melatonin or PBS (a vehicle control). (b) H&E staining and IHC staining for LSD1 were performed after administration of melatonin or PBS (as a vehicle control). Immunodetectable proteins are stained brown; nuclei are counterstained blue. Original magnification, 400×. Data are represented as mean ± SD. *P < 0.05 (Student t test).
Melatonin and LSD1 inhibitor suppressed tumor growth in oral cancer cells
To examine the significance of LSD1 in oral cancer cells, we used a pharmacological inhibitor of LSD1 activity, pargyline [24]. The treatment of oral cancer cells with melatonin or pargyline significantly reduced their proliferation in a dose-dependent manner (Figure 7). These results suggested that inhibition of LSD1 can potentially impair oral cancer cell proliferation.
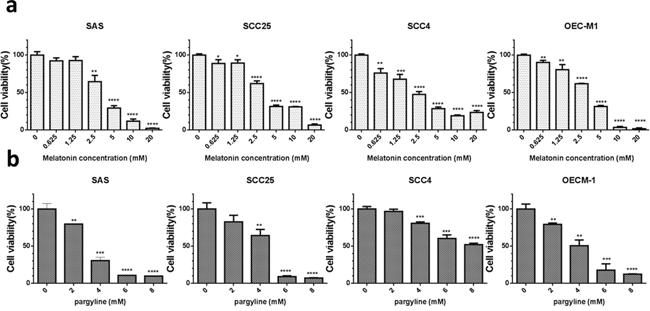
Figure 7: Melatonin inhibited oral cancer growth in vitro. (a) Assessment of cell proliferation and viability by using the methylene blue assay in four oral cancer cells treated with varying concentrations of melatonin (0–20 mM) or DMSO (1 μL/mL) for 24 h. (b) Assessment of cell proliferation and viability by using the MTT assay in four oral cancer cells treated with varying concentrations of pargyline (0–8 mM) or DMSO (1 μL/mL) for 24 h. Data are represented as mean ± SD. *P < 0.05 (Student’s t test).
Melatonin induced oral cancer cells G0/G1 arrest
Melatonin repressed LSD1 expression and inhibited oral cancer cell growth in vivo. To explore the anticancer mechanism of melatonin, we analyzed the cell cycle through flow cytometry. Our results revealed that melatonin induces G0/G1 cell cycle arrest in oral cancer cells (Figure 8).
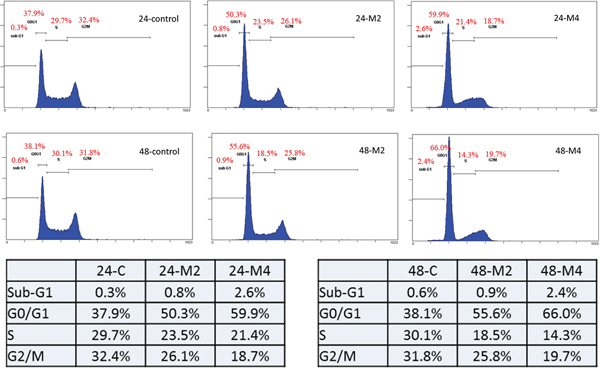
Figure 8: Melatonin induced oral cancer G0/G1 arrest. Flow cytometric cell cycle analysis of SAS cells treated with varying concentrations of melatonin (2–4 mM) or DMSO (1 μL/mL) for 24 and 48h.
Melatonin decreased LSD1 and cyclin D1 expression and increased p21 expression in oral cancer cells
Melatonin induces cell growth arrest by increasing p21 expression [25, 26]. Furthermore, LSD1 knockdown significantly increases p21 expression and induces cell cycle arrest [27]. Because LSD1 inhibition reduced oral cancer cell proliferation in our study, we examined whether melatonin reduces LSD1 expression in oral cancer cells. Melatonin substantially reduced LSD1 levels in both SAS and SCC25 cells (Figure 9a). Accordingly, western blot analysis revealed that treatment with melatonin significantly increased p21 and decreased cyclin D1 levels in SCC25 cells (Figure 9b). These studies suggested that blocking the LSD1 axis with melatonin can potentially reduce oral cancer cell proliferation and that melatonin likely exerts tumor-repressive effects by suppressing LSD1 expression.
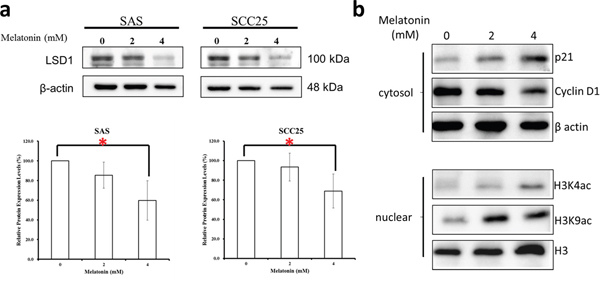
Figure 9: Melatonin repressed LSD1 expression in oral cancer cells. (a) Western blot analysis and densitometry for LSD1 after SAS and SCC25 cells treated with melatonin for 24 h. (b) Western blot analysis of cyclin D1 and p21 expression after SCC25 cells treated with melatonin for 24 hrs. Data are presented as mean ± SD. *P < 0.05 (Student t test). The presented blots are representatives of those from at least three independent experiments.
Melatonin induced H3K4/H3K9 acetylation
The current study revealed that melatonin induces H3 acetylation. To assess whether melatonin exerts an epigenetic role in oral cancer, we analyzed the H3 modification site in oral cancer. Our results revealed that melatonin induces H3 acetylation at H3K4 and H3K9 in oral cancer cells (Figure 9b).
DISCUSSION
Melatonin is a marvelous versatility hormone which is synthesized and secreted by the pineal gland and shows both a wide distribution within organisms from bacteria to humans [28–31]. Studies have demonstrated that melatonin embraces anti-oral cancer effects, including regulating cancer cell evading apoptosis, angiogenesis, metastasis, evading the immune system, and inflammation [28, 32–37]. Recent study demonstrated that melatonin can protect against radiation-induced oral mucositis (Supplementary Figure 4), which is a particularly severe and common acute side effect of chemotherapy or radiotherapy [34]. A surprising benefit of melatonin treatment is its few side effects; it relieved depressive symptoms, anxiety, insomnia, reduced cognitive function, and fatigue as observed in cancer patients being treated with other related therapies [15]. In this study, we evaluated the response of human oral cancer to melatonin treatment in patient-derived tumor xenograft, in vivo and in vitro. Melatonin effectively inhibited cell proliferation. Our results indicated that melatonin suppressed LSD1 expression (Figure 9a), reduced its mRNA levels, and induced H3K4 and H3K9 acetylation (Figure 9b). Melatonin also induced cell cycle arrest in the G0/G1 phase (Figure 8, Figure 9b). Melatonin can inhibit cancer cell growth by modulating epigenetic effects through DNA methylation and histone acetylation pathways [12, 17, 18, 38, 39]. Melatonin also induces H3 hyperacetylation in neural stem cells [18]. According to our results, melatonin can induce histone acetylation at H3K4 and H3K9 (Figure 9b). Moreover, H3K4 or H3K9 acetylation is typically associated with gene transcription [40, 41]. These findings indicate that the antioral cancer effect of melatonin is attributable to the alteration of the H3K4 or H3K9 acetylation.
In past decades, aberrant epigenetic dysregulation was considered the key indication of human cancer [3, 42]. Epigenetic alterations are generally reversible and dynamically regulated; thus, they may be novel therapeutic targets for cancer therapy [43, 44]. The increasing evidence has linked diverse histone modifications to cancer initiation and progression [45]. Moreover, H3K4 acetylation levels may facilitate diagnosis or therapy of oral cancer: low H3K4 acetylation levels in oral cancer indicate advanced tumor stage and poor prognosis [46]. LSD1, a H3K4 or H3K9 demethylase, regulates gene expression pathways by altering the epigenetic histone markers in gene promoters [47]. Epigenetic alterations in LSD1 have a major role in tumorigenicity [48, 49] and are associated with poor prognosis [7, 8, 50–55]. Inhibition of LSD1 activity or repression of LSD1 expression can inhibit tumor cell growth (Supplementary Figure 3) and induce histone acetylation [19]. High LSD1 expression levels in tongue cancer are associated with proliferation and poor prognosis [7]. Here, we verified LSD1 expression by using a clinical oral cancer tissue microarray (n = 78) and oral cancer cell lines (n = 4); we also assessed its clinicopathological significance in oral cancer patients. Our results indicated that LSD1 is aberrantly overexpressed in oral cancer, and this overexpression is associated with poor prognosis (Figure 1). The aggressive nature of LSD1-overexpressing oral cancer and lack of effective therapeutic options make clinical oral cancer treatment particularly challenging; thus, developing novel therapies is crucial. However, research in this area has been impeded by lack of clinically relevant experimental models.
Here, we determined the antitumor effects of melatonin in LSD1-overexpressing oral cancer PDTXs and oral cancer cell line xenograft models. To our knowledge, this is the first study of melatonin in PDTX models. Our results indicated that melatonin exerted a substantial antitumor effect on PDTX and traditional xenograft models of oral cancer (Figure 4). The PDTX mouse model accurately represents human tumors through increased diversity of molecular lesions and preservation of three-dimensional tumor-stromal cell components and interactions [56]. Hence, these models have been increasingly used as tools for the preclinical assessment of anticancer drugs. Some PDTX models (e.g., lung cancer [57–59], melanoma [60], colon cancer [61], breast cancer [62], hepatocellular carcinoma [63, 64], and gastrointestinal stromal tumor [65] have been established and used for evaluating antitumor compounds [66]; however, the availability of oral cancer PDTX models is limited. In this study, we used the NSG mice to establish oral cancer PDTX models. The tumor growth rate differed among our PDTX models. Both 127R and 134 PDTX models were obtained from clinical advanced oral cancer patients with lymphoid metastasis. We performed the histological and molecular characterization of the tissues from oral cancer patients and PDTX tissues through immunochemistry of cells from generations 4 and 7 of the 127R and 134 PDTX models, respectively. Our results showed that LSD1 is overexpressed in oral cancer tissues of both clinical patients and PDTX (Figure 3). As per the H&E staining results, histology of the PDTX models was similar among all cancer patient samples. We will establish more PDTX lines to further clarify the abundant tumor heterogeneity in future studies.
Our results revealed that melatonin has anticancer effects similar to 5-FU (Figure 4). Anticancer drugs cause severe side effects, including cardiotoxicity. For example, 5-FU and cisplatin are major chemotherapeutic agents in the treatment of head and neck cancer. Despite its proven therapeutic efficacy, 5-FU and cisplatin (Supplementary Figure 1) also possess several undesirable cardiotoxicities, including coronary vasospasm, coronary thrombosis, cardiomyopathy, and sudden cardiac death [67]. In contrast to many anticancer drugs on the cardiomyocytes, melatonin has low toxicity [68] and has beneficial effects against cardiotoxicity [69]. Based on previous studies, we compared the anticancer effects of 5-FU and melatonin on oral cancer PDTX models.
Our data suggested that melatonin is a potential therapeutic option for LSD1-overexpressing oral cancer, but the detailed action mechanism of melatonin in oral cancer requires further detailed investigation. Our in vitro study revealed that melatonin or an LSD1 inhibitor, pargyline, strongly inhibit the proliferation of tongue (SAS, SCC25, and SCC4) and gingival (OEC-M1) cancer cells. Our in vivo IHC assay demonstrated that melatonin strongly reduces LSD1 and Ki-67 expression (Figure 5a, 5b); consistent with this, in our in vitro study, melatonin significantly downregulated LSD1 (Figure 9a) and induced H3K4 methylation (Supplementary Figure 2). These data suggest that melatonin is a potential therapeutic option for LSD1-overexpressing oral cancer, but the detailed action mechanism of melatonin in oral cancer requires further detailed investigation.
In conclusion, we demonstrated that the anti-oral cancer effect of melatonin is accompanied by LSD1 downregulation in vitro, in vivo, and in our preclinical PDTX model. Thus, LSD1 can be a diagnostic marker and therapeutic target for oral cancer. Furthermore, melatonin can potentially be an effective chemotherapeutic agent for oral cancer. Finally, this study lays a foundation for further evaluation of the action mechanism of melatonin against oral cancer.
MATERIALS AND METHODS
Tissue microarray
Microarray slides of OSCC tissue were prepared using 78 paraffin-embedded primary OSCC tumors and 11 normal oral mucosal tissues (Figure 1A). Two core tissue samples were extracted from a representative area of each paraffin-embedded tumor tissue. Each representative core sample in the tissue microarray slide measured 2 mm in diameter. This microarray study was approved by the ethics review committees of Tri-Service General Hospital, Taiwan (IRB: TSGH-2-102-05-111 and TSGH-2-105-05-002). All patients were not received surgery, radiotherapy, or chemotherapy previously.
Histology and immunohistochemistry
Mice were sacrificed using CO2, and their tissues were fixed through perfusion by using 4% paraformaldehyde in 0.1 M phosphate buffer. Next, 5-μm-thick serial histological sections were taken on slides, deparaffinized in xylene, and rehydrated. After blocking endogenous peroxidase by using 3% hydrogen peroxide, the slides were incubated with the anti-LSD1 and anti-Ki-67 antibodies overnight at 4°C. Target protein expression was detected using an anti-mouse and anti-rabbit peroxidase complex, and peroxidase activity was observed using 3-amino-9-ethyl-carbazole. The slides were counterstained with hematoxylin (Sigma–Aldrich) and mounted using a mounting solution. The immunohistochemistry (IHC) calculations are described in the next section.
Evaluation of immunohistochemical staining
The intensity of tumor cell immunoreactivity was scored on a scale of 0–3: 0, no staining; 1, weak intensity; 2, moderate intensity; and 3, strong intensity. The percentage of tumor cells with nucleus staining for each intensity score was graded on a 5-point scale: 0, 0%; 1, 0- 25%; 2, 25%- 50%; 3, 50%- 75%; and 4, 75- 100% stained tumor cells. The immunostaining scores (ranged 0–12) were determined by multiplying scores based on the percentages of stained tumor cells (0–4) with the intensity scores (0–3). Definition of LSD1 high is samples that have IHC scores equate or above 4.
Cell culture and reagents
Human tongue squamous cell carcinoma cell-line SAS was provided by Dr. Jeng–Fan Lo [70]. SCC4 (CRL-1624; ATCC) and SCC25 (CRL-1628; ATCC) were obtained from the American Type Culture Collection. The OEC-M1 was established using primary tumors obtained from adult male OSCC patients from Taiwan with a history of betel quid chewing. SAS, SCC25, and OEC-M1 cells were cultured in RPMI 1640, whereas SCC4 cells were cultured in DMEM/F12. All cultured media were supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 2 mmol/L L-glutamine, and all cells were grown at 37°C in a humidified incubator with 5% CO2 atmosphere.
Melatonin (M5250, purity of ≥ 98% determined through high performance liquid chromatography; Sigma–Aldrich) was dissolved in ethanol (95%) to form a 0.1 g/mL stock and added to cells at indicated concentrations. 5-FU (F6627; purity of ≥ 99% determined through high performance liquid chromatography; Sigma-Aldrich) was dissolved in saline to form a 1.5 mg/mL stock as the positive control in the animal models.
Establishment of PDTX models and treatment protocol
Tumor specimens were obtained from oral squamous cell carcinoma (OSCC) patients with lymphatic metastases during initial surgery. The experiments were conducted according to the ethical guidelines of the institutional review board (TSGH-2-102-05-111) of the National Defense Medical Center, Taiwan. The patients had not received chemotherapy or radiotherapy before surgery. The histological type of all tumor specimens was T4aN2b, as per the World Health Organization criteria. The tumor samples were placed in RPMI 1640 under sterile conditions immediately after surgical resection and transported to the animal tissue culture facility.
The tumors were implanted subcutaneously into NOD/SCID/IL2R gamma null (or NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; NSG) mice. The NSG mice (aged 8–10 weeks) were obtained from Jackson Laboratory (USA) and maintained in the National Defense Medical Center, Taiwan. All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the National Defense Medical Center (IACUC 14–299 and 15–028). Xenograft growth was monitored at least two times weekly: lengths (longest diameters) and widths (shortest diameters) of the tumors were measured using Vernier calipers, and the tumor volume was calculated using the following formula: volume = 1/2 × length × width2. When a tumor volume was approximately 3000 mm3, the tumor was removed for serial transplantation.
When the tumor volume reached approximately 500 mm3, mice with fourth (127R)- or seventh (134)-generation PDTXs were randomized into three groups (127R PDTX line, n = 3 per group; 134 PDTX line, n = 5 per group) to receive melatonin (20 mg/kg/daily), vehicle control (phosphate-buffered saline, PBS), and 5-fluorouracil (5-FU; 15 mg/kg/daily; positive control) through intraperitoneal (i.p.) injection in late afternoon for 24 (127R model) and 42 (134 model) days. Mouse weight and tumor volume were measured at least two times weekly. The transplanted tumor size was measured using Vernier calipers two times weekly, and the tumor volume was calculated using the aforementioned formula. At the end of the treatment, the mice were sacrificed, and the tumors were removed, weighed, and visualized.
Xenograft tumor model
Six-week-old NOD.CB17 Prkdcscid/J mice (National Laboratory Animal Center, Taiwan) mice were maintained in a microisolator under pathogen-free conditions. Each mouse was subcutaneously injected with 2 × 106 SCC25 cells, and the mice were then divided into two groups (n = 5 per group). After 3 days, one group was treated with melatonin (20 mg/kg daily i.p.) in late afternoon, whereas the other was treated with a vehicle (PBS i.p.). The size of the transplanted tumors was measured using Vernier calipers two times weekly, and the tumor volume was calculated using the aforementioned formula. At the end of the treatment, the mice were sacrificed, and the tumors were removed, weighed, and visualized.
Cell growth inhibition assay
Cells (10,000/well in 24-well plates) were exposed to various concentrations of melatonin for 24–72 h. A methylene blue assay was used to evaluate the effect of melatonin on cell growth, as described previously [71]. The IC50 value, indicating the concentration of melatonin causing a 50% inhibition of cell growth, was calculated statistically by comparing it with that of controls.
Quantitative real-time PCR
Total RNA was extracted from oral cancer cells by using the TRIzol Reagent (Invitrogen) according to the manufacturer’s protocol. First-strand cDNA synthesis and amplification was performed using the Maxima H Minus First Strand cDNA Synthesis Kit (Thermo Scientific, Rockford, USA). The following quantitative real time PCR (Q-PCR) primers were designed using Primer3 (NCBI): LSD1, 5′-GCTCGGGGCTCTTATTCCTA-3′ (forward) and 5′-CCCAAAAACTGGTCTGCAAT-3′ (reverse); GAPDH, 5′-GGAAGGTGAAGGTCGGAGTCA-3′ (forward) and 5′-GTCATTGATGGCAACAATATCCACT-3′ (reverse). Q-PCR amplifications were performed on a real-time PCR system (Applied Biosystems 7500 Fast) by using 20-μL reaction volumes containing 15 μL of SYBR Green PCR Master Mix (Thermo Scientific). The changes in LSD1 expression were calculated using 7500 software (Version 2.0.6, Applied Biosystems).
Cell cycle analysis
The cell cycle was assayed by propidium iodide (PI) staining, followed by Cytomics FC500 Flow Cytometer CXP analysis. Cell cycle profiles were then determined using the CXP analysis software (Beckman Coulter Inc., Miami, FL, USA).
Western blot analysis
Cells were lysed directly in RIPA buffer containing 50 mM Tris (pH 7.8), 0.15 M NaCl, 5 mM EDTA, 0.5% Triton X-100, 0.5% NP-40, 0.1% sodium deoxycholate, a protease inhibitor mixture, and a phosphatase inhibitor mixture (Calbiochem, Billerica, MA, USA). The relative protein concentration in the supernatants was determined using a BCA protein assay kit (Thermo Scientific). For each lane of 10% SDS-PAGE, 30 μg of cell lysate protein was loaded, separated, and transferred onto a polyvinyldifluoride membrane (GE Healthcare, UK). The membranes were then probed using specific antibodies against LSD1 (A300-215A, Bethly), H3K4ac (07-539, Millipore), H3K9ac (ab4441, Abcam), H3K4me2 (ab32356, Abcam), H3 (H0164, Sigma-Aldrich), cyclin D1 (#1085-1, Epitomics), p21 (ab81283, Abcam) and β-actin (#3053-1, Epitomics).
Statistical analysis
Data (mean ± SD, 2–3 experiments) were analyzed for statistical significance by using unpaired, two-tailed Student t tests. P < 0.05 was considered statistically significant. We performed statistical analyses by using GraphPad Prism (GraphPad Software, San Diego, CA, USA). All cell-line experiments were repeated a minimum of three times, unless stated otherwise. Survival data were analyzed with the Kaplan-Meier method, tested with the log-rank method and further analyzed for multiple factors using the Cox regression model. P<0.05 was considered to indicate a statistically significant difference.
Author contributions
Cheng-Yu Yang contributed to experiment design, acquisition of data, and drafting of the manuscript. Chih-Kung Lin and Chang-Huei Tsao contributed to acquisition of data. Gu-Jiun Lin analyzed the data. Kuo-Hsing Ma, Cheng-Chih Hsieh and Huey-Kang Sytwu provided technical and material supports. Yi-Shing Shieh performed statistical analysis. Yuan-Wu Chen designed the experiments, and revised the manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the technical services provided by Instrument Center of National Defense Medical Center and the laboratory animal center of National Defense Medical Center.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This study was supported by research grants from Tri-Service General Hospital, Taiwan, Republic of China (grants No. TSGH-C104-008-S05, TSGH-C105-006-008-S05, TSGH-C106-004-006-008-S05, TSGH-C105-190, TSGH-C105-169, and National Science Council, Taiwan, Republic of China (grants No. NSC102-2314-B-016-018-MY3 and MOST 105-2314-B-016-021-MY3).
REFERENCES
1. Krishna Rao SV, Mejia G, Roberts-Thomson K, Logan R. Epidemiology of oral cancer in Asia in the past decade – an update (2000-2012). Asian Pacific journal of cancer prevention. 2013; 14:5567-5577.
2. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013; 63:11-30.
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
4. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004; 119:941-953.
5. Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005; 437:432-435.
6. Nicholson TB, Chen T. LSD1 demethylates histone and non-histone proteins. Epigenetics. 2009; 4:129-132.
7. Yuan C, Li Z, Qi B, Zhang W, Cheng J, Wang Y. High expression of the histone demethylase LSD1 associates with cancer cell proliferation and unfavorable prognosis in tongue cancer. J Oral Pathol Med. 2015; 44:159-165.
8. Wang M, Liu X, Guo J, Weng X, Jiang G, Wang Z, He L. Inhibition of LSD1 by Pargyline inhibited process of EMT and delayed progression of prostate cancer in vivo. Biochem Biophys Res Commun. 2015; 467:310-315.
9. Lan W, Zhang D, Jiang J. The roles of LSD1-mediated epigenetic modifications in maintaining the pluripotency of bladder cancer stem cells. Med Hypotheses. 2013; 81:823-825.
10. Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R, Solleder G, Bastian PJ, Ellinger J, Metzger E, Schule R, Buettner R. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006; 66:11341-11347.
11. Manchester LC, Coto-Montes A, Boga JA, Andersen LP, Zhou Z, Galano A, Vriend J, Tan DX, Reiter RJ. Melatonin: an ancient molecule that makes oxygen metabolically tolerable. J Pineal Res. 2015; 59:403-419.
12. Wei JY, Li WM, Zhou LL, Lu QN, He W. Melatonin induces apoptosis of colorectal cancer cells through HDAC4 nuclear import mediated by CaMKII inactivation. J Pineal Res. 2015; 58:429-438.
13. Xiang S, Dauchy RT, Hauch A, Mao L, Yuan L, Wren MA, Belancio VP, Mondal D, Frasch T, Blask DE, Hill SM. Doxorubicin resistance in breast cancer is driven by light at night-induced disruption of the circadian melatonin signal. J Pineal Res. 2015; 59:60-69.
14. Xin Z, Jiang S, Jiang P, Yan X, Fan C, Di S, Wu G, Yang Y, Reiter RJ, Ji G. Melatonin as a treatment for gastrointestinal cancer: a review. J Pineal Res. 2015; 58:375-387.
15. Hansen MV. Chronobiology, cognitive function and depressive symptoms in surgical patients. Dan Med J. 2014; 61:B4914.
16. Yousaf F, Seet E, Venkatraghavan L, Abrishami A, Chung F. Efficacy and safety of melatonin as an anxiolytic and analgesic in the perioperative period: a qualitative systematic review of randomized trials. Anesthesiology. 2010; 113:968-976.
17. Niles LP, Pan Y, Kang S, Lacoul A. Melatonin induces histone hyperacetylation in the rat brain. Neurosci Lett. 2013; 541:49-53.
18. Sharma R, Ottenhof T, Rzeczkowska PA, Niles LP. Epigenetic targets for melatonin: induction of histone H3 hyperacetylation and gene expression in C17.2 neural stem cells. J Pineal Res. 2008; 45:277-284.
19. Huang Y, Vasilatos SN, Boric L, Shaw PG, Davidson NE. Inhibitors of histone demethylation and histone deacetylation cooperate in regulating gene expression and inhibiting growth in human breast cancer cells. Breast Cancer Res Treat. 2012; 131:777-789.
20. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, Zhang C, Schnell C, Yang G, Zhang Y, Balbin OA, Barbe S, Cai H, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015; 21:1318-1325.
21. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012; 9:338-350.
22. Lokken AA, Zeleznik-Le NJ. Breaking the LSD1/KDM1A addiction: therapeutic targeting of the epigenetic modifier in AML. Cancer Cell. 2012; 21:451-453.
23. Lin GJ, Huang YS, Lin CK, Huang SH, Shih HM, Sytwu HK, Chen YW. Daxx and TCF4 interaction links to oral squamous cell carcinoma growth by promoting cell cycle progression via induction of cyclin D1 expression. Clinical oral investigations. 2015.
24. Cortez V, Mann M, Tekmal S, Suzuki T, Miyata N, Rodriguez-Aguayo C, Lopez-Berestein G, Sood AK, Vadlamudi RK. Targeting the PELP1-KDM1 axis as a potential therapeutic strategy for breast cancer. Breast Cancer Res. 2012; 14:R108.
25. Proietti S, Cucina A, Dobrowolny G, D'Anselmi F, Dinicola S, Masiello MG, Pasqualato A, Palombo A, Morini V, Reiter RJ, Bizzarri M. Melatonin down-regulates MDM2 gene expression and enhances p53 acetylation in MCF-7 cells. J Pineal Res. 2014; 57:120-129.
26. Hong Y, Won J, Lee Y, Lee S, Park K, Chang KT, Hong Y. Melatonin treatment induces interplay of apoptosis, autophagy, and senescence in human colorectal cancer cells. J Pineal Res. 2014; 56:264-274.
27. Sareddy GR, Nair BC, Krishnan SK, Gonugunta VK, Zhang QG, Suzuki T, Miyata N, Brenner AJ, Brann DW, Vadlamudi RK. KDM1 is a novel therapeutic target for the treatment of gliomas. Oncotarget. 2013; 4:18-28. doi: 10.18632/oncotarget.725.
28. Calvo JR, Gonzalez-Yanes C, Maldonado MD. The role of melatonin in the cells of the innate immunity: a review. J Pineal Res. 2013; 55:103-120.
29. Manchester LC, Poeggeler B, Alvares FL, Ogden GB, Reiter RJ. Melatonin immunoreactivity in the photosynthetic prokaryote Rhodospirillum rubrum: implications for an ancient antioxidant system. Cell Mol Biol Res. 1995; 41:391-395.
30. Dubbels R, Reiter RJ, Klenke E, Goebel A, Schnakenberg E, Ehlers C, Schiwara HW, Schloot W. Melatonin in edible plants identified by radioimmunoassay and by high performance liquid chromatography-mass spectrometry. J Pineal Res. 1995; 18:28-31.
31. Vaughan GM, Pelham RW, Pang SF, Loughlin LL, Wilson KM, Sandock KL, Vaughan MK, Koslow SH, Reiter RJ. Nocturnal elevation of plasma melatonin and urinary 5-hydroxyindoleacetic acid in young men: attempts at modification by brief changes in environmental lighting and sleep and by autonomic drugs. J Clin Endocrinol Metab. 1976; 42:752-764.
32. Cutando A, López-Valverde A, De Vicente J, Gimenez JL, Carcía IA, De Diego RG. Action of melatonin on squamous cell carcinoma and other tumors of the oral cavity (Review). Oncol Lett. 2014; 7:923-926.
33. Lin FY, Lin CW, Yang SF, Lee WJ, Lin YW, Lee LM, Chang JL, Weng WC, Lin CH, Chien MH. Interactions between environmental factors and melatonin receptor type 1A polymorphism in relation to oral cancer susceptibility and clinicopathologic development. PLoS One. 2015; 10:e0121677.
34. Ortiz F, Acuna-Castroviejo D, Doerrier C, Dayoub JC, Lopez LC, Venegas C, Garcia JA, Lopez A, Volt H, Luna-Sanchez M, Escames G. Melatonin blunts the mitochondrial/NLRP3 connection and protects against radiation-induced oral mucositis. J Pineal Res. 2015; 58:34-49.
35. Reiter RJ, Rosales-Corral SA, Liu XY, Acuna-Castroviejo D, Escames G, Tan DX. Melatonin in the oral cavity: physiological and pathological implications. J Periodontal Res. 2015; 50:9-17.
36. Yeh CM, Lin CW, Yang JS, Yang WE, Su SC, Yang SF. Melatonin inhibits TPA-induced oral cancer cell migration by suppressing matrix metalloproteinase-9 activation through the histone acetylation. Oncotarget. 2016; 7:21952-21967. doi: 10.18632/oncotarget.8009.
37. Goncalves Ndo N, Rodrigues RV, Jardim-Perassi BV, Moschetta MG, Lopes JR, Colombo J, Zuccari DA. Molecular markers of angiogenesis and metastasis in lines of oral carcinoma after treatment with melatonin. Anticancer Agents Med Chem. 2014; 14:1302-1311.
38. Fan C, Pan Y, Yang Y, Di S, Jiang S, Ma Z, Li T, Zhang Z, Li W, Li X, Reiter RJ, Yan X. HDAC1 inhibition by melatonin leads to suppression of lung adenocarcinoma cells via induction of oxidative stress and activation of apoptotic pathways. J Pineal Res. 2015; 59:321-333.
39. Jung-Hynes B, Reiter RJ, Ahmad N. Sirtuins, melatonin and circadian rhythms: building a bridge between aging and cancer. J Pineal Res. 2010; 48:9-19.
40. Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nature genetics. 2008; 40:897-903.
41. Nishida H, Suzuki T, Kondo S, Miura H, Fujimura Y, Hayashizaki Y. Histone H3 acetylated at lysine 9 in promoter is associated with low nucleosome density in the vicinity of transcription start site in human cell. Chromosome research. 2006; 14:203-211.
42. Baudino TA. Targeted Cancer Therapy: The next generation of cancer treatment. Curr Drug Discov Technol. 2015; 12:3-20.
43. Yen CY, Huang HW, Shu CW, Hou MF, Yuan SS, Wang HR, Chang YT, Farooqi AA, Tang JY, Chang HW. DNA methylation, histone acetylation and methylation of epigenetic modifications as a therapeutic approach for cancers. Cancer Lett. 2016; 373:185-192.
44. Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotech. 2010; 28:1069-1078.
45. Hojfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov. 2013; 12:917-930.
46. Chen YW, Kao SY, Wang HJ, Yang MH. Histone modification patterns correlate with patient outcome in oral squamous cell carcinoma. Cancer. 2013; 119:4259-4267.
47. Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005; 437:436-439.
48. Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, Field HI, Neal DE, Yamaue H, Ponder BA, Nakamura Y, Hamamoto R. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer. 2011; 128:574-586.
49. Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X, Song Y. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS One. 2012; 7:e35065.
50. Niebel D, Kirfel J, Janzen V, Holler T, Majores M, Gutgemann I. Lysine-specific demethylase 1 (LSD1) in hematopoietic and lymphoid neoplasms. Blood. 2014; 124:151-152.
51. Ding J, Zhang ZM, Xia Y, Liao GQ, Pan Y, Liu S, Zhang Y, Yan ZS. LSD1-mediated epigenetic modification contributes to proliferation and metastasis of colon cancer. Br J Cancer. 2013; 109:994-1003.
52. Qin Y, Zhu W, Xu W, Zhang B, Shi S, Ji S, Liu J, Long J, Liu C, Liu L, Xu J, Yu X. LSD1 sustains pancreatic cancer growth via maintaining HIF1alpha-dependent glycolytic process. Cancer Lett. 2014; 347:225-232.
53. Zhang H, Diab A, Fan H, Mani SK, Hullinger R, Merle P, Andrisani O. PLK1 and HOTAIR accelerate proteasomal degradation of SUZ12 and ZNF198 during Hepatitis B virus-induced liver carcinogenesis. Cancer Res. 2015; 75:2363-2374.
54. Wu J, Hu L, Du Y, Kong F, Pan Y. Prognostic role of LSD1 in various cancers: evidence from a meta-analysis. Onco Targets Ther. 2015; 8:2565-2570.
55. Zhao ZK, Dong P, Gu J, Chen L, Zhuang M, Lu WJ, Wang DR, Liu YB. Overexpression of LSD1 in hepatocellular carcinoma: a latent target for the diagnosis and therapy of hepatoma. Tumour Biol. 2013; 34:173-180.
56. Garber K. From human to mouse and back: 'tumorgraft' models surge in popularity. Journal of the National Cancer Institute. 2009; 101:6-8.
57. Cutz JC, Guan J, Bayani J, Yoshimoto M, Xue H, Sutcliffe M, English J, Flint J, LeRiche J, Yee J, Squire JA, Gout PW, Lam S, Wang YZ. Establishment in severe combined immunodeficiency mice of subrenal capsule xenografts and transplantable tumor lines from a variety of primary human lung cancers: potential models for studying tumor progression-related changes. Clinical cancer research. 2006; 12:4043-4054.
58. Zhang XC, Zhang J, Li M, Huang XS, Yang XN, Zhong WZ, Xie L, Zhang L, Zhou M, Gavine P, Su X, Zheng L, Zhu G, Zhan P, Ji Q, Wu YL. Establishment of patient-derived non-small cell lung cancer xenograft models with genetic aberrations within EGFR, KRAS and FGFR1: useful tools for preclinical studies of targeted therapies. J Transl Med. 2013; 11:168.
59. Fichtner I, Rolff J, Soong R, Hoffmann J, Hammer S, Sommer A, Becker M, Merk J. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clinical cancer research. 2008; 14:6456-6468.
60. Nemati F, Sastre-Garau X, Laurent C, Couturier J, Mariani P, Desjardins L, Piperno-Neumann S, Lantz O, Asselain B, Plancher C, Robert D, Peguillet I, Donnadieu MH, et al. Establishment and characterization of a panel of human uveal melanoma xenografts derived from primary and/or metastatic tumors. Clinical cancer research. 2010; 16:2352-2362.
61. Dangles-Marie V, Pocard M, Richon S, Weiswald LB, Assayag F, Saulnier P, Judde JG, Janneau JL, Auger N, Validire P, Dutrillaux B, Praz F, Bellet D, Poupon MF. Establishment of human colon cancer cell lines from fresh tumors versus xenografts: comparison of success rate and cell line features. Cancer Res. 2007; 67:398-407.
62. Marangoni E, Vincent-Salomon A, Auger N, Degeorges A, Assayag F, de Cremoux P, de Plater L, Guyader C, De Pinieux G, Judde JG, Rebucci M, Tran-Perennou C, Sastre-Garau X, et al. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clinical cancer research. 2007; 13:3989-3998.
63. Huynh H, Soo KC, Chow PK, Tran E. Targeted inhibition of the extracellular signal-regulated kinase kinase pathway with AZD6244 (ARRY-142886) in the treatment of hepatocellular carcinoma. Molecular cancer therapeutics. 2007; 6:138-146.
64. Huynh H, Chow PK, Soo KC. AZD6244 and doxorubicin induce growth suppression and apoptosis in mouse models of hepatocellular carcinoma. Molecular cancer therapeutics. 2007; 6:2468-2476.
65. Huynh H, Lee JW, Chow PK, Ngo VC, Lew GB, Lam IW, Ong HS, Chung A, Soo KC. Sorafenib induces growth suppression in mouse models of gastrointestinal stromal tumor. Molecular cancer therapeutics. 2009; 8:152-159.
66. Fujimoto-Ouchi K, Sekiguchi F, Yasuno H, Moriya Y, Mori K, Tanaka Y. Antitumor activity of trastuzumab in combination with chemotherapy in human gastric cancer xenograft models. Cancer chemotherapy and pharmacology. 2007; 59:795-805.
67. Sorrentino MF, Kim J, Foderaro AE, Truesdell AG. 5-fluorouracil induced cardiotoxicity: review of the literature. Cardiology journal. 2012; 19:453-458.
68. Andersen LP, Gogenur I, Rosenberg J, Reiter RJ. The Safety of Melatonin in Humans. Clin Drug Investig. 2016; 36:169-175.
69. Lissoni P, Barni S, Mandala M, Ardizzoia A, Paolorossi F, Vaghi M, Longarini R, Malugani F, Tancini G. Decreased toxicity and increased efficacy of cancer chemotherapy using the pineal hormone melatonin in metastatic solid tumour patients with poor clinical status. Eur J Cancer. 1999; 35:1688-1692.
70. Lo JF, Yu CC, Chiou SH, Huang CY, Jan CI, Lin SC, Liu CJ, Hu WY, Yu YH. The epithelial-mesenchymal transition mediator S100A4 maintains cancer-initiating cells in head and neck cancers. Cancer Res. 2011; 71:1912-1923.
71. Chen YW, Lin GJ, Chia WT, Lin CK, Chuang YP, Sytwu HK. Triptolide exerts anti-tumor effect on oral cancer and KB cells in vitro and in vivo. Oral Oncol. 2009; 45:562-568.