
Introduction
Numerous strategies have been developed to stimulate the patients' immune system to reject cancer cells. Of these, immunization with ex vivo generated dendritic cells (DCs) loaded with tumor-associated antigens (TAAs) has shown promise [1-3]. In order to perform their stimulatory function, these DCs are further activated, for which a multitude of stimuli are available [4]. A major question that remains is whether these ex vivo generated DCs resemble a specific DC-subset that can be found in vivo and linked herewith whether these are the best equipped for cancer immunotherapy purposes [5, 6].
Several DC-subsets have been described in mice. Simplified, DCs are subdivided in conventional DCs (cDCs), plasmacytoid DCs (pDCs) and certain tissue-specific populations such as Langerhans’ cells (LCs) in the epidermis. In addition, cDCs are usually subdivided into CD8alfa- and CD8afla+ cDCs [6, 7]. These DC-subsets are endowed with distinct functions and it is believed that cDCs, in particular CD8alfa+ cDCs are key players in the activation of cancer-specific immunity. They produce large amounts of IL-12, as such enabling the polarization of naive CD4+ T cells towards a T helper 1 (TH1) phenotype [8]. This is critical as these TH1 cells have three main functions in the anti-cancer immune response: (1) DC licensing, (2) supporting CD8+ cytotoxic T lymphocyte (CTL) responses and (3) aiding directly in tumor rejection [9]. Moreover, CD8alfa+ cDCs are involved in cross-presentation, a critical process in spontaneous tumor cell rejection. Although subtle differences exist between the human and mouse immune system, it needs to be highlighted that the aforementioned DC-subsets are also found in humans. Here pDCs are characterized by the expression of blood-DC antigen (BDCA) 2 and 4, whereas cDCs either express BDCA1 (CD1c) or BDCA3 (CD141) [6]. Formerly, BDCA3+ cDCs were seen as the counterparts of the mouse CD8alfa+ cDCs as they efficiently cross-present TAAs to CTLs and as both depend on transcription factor BATF3 for their generation [10]. However, recently the BDCA1+ cDCs were shown to closer resemble mouse CD8alfa+ cDCs in terms of IL-12 secretion and cross-presentation [11]. Since it remains a major challenge to generate ex vivo high numbers of DCs that resemble a certain subset, an attractive alternative would be to target DC-subsets in vivo.
Various TAA-delivery systems have been developed, including autologous and allogeneic tumor cell lysates, proteins, peptides, DNA- and mRNA-based formulations. In general these are all safe but as no intrinsic immunogenic factors are present, the inclusion of adjuvants or immunomodulators are crucial for optimal DC activation as otherwise tolerance could be induced [12]. Recently, the development of particulate delivery systems was introduced, in which TAAs are targeted to DCs along with adjuvants. These delivery systems have been extensively reviewed elsewhere [13]. However, TAA-derived proteins and peptides have poor pharmacokinetic properties and are rapidly cleaved. Moreover, as these are exogenous antigens, only cross-presentation can lead to MHC I-mediated CTL induction. Therefore the use of TAA-encoding plasmid DNA or mRNA would be more interesting.
An attractive way to introduce nucleic acids in antigen presenting cells (APCs) is through viral transduction. In this regard, lentivectors (LVs) represent excellent tools for the delivery of viral RNA to APCs [14]. In addition, they ensure persistent transgene expression in the transduced APC, which leads to continual antigen presentation and immunization [15]. Moreover, LVs are intrinsically immunogenic, which leads to activation of innate viral sensing pathways such as Toll like receptors (TLRs), which consequently leads to the induction of strong adaptive immunity [16-18]. In comparative studies of in vivo administration of LVs to ex vivo transduced DCs, peptide or DNA vaccination strategies, stronger TAA-specific immune responses were elicited with increased protection to tumor challenge and survival when immunization was performed with LVs [19-21]. This could be partially explained by the observation that cytokine driven DCs are less potent than DCs activated through microbial/viral signals in the generation of adaptive immunity [22]. Another important advantage of LVs is the ease with which their envelope can be engineered to alter their tropism. This process is called pseudotyping and enables targeting of specific DC-subsets. The latter is advantageous as it reduces the risk of insertional mutagenesis, since proviral DNA is only inserted in the genome of terminally differentiated APCs, which are short-lived after activation.
To answer the purpose of DC-subset specific targeting, we developed the Nanobody (Nb) display technology in which DC-specific Nbs are used as targeting moieties. These are inserted in the LV envelope together with a truncated version of the vesicular stomatitis virus glycoprotein (VSV.GS). This envelope initiates fusion after the Nb binds its antigen on the target cell. Previously we published the proof-of-principle of this technology in which we demonstrated that it is possible to produce Nb displaying LVs at high titers and that these allow in vivo targeting of APCs [23]. We used Nb DC2.1, which was described by De Groeve et al. to recognize a wide range of myeloid cells [24]. In the study, they also reported on Nb DC1.8, which specifically binds to immature bone marrow–derived DCs in vitro. In the present study we compared the in vivo transduction profile and immune stimulatory potential of broad tropism LVs to non-infectious BCII10, APC-targeted DC1.8 and DC2.1 displaying LVs, referred to as VSV.G-, BCII10-, DC1.8- and DC2.1-LVs respectively. We report on the possibility to exclusively transduce cDCs by DC1.8-LVs while also macrophages and pDCs are transduced by DC2.1-LVs. This difference in transduction profile was reflected in their potential to stimulate both antigen-specific CD8+ and CD4+ T cells. Surprisingly, the APC-targeted LVs as well as the non-infectious BCII10-LVs showed a similar therapeutic potential to VSV.G-LVs but for the former LV-types, this depended entirely on type I interferon (IFN).
Results
DC1.8- and DC2.1-LVs target distinct mouse APC-subsets
Recently, we delivered a proof-of-principle on the Nb display technology, demonstrating high titer production of DC2.1-LVs and their ability to target mouse DCs and macrophages in vivo [23]. In this study we additionally utilized Nb DC1.8, which was shown to exclusively bind immature mouse DCs [24], demonstrating that also DC1.8-LVs can be produced at high titers (data not shown).
To compare the transduction profile of DC1.8-LVs to that of VSV.G-, BCII10- and DC2.1-LVs in vivo, Thy1.1 encoding LVs were administered intranodally in C57BL/6 mice. Thirty-six hours later, LNs were resected and Thy1.1 expressing cells characterized by flow cytometry (Figure 1A), demonstrating that VSV.G-LVs transduced B and T cells, macrophages, cDCs and pDCs. In contrast, BCII10-LVs did not infect these cell types. Importantly, DC1.8-LVs specifically transduced CD8alfa+ and CD8alfa- cDCs, while DC2.1-LVs additionally transduced macrophages and pDCs (Figure 1B). In vivo bioluminescence imaging was performed at several time points after intranodal delivery of FLuc encoding LVs to evaluate the persistence of modified cells. Thirty-six hours after administration of infectious LVs, expression of FLuc was observed as a luminescent signal, while no signal was observed after BCII10-LV injection. Luminescence was detectable for over 20 days when VSV.G-LVs were administered. In contrast, FLuc expression decreased to undetectable levels over a period of four days when DC1.8- or DC2.1-LVs were used (Figure 1C).
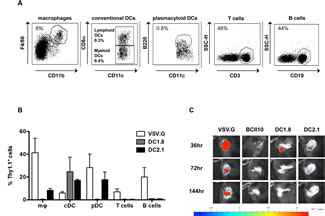
Figure 1: Targeted transduction of murine APCs in situ. (A) To evaluate Thy1.1 expression in macrophages, cDCs, pDCs, B and T cells, LN cells were stained with an anti-Thy1.1 antibody in combination with antibodies directed against CD11b, F4/80, CD11c, B220, CD19 and CD3, respectively. (B) To track the transduced LN cells, we administered 15 μl of PBS (mock) or 10E6 TUs of the different LV pseudotypes encoding Thy1.1 to the inguinal LN of Thy1.2+ C57BL/6 mice. After 36 hours the injected LNs were isolated and flow cytometry was performed on single cell suspensions. The graph summarizes the results of 3 independent experiments as mean ± SEM (n = 3, 1 mouse per experiment). (C) To evaluate the transduction profile and kinetics of VSV.G-, BCII10-, DC1.8- and DC2.1-LVs in vivo, we injected 10E6 TUs of each LV stock encoding Fluc in the inguinal LN of C57Bl/6 mice. Thirty-six, 72 and 144 hours later, in vivo bioluminescence imaging was performed to obtain bioluminescent pseudo-color images superimposed on gray-scale photographs in which high luminescence is shown in red and weak luminescence in blue (n = 3, 1 mouse per experiment). The color scale underneath the images represents the correlation between the luminescent signal and the absolute amount of counts (light units).
Immunization with targeted LVs elicits CD8+ T lymphocyte responses, be it to a lesser extent than immunization with broad tropism LVs
Since CD8+ CTLs play a critical role in the control of cancer, we first evaluated the proliferation of CD8+ T cells upon immunization with ovalbumin encoding LVs. We transferred CD45.2+ OT-I cells to CD45.1+ mice after which these were immunized intranodally. Five days later, the injected LNs were isolated to evaluate OT-I proliferation in flow cytometry. Although proliferation of CD8+ T cells was observed in mice immunized with BCII10-, DC1.8- or DC2.1-LVs, it did not reach the levels observed in mice immunized with VSV.G-LVs (Figure 2A-B). To further investigate the ability of the targeted LVs to elicit functional CD8+ T cell responses, the percentage of IFN-gamma+ CD8+ T cells was evaluated by enriching the CD45.2+ OT-I cells from the injected LN and spleen, and in vitro restimulating them for 72 hours. This experiment demonstrated comparable percentages for VSV.G-, DC1.8- and DC2.1-LVs while this percentage decreased by half for BCII10-LVs (Figure C-D).
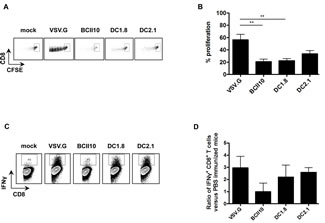
Figure 2: Intranodal delivery of targeted LVs encoding ovalbumin results in stimulation of ovalbumin-specific CD8+ T cells. (A and B) CFSE-labeled OT-I cells were adoptively transferred to C57BL/6 mice one day prior to intranodal administration of PBS, 5x10E5 TUs VSV.G-, BCII10-, DC1.8- or DC2.1-LVs encoding ovalbumin. Five days later, LNs were resected, single cell suspensions prepared, after which induction of OT-I proliferation was evaluated by flow cytometry. Average values of the PBS group were substracted from the other values (n = 3, 2 mice per experiment). (C and D) To recover a reliable amount of CD45.2+ OT-I cells we adoptively transferred them to CD45.1+ C57BL/6 mice in which the CD45 marker served as a tool to distinguish which CD8+ T cells were OT-I and which were endogenous cells. Five days after immunization, the CD45.2+ fraction was isolated from single cell suspensions, which were subsequently restimulated with peptide loaded CD45.1+ splenocytes. After 72 hours, the amount of IFN-gamma + CD8+ T cells was evaluated in flow cytometry. Average values of the PBS group were substracted from the other values (n = 2, 2 mice per experiment).
The generation of antigen-specific CTLs upon immunization with targeted LVs depends on type I IFNs
To evaluate the cytotoxic potential of antigen-specific CD8+ T cells, we performed an in vivo cytotoxicity assay. Although lysis of target cells was observed in mice immunized with DC1.8- or DC2.1-LVs, it did not reach the levels observed in mice immunized with VSV.G-LVs. In addition, as observed in the OT-I proliferation assay, immunization with BCII10-LVs also resulted in a certain degree of CTL induction (Figure 3A-B). Since pDCs and cDCs produce type I IFNs upon infection with VSV.G-LVs [11, 17], we addressed their role in the induction of ovalbumin-specific CTLs. Therefore we performed a CTL assay in IFNAR ko mice. The CTL response after administration of VSV.G-LVs was not significantly affected, whereas the CTL response after administration of DC1.8- and DC2.1-LVs as well as BCII10-LVs was dependent on type I IFNs (Figure 3C). We hypothesized that this might be explained by a different activation of the innate immune system by the broad tropism LVs. Therefore we evaluated the induction of TNF-alfa as this cytokine was previously described to be upregulated upon systemic delivery of broad tropism LVs. We observed that the production of TNF-alfa was most pronounced upon delivery of broad tropism LVs, which strengthens the hypothesis that broad tropism and targeted LVs use other innate cytokines such as TNF-alfa and type I IFN, respectively (Figure 3D).
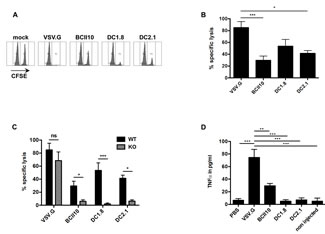
Figure 3: Intranodal delivery of targeted LVs encoding ovalbumin results in stimulation of cytotoxic ovalbumin specific CD8+ T cells and depends entirely on type I IFN. Ovalbumin-specific cytotoxicity of the induced CD8+ T cells was evaluated via an in vivo CTL assay. This assay was performed in wild type (A and B) or in IFN type I receptor (IFNAR) ko mice (C), five days after intranodal administration of PBS, 5x10E5 TUs VSV.G-, BCII10-, DC1.8- or DC2.1-LVs encoding ovalbumin. Average values of the PBS group were substracted from the other values (n = 2, 3 mice per experiment). To evaluate the induction of the innate cytokine TNF-alfa, LNs were isolated and placed in culture three hours after they were injected with nothing, PBS, broad tropism, BCII10- or targeted-LVs. Twenty hours later, the supernatants of the crushed LNs were screened for the presence of TNF-alfa via ELISA (D) (n=2, 2 mice per experiment).
Immunization with targeted LVs elicits distinct antigen-specific CD4+ T cell responses
Although CTLs are considered to be the main effector cells in anti-tumor immunity, accumulating evidence shows that CD4+ T cells are another critical component [25, 26]. Therefore, we evaluated the expansion of ovalbumin-specific CD4+ T cells upon intranodal immunization. Proliferation of OT-II cells was strongest after immunization with VSV.G-LVs. Notably, the proliferation of OT-II cells was comparable after administration of targeted as well as BCII10-LVs (Figure 4A). To ensure the specificity of this response, we immunized mice with VSV.G-LVs encoding Trp2, demonstrating a lack of proliferation. As BCII10-LVs are unable to transduce APCs, we hypothesized that these viral particles were engulfed via an alternative route, resulting in the uptake of the ovalbumin encoding viral genome or possibly plasmid DNA or proteins [27]. To evaluate this hypothesis, we produced BCII10-LVs encoding Trp2 in the presence of an ovalbumin encoding eukaryotic expression vector. This allows non-specific packaging of plasmid DNA and proteins [28]. Subsequently, mice were immunized with these LVs. Strong proliferation of OT-II cells was induced (Figure 4B). Next we scrutinized whether this proliferation was due to the presence of plasmid DNA or protein. Therefore, mice were immunized with LVs that were pre-treated with DNase or proteinase K to remove plasmid DNA or proteins respectively. To investigate the role of the viral genome, mice were injected 24 hours prior to immunization with the RT inhibitor Truvada [29]. Proliferation of ovalbumin-specific CD4+ OT-II cells was observed in all conditions, except when LVs were pre-treated with proteinase K suggesting that the presence of protein within the LV stock is the main inducer of OT-II proliferation (Figure 4C).
Since administration of BCII10-LVs was sufficient to induce antigen-specific CD4+ T cell proliferation, but had only minor effects on CD8+ T cell induction when compared to targeted LVs, we addressed which CD4+ T cell phenotype was stimulated by the LVs. Therefore we evaluated the secretion of cytokines by CD4+ T cells that were restimulated in vitro. The CD4+ T cells from mice immunized with VSV.G- and DC2.1-LVs induced a similar TH1 profile with similar amounts of IFN-gamma, IL-2, and IL-10. To our surprise, mice immunized with DC1.8-LVs produced less of these cytokines, which made their profile similar to the one induced by the BCII10-LVs (Figure 4D).
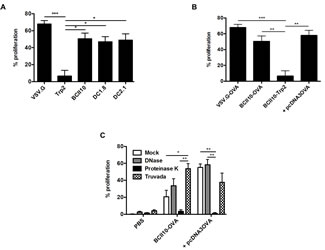
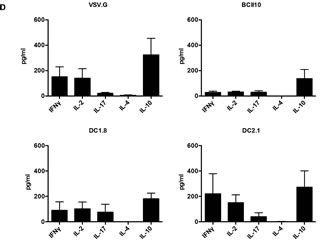
Figure 4: Intranodal delivery of targeted as well as BCII10-LVs encoding ovalbumin results in stimulation of ovalbumin-specific CD4+ T cells. (A-C) CFSE labeled OT-II cells were adoptively transferred one day prior to intranodal administration of PBS or 5x10E5 TUs of the respective LVs. Five days later, LNs were resected and single cell suspensions prepared to evaluate the induction of OT-II proliferation by flow cytometry. Average values of the PBS group were substracted from the other values (n = 2, 2 mice per experiment). (D) Alternatively, CD4+ T cells were sorted out of the LN-derived single cell suspensions and restimulated with ISQAVHAAHAEINEAGR peptide-loaded bone marrow-derived DCs for 48 hours after which supernatants were analyzed for their presence of IFN-gamma, IL-2, IL-17, IL-4 and IL-10 in ELISA. Average values of the PBS group were substracted from the other values (n = 3, 4 mice pooled in two groups per experiment). More specifically in (A) VSV.G-, BCII10-, DC1.8- or DC2.1-LVs encoding ovalbumin as well as VSV.G-LVs encoding Trp2 were injected, in (B) VSV.G- and BCII10-LVs encoding ovalbumin, BCII10-LVs encoding Trp2 or BCII10-LVs encoding Trp2 produced in the presence of an ovalbumin encoding plasmid (depicted as + pcDNA3OVA) were administered and in (E) BCII10-LVs encoding ovalbumin or Trp2 + pcDNA3OVA and pretreated with 10 U/ml DNase, 100 μg/ml Proteinase K were used or mice were injected intraperitoneally 24 hours earlier with 5 ng of the RT inhibitor, Truvada.
Immunization with broad tropism or targeted LVs results in similar therapeutic efficacy
To evaluate the therapeutic potential of the LVs, mice bearing E.G7-OVA tumors were immunized twice at a seven-day interval. The median survival of mice treated with PBS was 14 days while this was prolonged to 19, 20, 17 and 23 days upon immunization with ovalbumin encoding BCII10-, DC1.8-, DC2.1- and VSV.G-LVs respectively. Of note, although DC2.1-LVs induced the lowest median survival in days, they did show the longest dispersion in time with the last mouse surviving till day 32 (Figure 5).
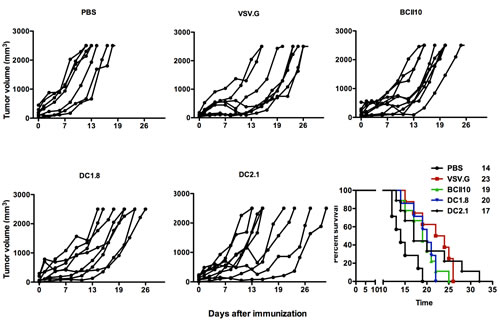
Figure 5: Intranodal vaccination of targeted LVs encoding ovalbumin results in prolonged survival in an E.G7-OVA model. To evaluate the therapeutic potential of the targeted LVs, C57BL/6 mice were challenged on day 0 with 3x10E5 E.G7-OVA cells. When palpable tumors were present mice were immunized intranodally with PBS or 10E6 TUs VSV.G-, BCII10-, DC1.8- or DC2.1-LVs encoding ovalbumin. Seven days later, the treatment was repeated. Tumor growth and survival were examined every two days. The results shown are representative for two independent experiments (n = 2, 4 mice per experiment).
Discussion
Targeting APCs is believed to be key to develop safe and efficacious LV-based anti-cancer vaccines [30-34]. Here we report on the Nb display technology to target LVs to mouse APC-subsets.
We compared the transduction profile of broad tropism LVs to LVs harboring the β-lactamase-specific Nb BCII10, or the APC-targeting Nbs DC1.8 or DC2.1. We chose to deliver these LVs to the LN as this organ contains a relative high number of DCs [35] and as it has been described that lower antigen doses are required to achieve similar immune responses [36]. Tracking the transduced cells within the LN demonstrated the exclusive transduction of cDCs by DC1.8-LVs, while DC2.1-LVs transduced cDCs, pDCs and macrophages. Of note, both the CD8alfa+ and CD8alfa- cDC fractions were transduced by these LVs. The VSV.G-LVs additionally transduced B and T cells which is expected as VSV.G is the envelope of choice for efficient transduction of a broad range of cells [37]. Of note, it has been postulated that targeting LVs would increase their transduction efficiency [38]. However, similar to the observations of Ageichik et al., who targeted LVs to MHC II-expressing cells [33], we observed a lower transduction efficiency with DC1.8- and DC2.1-LVs compared to VSV.G-LVs. In contrast, Yang et al. observed that LVs targeted to DC-SIGN were more efficient in transduction of CD11c+ cells than VSV.G-LVs [39]. The latter might be explained by the type of antigen targeted, both in terms of its expression level and function. For instance, DC-SIGN is a C-type lectin, involved in the capture of HIV-1 and furthermore protects it from degradation in early endosomes [40]. The results from the in vivo bioluminescence imaging upon delivery of FLuc encoding LVs, support the notion that only short-lived APCs are transduced by DC1.8- and DC2.1-LVs. As expected, the luminescence signal lasted for more than 20 days after VSV.G-LV delivery, implying the transduction of long-lived, possibly non-APCs, which increases the risk of insertional mutagenesis [41].
Because DC1.8- and DC2.1-LVs deliver their cargo to different APC-subsets, we evaluated and compared their immunogenicity. It is generally accepted that cDCs within the LNs are required to present antigens to CD8+ T cells, while migrated DCs and pDCs are insufficient to prime T cells [42, 43]. Moreover within the cDC population it was found that the CD8alfa+ DEC205+ DC population surpasses other APC-subsets in cross-presenting skills [44-47]. Fuertes et al. further demonstrated that host type I IFNs are critical for the innate immune recognition of a growing tumor through signalling on the CD8alfa+ DC fraction [48]. In line with this observation, it was described that cDCs in a human setting need type I IFN and TNF-alfa, released from non-replicating HIV-1 activated pDCs, to induce their full bystander maturation [49, 50]. Although cDCs seem to have a prominent role, the latter study stresses the importance of the pDCs. However, it was also shown in certain tumor models that tumor draining LN-resident pDCs expressed the enzyme indoleamine 2,3-dioxygenase, which induces the generation of regulatory T cells [51, 52]. The role of LN-resident macrophages in T cell stimulation is less defined. Although it was shown that CD8+ T cells interact with virally infected macrophages and DCs, macrophages were not shown to fully prime anti-viral CD8+ T cells [35]. Moreover, Albert et al. showed that human DCs but not macrophages efficiently presented antigens from apoptotic cells to CD8+ T cells [53]. Nonetheless, some studies report that macrophages can cross-prime CD8+ T cells [54]. In order to shed light on the importance of macrophages and pDCs in the stimulation of T cell responses, we evaluated the expansion of CD4+ and CD8+ T cells as well as their functionality upon immunization with DC1.8-, DC2.1- and VSV.G-LVs. Although immunization with VSV.G-LVs elicited stronger CD8+ T cell responses, we demonstrated that DC1.8- and DC2.1-LVs were equally potent in the induction of CD8+ T cells with lytic and more importantly therapeutic efficacy. Interestingly, we observed that the induced CD4+ cytokine profile of DC2.1-LVs closely resembled that of VSV.G-LVs as both elicited a similar amount of IFN-gamma, IL-2 and IL-10. These cytokines are typical for a TH1 response and therefore crucial for a potent antitumor immune response. In contrast, DC1.8-LVs induced less of these cytokines while its profile resembled more that of the non-infectious BCII10 LVs, suggesting that presentation of antigens by cDCs alone is probably not as adequate as presentation by a broad range of APCs. Another striking observation was the complete abrogation of CTL inducing capacity of DC1.8- and DC2.1-LVs in IFNAR ko mice. The latter was unexpected, as it was described that pDCs and LN-resident macrophages are the main producers of type I IFNs upon LV transduction [35, 55]. Therefore, we only expected to observe a reduction in immunogenicity in IFNAR ko mice for VSV.G- and DC2.1-LVs. However, the immunogenicity of VSV.G-LVs was unaffected, whereas that of DC2.1- and DC1.8-LVs was compromised. The latter can in part be explained by our previous observations that LVs activate TLR3/7 and as such induce type I IFN production by DCs [17]. The ability of VSV.G-LVs to elicit CTL responses in IFNAR ko mice suggests that these activate other innate pathways that are able to bypass the need for type I IFNs. The latter hypothesis was strengthened by the observation that VSV.G-LVs triggered the highest production of the pro-inflammatory cytokine TNF-alfa [49, 56].
Another surprise was the ability of the BCII10-LVs to induce immune responses, despite their inability to transduce cells. The latter was explained by the presence of protein contaminants within the LV preparation and highlights the need for the development of easy to use and high efficiency LV-purification systems. Although, immunization with BCII10-LVs resulted in T cell responses, it is important to note that the CD4+ and CD8+ T cell responses upon immunization with infectious LVs were stronger. These observations are in line with the observations of Ageichik et al., who compared the induction of IFN-gamma+ CD8+ T cells upon administration of LVs targeted to MHC II or carcinoembryonic antigen [33]. However, in therapy, no striking differences were observed between BCII10-, DC1.8-, DC2.1- and VSV.G-LVs. The latter has not been described but this could be explained by the fact that previous reports on the use of targeted LVs for anti-cancer vaccination only compared LVs encoding the tumor antigen versus LVs encoding an irrelevant antigen [39, 57]. Importantly, studies using nanoparticles demonstrate that the strength of the immune responses induced is co-determined by the cell population that engulfs the particle as well as the cargo of these particles [58, 59]. We delivered the BCII10-LVs intranodally, which can be considered as an anatomical targeting of virus like particles [31]. It is therefore likely that BCII10-LVs have been engulfed by DCs and as such deliver proteins as well as viral components, ie activation signals which ultimately led to the induction of a small amount of TNF-alfa, IFN type I and IL-10.
In conclusion, we demonstrate that LV-targeting to distinct DC-subsets via the Nb display
technology is feasible and induces qualitatively distinct T cell responses that are equally potent in the control of cancer. Furthermore, this targeting strategy increases the LV-based vaccines’ safety through faster LV-clearance. In the future, experiments with tumor relevant antigens like MageA3, Trp2, p62 and brachyury, have to confirm the observed results with the egg white derived protein ovalbumin [60-63]. Furthermore the possible relief of side effects like vitiligo [64] with APC-targeted LVs has to be evaluated. Since we possess Nbs that target human cDCs alone [65] or human cDCs, pDCs and macrophages [23], this method represents a good option to produce LV-based vaccines for preclinical studies in cancer.
Materials and Methods
Mice and cell cultures
Six to 12 week old female CD45.2+/Thy1.2+ C57BL/6 mice were purchased from Harlan. The CD45.1+/Thy1.2+ C57BL/6 mice were obtained from J. Van Ginderachter (Vrije Universiteit Brussel). Transgenic OT-I and OT-II mice were purchased from Charles River. Mice lacking the IFN type I receptor (IFNAR ko) were obtained from C. Libert (Universiteit Gent). All animals were handled according to the institutional guidelines and experiments were approved by the Ethical Committee for use of laboratory animals of the Vrije Universiteit Brussel. The human embryonal kidney 293T cell line (HEK293T) and T-cell lymphoma cell line E.G7-OVA were cultured as recommended by the American Type Culture Collection (ATCC). Single cell suspensions were prepared from spleens and lymph nodes (LNs) as described [23].
Lentiviral vector production
Plasmids. The packaging plasmid pCMVΔR8.9 and VSV.G encoding plasmid pMD.G were a gift from Dr. D. Trono (University of Geneva). The plasmid pUB6-VSV.GS, which encodes the binding-defective, but fusion-competent VSV.G was described by Zhang et al. [66]. The plasmids pSIN-Thy1.1, pHR trip CMV luc2-Ires-tNGFR SIN, pHR’ trip CMV Ii80tOVA-Ires-tNGFR SIN, pcDNA1 Ii80tOVA, pHR’ trip CMV Nb BCII10 SIN and pHR’ trip CMV Nb DC2.1 SIN were described [17, 23, 28, 67]. The sequence encoding the membrane bound form of Nb DC1.8 was cloned into the pHR’ trip CMV SIN vector following the same strategy as described for Nb BCII10 and DC2.1 [23].
Virus production and characterization. Production of broad tropism and targeted LVs as well as their characterization was performed as described [23].
In vivo bioluminescence imaging
In vivo bioluminescence imaging was performed at the indicated time points to visualize in situ transduction of cells after intranodal delivery of 10E6 transducing units (TUs) of FLuc encoding LVs. The procedure was performed as described [68].
In vivo tracking of lentivirally transduced cells
To evaluate the transduction profile of the LVs, C57BL/6 mice were injected in the inguinal LN with 10E6 TUs of Thy1.1 encoding LVs. Thirty-six hours later, mice were euthanized, injected LNs isolated, single cell suspensions prepared and cells characterized by flow cytometry [23].
In vivo OT-I and OT-II stimulation with ELISA on restimulated OT-II cells
One day prior to immunization, 1-2 x 10E6 purified and carboxyfluorescein diacetate succinimidine ester (CFSE, Invitrogen) labeled CD8+ OT-I or CD4+ OT-II spleen cells were adoptively transferred to mice by intravenous injection. Mice were immunized through single administration of 5 x 10E5 TUs of broad tropism or Nb-targeted LVs encoding ovalbumin into the inguinal LN. Administration of PBS or BCII10-LVs served as a control. Five days post-immunization, proliferation of T cells was analyzed. Therefore, LNs were isolated, single cell suspensions prepared, stained with a peridinin-chlorophyll protein-Cy5.5 (PerCP-Cy5.5) conjugated antibody against CD8 or CD4 (BD) and analyzed via flow cytometry as described [20]. When mice were adoptively transferred with OT-II cells, part of the single cell suspensions were used to sort and restimulate the CD4+ T cells. Therefore, 5 x 10E5 of CD4+ T cells were plated in a 48-well plate in 500 µl of culture medium and restimulated for 48 hours with bone marrow-derived DCs that were loaded for two hours with 5 μM ISQAVHAAHAEINEAGR peptide (Eurogentec) at a 10:1 ratio. Supernatants were screened in enzyme-linked immunosorbent assay (ELISA) for the presence of IFN-gamma, IL-2, IL-17, IL-4 and IL-10 (eBioscience).
Intracytoplasmic staining of IFN-gamma
One day prior to immunization, CD45.1+ C57BL/6 mice were injected with 5 x 10E6 CD45.2+ OT-I cells. Mice were immunized as described above. Five days post-immunization, spleens and LNs were isolated and CD45.2+ OT-I cells sorted using anti-CD45.2 beads (Miltenyi). Subsequently, 5 x 10E5 of these cells were plated in a 48-well plate in 500 µl of RPMI+ (RPMI-1640 medium supplemented with 5% FCI, 5% supplements and 50 μmol/L β-mercaptoethanol) and restimulated with SIINFEKL loaded CD45.1 splenocytes at a 10:1 ratio. Two days later Golgi plug was added (1mg/ml, BD) and intracytoplasmic expression of IFN-gamma was evaluated the next day by intracellular staining with a phycoerythrin-Cy7 (PE-Cy7) conjugated anti-IFN-gamma antibody (eBioscience) followed by flow cytometric analysis.
In vivo cytotoxic T lymphocyte assay
Mice were immunized through intranodal administration of 5 x 10E5 TUs of broad tropism or targeted ovalbumin encoding LVs. Administration of PBS or BCII10-LVs served as a control. The in vivo cytotoxicity assay was performed five days post-immunization as described [20].
Evaluation of TNF-alfa secretion after intranodal immunization
Mice were injected intranodally with 5 x 10E5 TUs of broad tropism or targeted ovalbumin encoding LVs. No injection, PBS or BCII10-LVs served as control. Three hours later, LNs were resected and transferred to a 24-well plate with 300 µl of RPMI+. Twenty hours later, supernatants were screened in ELISA for the presence of TNF-alfa (eBioscience).
Flow cytometry
Staining of surface markers was performed as described [23]. Data were collected using a FACS Canto flow cytometer (BD) and analyzed using FACSDivaTM (BD) or FlowJoTM (Tristar Inc.) software.
Tumor rejection experiments
Mice were grafted subcutaneously with 3 x 10E5 E.G7-OVA tumor cells at the tail base. Mice bearing palpable tumors were immunized twice with 10E6 TUs of LVs encoding ovalbumin at a seven-day interval. Tumor growth was monitored. Mice were killed when the tumor reached a volume of 2500 mm3.
Statistical analyses
A one-way ANOVA followed by a Bonferroni’s multiple comparison test was performed. Sample sizes and number of times experiments were repeated are indicated in the figure legends. Number of asterisks in the figures indicates the level of statistical significance as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. The results are shown in a column graph or table as the mean ± SEM. Survival was visualized in a Kaplan-Meier plot. Differences in survival were analyzed by the log-rank test.
Acknowledgements
We thank Elsy Vaeremans, Petra Roman and Xavier Debaere for technical assistance. We furthermore thank Prof. Tony Lahoutte for the use of the imaging facilities and Prof. Claude Libert for providing the IFNAR ko mice.
This research was performed with the financial support of the Research foundation Flanders (FWO-V), the Agency of Innovation by Science and Technology (IWT-SBO), the Interuniversity Attraction Poles Program (IUAP), the Belgian State-Belgian Science Policy and the research committee of the VUB (OZR). CG and KB are funded by the FWO-V.
Authorship Contributions
Cleo Goyvaerts: contributed to the design of the study, performed and analyzed most experiments, and assisted in the writing of the manuscript
De Groeve Kurt: generated and characterized the Nbs used in this study
Sandra Van Lint: performed and analyzed certain experiments, and assisted in the writing of the manuscript
Carlo Heirman: generated several of the lentiviral transfer vectors used in this manuscript and assisted in the writing of the manuscript
Jo A. Van Ginderachter: bred, characterized and provided transgenic mouse strains and assisted in the writing of the manuscript
Patrick De Baetselier: generated and characterized the Nbs used in this study, contributed to the design of the study and assisted in the writing of the manuscript
Geert Raes: generated and characterized the Nbs used in this study, contributed to the design of the study and assisted in the writing of the manuscript
Kris Thielemans: contributed to the design of the study and assisted in the writing of the manuscript
Karine Breckpot: contributed to the design of the study, performed and analyzed certain experiments, and assisted in the writing of the manuscript
Disclosure of Conflicts of Interest
The authors and their institutions have no financial or other relationship with other people or organizations that may inappropriately influence this work.
Reference
1. Van Tendeloo VF, Van de Velde A, Van Driessche A, Cools N, Anguille S, Ladell K, Gostick E, Vermeulen K, Pieters K, Nijs G, Stein B, Smits EL, Schroyens WA, et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc Natl Acad Sci USA. 2010; 107:13824–13829.
2. Akiyama Y, Oshita C, Kume A, Iizuka A, Miyata H, Komiyama M, Ashizawa T, Yagoto M, Abe Y, Mitsuya K, Watanabe R, Sugino T, Yamaguchi K, et al. α-type-1 polarized dendritic cell-based vaccination in recurrent high-grade glioma: a phase I clinical trial. BMC Cancer. 2012; 12:623.
3. Wilgenhof S, Van Nuffel AM, Benteyn D, Corthals J, Aerts C, Heirman C, Van Riet I, Bonehill A, Thielemans K, Neyns B. A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann Oncol. 2013; 24(10):2686–93.
4. Tuyaerts S, Aerts JL, Corthals J, Neyns B, Heirman C, Breckpot K, Thielemans K, Bonehill A. Current approaches in dendritic cell generation and future implications for cancer immunotherapy. Cancer Immunol Immunother. 2007; 56:1513–1537.
5. Gallois A and Bhardwaj N. A needle in the “cancer vaccine” haystack. Nat Med. 2010; 16(8):854–6.
6. Palucka K, Banchereau J, Mellman I. Designing vaccines based on biology of human dendritic cell subsets. Immunity. 2010; 33:464–478.
7. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013; 31:563–604.
8. Maldonado-López R, De Smedt T, Michel P, Godfroid J, Pajak B, Heirman C, Thielemans K, Leo O, Urbain J, Moser M. CD8alpha+ and CD8alpha- subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J Exp Med. 1999; 189:587–592.
9. Bonehill A, Heirman C, Tuyaerts S, Michiels A, Breckpot K, Brasseur F, Zhang Y, Van Der Bruggen P, Thielemans K. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J Immunol. 2004; 172:6649–6657.
10. P Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, Keller AM, Joffre O, Zelenay S, Nye E, Le Moine A, Faure F, Donckier V, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med. 2010; 207:1261–1271.
11. Nizzoli G, Krietsch J, Weick A, Steinfelder S, Facciotti F, Gruarin P, Bianco A, Steckel B, Moro M, Crosti M, Romagnani C, Stölzel K, Torretta S, et al. Human CD1c+ dendritic cells secrete high levels of IL-12 and potently prime cytotoxic T cell responses. Blood. 2013; 932–942.
12. Melief C. Cancer immunotherapy by dendritic cells. Immunity. 2008; 29:372–383.
13. Joshi M, Unger W, Storm G. Targeting tumor antigens to dendritic cells using particulate carriers. J Control Release. 2012; 161:25–37.
14. Breckpot K, Aerts JL, Thielemans K. Lentiviral vectors for cancer immunotherapy: transforming infectious particles into therapeutics. Gene Ther. 2007; 14:847–862.
15. Arce F, Rowe HM, Chain B, Lopes L, Collins MK. Lentiviral vectors transduce proliferating dendritic cell precursors leading to persistent antigen presentation and immunization. Mol Ther. 2009; 17:1643–1650.
16. Breckpot K, Emeagi PU, Thielemans K. Lentiviral vectors for anti-tumor immunotherapy. Curr Gene Ther. 2008; 8:438–448.
17. Breckpot K, Escors D, Arce F, Lopes L, Karwacz K, Van Lint S, Keyaerts M, Collins M. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J Virol. 2010; 84:5627–5636.
18. Garaude J and Blander JM. Attacking tumor cells with a dual ligand for innate immune receptors. Oncotarget. 2012; 3(4):361–2.
19. Chapatte L, Colombetti S, Cerottini J-C, Lévy F. Efficient induction of tumor antigen-specific CD8+ memory T cells by recombinant lentivectors. Cancer Res. 2006; 66:1155–1160.
20. Dullaers M, Van Meirvenne S, Heirman C, Straetman L, Bonehill A, Aerts JL, Thielemans K, Breckpot K. Induction of effective therapeutic antitumor immunity by direct in vivo administration of lentiviral vectors. Gene Ther. 2006; 13:630–640.
21. Rusakiewicz S, Dosset M, Mollier K, Souque P, Charneau P, Wain-Hobson S, Langlade-Demoyen P, Adotévi O. Immunogenicity of a recombinant lentiviral vector carrying human telomerase tumor antigen in HLA-B*0702 transgenic mice. Vaccine. 2010; 28:6374–6381.
22. Joffre O, Nolte MA, Spörri R, Reis e Sousa C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev. 2009; 227:234–247.
23. Goyvaerts C, De Groeve K, Dingemans J, Van Lint S, Robays L, Heirman C, Reiser J, Zhang XY, Thielemans K, De Baetselier P, Raes G, Breckpot K. Development of the Nanobody display technology to target lentiviral vectors to antigen-presenting cells. Gene Ther. 2012; 1–8.
24. De Groeve K, Deschacht N, De Koninck C, Caveliers V, Lahoutte T, Devoogdt N, Muyldermans S, De Baetselier P, Raes G. Nanobodies as tools for in vivo imaging of specific immune cell types. J Nucl Med. 2010; 51:782–789.
25. Velders MP, Markiewicz MA, Eiben GL, Kast WM. CD4+ T cell matters in tumor immunity. Int Rev Immunol. 2003; 22:113–140.
26. Sun JZ, Xiao WH, Yu L. [Recent research advances on function of CD4+T lymphocytes]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2010; 18:544–548.
27. Merten OW, Charrier S, Laroudie N, Fauchille S, Dugué C, Jenny C, Audit M, Zanta-Boussif MA, Chautard H, Radrizzani M, Vallanti G, Naldini L, Noguiez-Hellin P. Large-scale manufacture and characterization of a lentiviral vector produced for clinical ex vivo gene therapy application. Hum Gene Ther. 2011; 22:343–356.
28. Breckpot K, Dullaers M, Bonehill A et al. Lentivirally transduced dendritic cells as a tool for cancer immunotherapy. J. Gene Med. 2003; 5:654–667.
29. Denton PW, Krisko JF, Powell DA, Mathias M, Kwak YT, Martinez-Torres F, Zou W, Payne DA, Estes JD, Garcia JV. Systemic administration of antiretrovirals prior to exposure prevents rectal and intravenous HIV-1 transmission in humanized BLT mice. PLoS One. 2010; 5:e8829.
30. Cubillos-Zapata C, Guzman E, Turner A, Gilbert SC, Prentice H, Hope JC, Charleston B. Differential effects of viral vectors on migratory afferent lymph dendritic cells in vitro predict enhanced immunogenicity in vivo. J Virol. 2011; 85:9385–9394.
31. Paulis LE, Mandal S, Kreutz M, Figdor CG. Dendritic cell-based nanovaccines for cancer immunotherapy. Curr Opin Immunol. 2013; 25:389–95.
32. Bryson PD, Zhang C, Lee C-L, Wang P. A tetracycline-regulated cell line produces high-titer lentiviral vectors that specifically target dendritic cells. J Vis Exp. 2013. doi:10.3791/50606.
33. Ageichik A, Buchholz CJ, Collins MK. Lentiviral vectors targeted to MHC II are effective in immunization. Hum Gene Ther. 2011; 22(10):1249–54.
34. Blander JM and Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006; 440:808–812.
35. Hickman HD, Li L, Reynoso GV, Rubin EJ, Skon CN, Mays JW, Gibbs J, Schwartz O, Bennink JR, Yewdell JW. Chemokines control naive CD8+ T cell selection of optimal lymph node antigen presenting cells. J Exp Med. 2011; 208:2511–2524.
36. Jonuleit H, Giesecke-Tuettenberg A, Tüting T, Thurner-Schuler B, Stuge TB, Paragnik L, Kandemir A, Lee PP, Schuler G, Knop J, Enk AH. A comparison of two types of dendritic cell as adjuvants for the induction of melanoma-specific T-cell responses in humans following intranodal injection. Int J Cancer. 2001; 93(2):243–51.
37. Cronin J, Zhang X-Y, Reiser J. Altering the tropism of lentiviral vectors through pseudotyping. Curr Gene Ther. 2005; 5:387–398.
38. Maurice M, Verhoeyen E, Salmon P, Trono D, Russell SJ, Cosset FL. Efficient gene transfer into human primary blood lymphocytes by surface-engineered lentiviral vectors that display a T cell-activating polypeptide. Blood. 2002; 99:2342–2350.
39. Yang L, Yang H, Rideout K, Cho T, Joo KI, Ziegler L, Elliot A, Walls A, Yu D, Baltimore D, Wang P. Engineered lentivector targeting of dendritic cells for in vivo immunization. Nat Biotechnol. 2008; 26:326–334.
40. Geijtenbeek TBH, Engering A, Van Kooyk Y. DC-SIGN, a C-type lectin on dendritic cells that unveils many aspects of dendritic cell biology. J Leukoc Biol. 2002; 71:921–931.
41. Barry MA, Ikeda Y, Sakuma T. Lentiviral vectors: basic to translational. Biochem J. 2012; 443:603–618.
42. Goold HD, Escors D, Conlan TJ, Chakraverty R, Bennett CL. Conventional dendritic cells are required for the activation of helper-dependent CD8 T cell responses to a model antigen after cutaneous vaccination with lentiviral vectors. J Immunol. 2011; 186:4565–4572.
43. Flacher V, Tripp CH, Haid B, Kissenpfennig A, Malissen B, Stoitzner P, Idoyaga J, Romani N. Skin langerin+ dendritic cells transport intradermally injected anti-DEC-205 antibodies but are not essential for subsequent cytotoxic CD8+ T cell responses. J Immunol. 2012; 188:2146–55.
44. Den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. 2000; 192(12):1685–96.
45. Iyoda T, Shimoyama S, Liu K, Omatsu Y, Akiyama Y, Maeda Y, Takahara K, Steinman RM, Inaba K. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J Exp Med. 2002; 195:1289–1302.
46. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee HW, Park CG, Steinman RM, Nussenzweig MC. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007; 315:107–111.
47. Suleman M, Galea S, Gavard F, Merillon N, Klonjkowski B, Tartour E, Richardson J. Antigen encoded by vaccine vectors derived from human adenovirus serotype 5 is preferentially presented to CD8+ T lymphocytes by the CD8α+ dendritic cell subset. Vaccine. 2011; 29:5892–5903.
48. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011; 208(10):2005–16.
49. Rossetti M, Gregori S, Hauben E, Brown BD, Sergi LS, Naldini L, Roncarolo MG. HIV-1-derived lentiviral vectors directly activate plasmacytoid dendritic cells, which in turn induce the maturation of myeloid dendritic cells. Hum Gene Ther. 2011; 22:177–188.
50. Fonteneau JF, Larsson M, Beignon AS, McKenna K, Dasilva I, Amara A, Liu YJ, Lifson JD, Littman DR, Bhardwaj N. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J Virol. 2004; 78:5223–5232.
51. Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler P, Mellor AL, He Y, Munn DH. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood. 2009; 113:6102–6111.
52. Coquerelle C and Moser M. DC subsets in positive and negative regulation of immunity. Immunol Rev. 2010; 234:317–334.
53. Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998; 392:86–89.
54. Schliehe C, Redaelli C, Engelhardt S, Fehlings M, Mueller M, van Rooijen N, Thiry M, Hildner K, Weller H, Groettrup M. CD8- Dendritic Cells and Macrophages Cross-Present Poly(D,L-lactate-co-glycolate) Acid Microsphere-Encapsulated Antigen In Vivo. J Immunol. 2011; 187:2112–2121.
55. Brown BD, Sitia G, Annoni A, Hauben E, Sergi LS, Zingale A, Roncarolo MG, Guidotti LG, Naldini L. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood. 2007; 109:2797–2805.
56. Vandendriessche T, Thorrez L, Acosta-Sanchez A, Petrus I, Wang L, Ma L, DE Waele L, Iwasaki Y, Gillijns V, Wilson JM, Collen D, Chuah MK. Efficacy and safety of adeno-associated viral vectors based on serotype 8 and 9 vs. lentiviral vectors for hemophilia B gene therapy. J Thromb Haemost. 2007; 5:16–24.
57. Gennari F, Lopes L, Verhoeyen E, Marasco W, Collins MK. Single-chain antibodies that target lentiviral vectors to MHC class II on antigen-presenting cells. Hum Gene Ther. 2009; 20:554–562.
58. Kreutz M, Giquel B, Hu Q, Abuknesha R, Uematsu S, Akira S, Nestle FO, Diebold SS. Antibody-Antigen-Adjuvant Conjugates Enable Co-Delivery of Antigen and Adjuvant to Dendritic Cells in Cis but Only Have Partial Targeting Specificity. PLoS One. 2012; 7:e40208.
59. Thomann-Harwood LJ, Kaeuper P, Rossi N, Milona P, Herrmann B, McCullough KC. Nanogel vaccines targeting dendritic cells: Contributions of the surface decoration and vaccine cargo on cell targeting and activation. J Control Release 2013; 166:95–105.
60. Gottschalk S, Yu F, Ji M, Kakarla S, Song XT. A vaccine that co-targets tumor cells and cancer associated fibroblasts results in enhanced antitumor activity by inducing antigen spreading. PLoS One. 2013; 8:e82658.
61. Kruit WH, Suciu S, Dreno B, Mortier L, Robert C, Chiarion-Sileni V, Maio M, Testori A, Dorval T, Grob JJ, Becker JC, Spatz A, Eggermont AM et al. Selection of immunostimulant AS15 for active immunization with MAGE-A3 protein: results of a randomized phase II study of the European Organisation for Research and Treatment of Cancer Melanoma Group in Metastatic Melanoma. J Clin Oncol. 2013; 31(19):2413–20.
62. Venanzi F, Shifrin V, Sherman M, Gabai V, Kiselev O, Komissarov A, Grudinin M, Shartukova M, Romanovskaya-Romanko EA, Kudryavets Y, Bezdenezhnykh N, Lykhova O, Semesyuk N et al. Broad-spectrum anti-tumor and anti-metastatic DNA vaccine based on p62-encoding vector. Oncotarget. 2013; 4(10):1–7.
63. Hamilton DH, Litzinger MT, Jales A, Huang B, Fernando RI, Hodge JW, Ardiani A, Apelian D, Schlom J, Palena C. Immunological targeting of tumor cells undergoing an epithelial- mesenchymal transition via a recombinant brachyury-yeast vaccine. Oncotarget. 2013; 4(10):1777-90.
64. Byrne KT and Turk MJ. New Perspectives on the Role of Vitiligo in Immune Responses to Melanoma. Oncotarget. 2011; 2(9):684–694.
65. Goyvaerts C, Dingemans J, De Groeve K, Heirman C, Van Gulck E, Vanham G, De Baetselier P, Thielemans K, Raes G, Breckpot K. Targeting of human antigen-presenting cell subsets. J Virol. 2013; 87:11304–8.
66. Zhang XY, Kutner RH, Bialkowska A, Marino MP, Klimstra WB, Reiser J. Cell-specific targeting of lentiviral vectors mediated by fusion proteins derived from Sindbis virus, vesicular stomatitis virus, or avian sarcoma/leukosis virus. Retrovirology. 2010; 7:3.
67. Van Meirvenne S, Straetman L, Heirman C, Dullaers M, De Greef C, Van Tendeloo V, Thielemans K. Efficient genetic modification of murine dendritic cells by electroporation with mRNA. Cancer Gene Ther. 2002; 9:787–797.
68. Keyaerts M, Verschueren J, Bos TJ, Tchouate-Gainkam LO, Peleman C, Breckpot K, Vanhove C, Caveliers V, Bossuyt A, Lahoutte T. Dynamic bioluminescence imaging for quantitative tumour burden assessment using IV or IP administration of D: -luciferin: effect on intensity, time kinetics and repeatability of photon emission. Eur J Nucl Med Mol Imaging. 2008; 35:999–1007.