
INTRODUCTION
Gastric cancer is the third most frequent cause of cancer deaths in the world [1]. Infection with Helicobacter pylori (H. pylori) is considered to be an important factor for the development of intestinal-type noncardia gastric cancer [2]. Gastric cancer is the result of a cascade of inflammatory events leading from normal epithelia to non-atrophic gastritis (NAG), multifocal atrophic gastritis without intestinal metaplasia (MAG), intestinal metaplasia (IM), dysplasia and cancer [3-5]. Even though the architecture of each lesion is well known, the molecular mechanisms associated with specific stages are not well characterized. Many factors, including those from the host (i.e. genetic background, gastric juice acidity, diet) and the bacterium (i.e. CagA and VacA virulence factors) may modify the risk of gastric cancer [6, 7]. We have previously shown that variation of single nucleotide polymorphisms (SNPs) in cytokine genes is associated with differential risk of gastric precancerous lesions in African American and Caucasian individuals [8-10]. Some of these SNPs have been also associated with gastric cancer risk in several populations [11-14].
The gene Deleted in malignant brain tumors 1 (DMBT1) was first proposed as a candidate tumor suppressor gene because of its inactivation in several medulloblastoma cell lines, as compared with normal cells [15]. DMBT1 encodes a 340KD glycoprotein containing 14 repeats of the scavenger receptor cysteine-rich (SRCR) domains separated by short serine- and threonine-rich amino acid domains (SIDs), 2 C1r/C1s-Uegf-BMp1 domains, and a carboxy-terminal zona pellucida domain [16, 17]. Thus, DMBT1, also known as Gp-340, muclin, and agglutinin, is composed of motifs that are known to mediate protein-protein interactions and that function as a pattern recognition molecule for Gram-positive and -negative bacteria. In fact, DMBT1 has been shown to bind a wide range of bacteria including H. pylori, Streptococcus mutans, Staphylococcus aureus, and Lactobacillus casei, as well as some viruses [18-22]. It also interacts with other endogenous molecules involved in innate immune response, such as SpA and SpD [16, 23]. As a molecule that participates in host defense, DMBT1 is expressed on various mucosal surfaces such as the gastrointestinal mucosa, the trachea and lungs, and the vaginal mucosa [22, 24-26]. DMBT1 expression is upregulated following infection and/or inflammation [27-31]. DMBT1 may also serve as an epithelial differentiating factor and has been implicated in polarization of epithelial cells [32-34].
Studies on the expression of DMBT1 in the gastric mucosa have reported varied results. Initial reports described DMBT1 expression in normal mucosa as well as in gastric tumors [32, 33, 35]. Kang et al. reported induction of DMBT1 in 100% of intestinal type gastric adenocarcinomas but only in 20% of the diffuse type [32]. Conde et al. described upregulation of DMBT1 in 62% of gastric adenocarcinomas [35]). Recent work has shown that DMBT1 expression is practically non-existent in normal gastric mucosa and that is upregulated in areas of intestinal metaplasia and gastric carcinomas as compared to normal tissues [36]. DMBT1 is involved in the modulation of immune responses during inflammatory and infectious processes [37, 38]; however no studies have determined the effect of the lack of this gene in H. pylori infection. The goal of the present study was to identify genes associated with the presence of advanced gastric lesions across ethnicities. We report that mucosal DMBT1 expression is positively correlated with more advanced gastric precancerous lesions in three different ethnic populations; we also show that our mouse model lacking Dmbt1 (Dmbt1-/-) and infected with H. pylori develops more severe inflammation and more extensive mucous metaplasia, which is paralleled with increased cell proliferative rates. Genomic analysis of these H. pylori-infected Dmbt1 knock-out mice revealed reduced expression of Il33 mRNA levels and ERK phosphorylation when compared to wild type counterparts. This is interesting because previous research suggest that IL-33 is one of the so called alarmins which initiate tissue recovery after damage or infection (reviewed in [39]). In addition, it has been shown that IL-33 signals through ERK [40, 41]. Our results in mice highlight the role of DMBT1 in the modulation of the inflammatory cascade and its association with gastric precancerous lesions in response to H. pylori infection.
RESULTS
Microarray analysis identifies DMBT1 as a gene associated with advanced precancerous lesions
The goal of this study was to identify genes associated with advanced gastric precancerous lesions. As presented in Supplementary Figure 1A, the scatterplot comparison between MAG and dysplasia samples showed 16 genes with at least 30% change between the two stages (blue dots). Five genes encoding for a transcription factor (E2F3), a collagen type I (COL1A1), a putative tumor suppressor (TSC2), a fibroblast receptor (FGFR1) and a sensor of stress-induced growth arrest (GADD45A) were overexpressed in MAG.On the other hand, genes genes overexpressed in dysplasia included a protein tyrosine phosphatase (PTPRH), a member of the TNF-receptor superfamily (TNFRS1A), the retinoic acid receptor alpha (RARA), the pleiotropic cytokine leukemia inhibitory factor (LIF), the isoform 1 of the superoxide dismutase (SOD1), the cyclin-dependent kinase 9 (CDK9), a member of the lipocalin family (LCN2), a gene encoding for a protein that binds to BCL2 (BAG1), a member of the fibroblast growth factor receptor (FGFR3), the damage specific DNA binding protein 2 (DDB2), and the deleted in malignant brain tumor (DMBT1). Interestingly, only the expression of DMBT1 was significantly different between the two stages (Table 1). We identified E2F3 and DMBT1 as the genes with the largest change in expression when dysplasia samples were compared to MAG (29% reduction for E2F3 and 2.5 -fold increase for DMBT1, Table 1). Using these two genes, we generated heatmaps to determine how well those two genes separated samples from the dysplasia vs MAG stages (Supplementary Figure 1B). Eleven out of 12 dysplasia samples grouped together in one branch of the dendrogram (identified with red arrows), while 29 out of 37 MAG samples grouped together in the other branch. Our results suggest that a significant difference in the expression of DMBT1 could be used to separate MAG from dysplasia cases.
Table 1: Genes with at least 30% fold change expression between dysplasia and MAG
SYMBOL |
MAG N=37 |
Dysplasia N=12 |
p-value |
Dysplasia/MAG Ratio |
|---|---|---|---|---|
E2F3 |
3263.9±179.5* |
2306.9±139.9 |
0.81495 |
0.71 |
COL1A1 |
3068.8±116.9 |
2272.6±215.5 |
0.73468 |
0.74 |
TSC2 |
4061.5±217.5 |
3051.6±217.0 |
0.60963 |
0.75 |
FGFR1 |
2805.6±198.6 |
2139.5±195.7 |
0.89221 |
0.76 |
GADD45A |
3547.7±354.9 |
2709±318.2 |
0.88833 |
0.76 |
PTPRH |
3807.4±263.4 |
4980.2±511.8 |
0.87948 |
1.31 |
TNFRSF1A |
3883.7±275.7 |
5102.1±505.0 |
0.8205 |
1.31 |
RARA |
2814.2±116.1 |
3733.4±355.1 |
0.72687 |
1.33 |
LIF |
2349.9±160.8 |
3134±280.2 |
0.6291 |
1.33 |
SOD1 |
4413.2±224.9 |
5901.1±428.9 |
0.45848 |
1.34 |
CDK9 |
2692.9±104.5 |
3767.4±749.3 |
0.83884 |
1.4 |
LCN2 |
3867.5±267.9 |
5435.4±704.2 |
0.77509 |
1.41 |
BAG1 |
2603.9±101.2 |
3700.1±749.3 |
0.83795 |
1.42 |
FGFR3 |
3264.7±220.9 |
4690.8±647.8 |
0.74522 |
1.44 |
DDB2 |
2333.5±105.8 |
3392.4±802.5 |
0.86253 |
1.45 |
DMBT1 |
2169.3±174.0 |
5371.1±665.1 |
0.00384 |
2.48 |
*Mean±SE.
DMBT1 expression is higher in gastric precancerous lesions in three different populations
To confirm the results of the microarray analysis and to determine whether the same differences were observed in other populations, we performed real-time PCR in gastric biopsies from Hispanic, African American and Caucasian individuals with various gastric precancerous lesions. Hispanic individuals (Figure 1A) with dysplasia had significantly higher levels of DMBT1 expression when compared to MAG patients (p=0.0051). We also found that DMBT1 expression was up-regulated in NAG, MAG and IM in both African and Caucasian Americans (Figure 1B), suggesting that this association is conserved across populations. Immunohistochemistry (IHC) staining for DMBT1 showed that African Americans had increased percentage of DMBT1+ epithelial cells in the gastric epithelium when compared with Caucasian individuals (p<0.0001; Table 2). Representative images of DMBT1 IHC are shown in Figure 1C. Since DMBT1 is a known pattern recognition protein, we analyzed its expression according to H. pylori infection. As can be seen in Figure 2, African Americans with precancerous gastric lesions (MAG and IM) and infected with H. pylori showed the highest DMBT1 expression, as compared to the other diagnoses (p=0.007 compared to normal mucosa), suggesting an association between the expression of this gene and the infection with H. pylori, as has been shown for this protein in other models of infection [42-44]. Taken together, these data show that DMBT1 up-regulation starts very early in the precancerous cascade and increases during the development of the gastric precancerous lesions, suggesting that DMBT1 expression may indicate the transition from inflammatory states to precancerous stages in the cascade of events that lead to cancer.
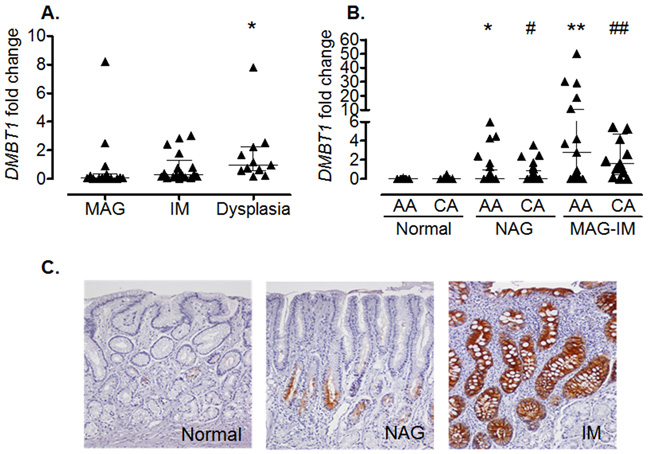
Figure 1: Increased DMBT1 expression in gastritis and precancerous lesions. (A) Hispanic individuals with dysplasia have increased levels of DMBT1 when compared to individuals with MAG (*p=0.0051). (B) African American (AA) and Caucasian (CA) individuals with NAG or MAG-IM (combined MAG and IM) show increased gastric levels of DMBT1 mRNA when compared to individuals with normal gastric mucosa from either ethnic group. *p=0.0058; **p<0.0001 when compared to AA normal; #p=0.0001; ##p<0.001 when compared to CA normal. Median and interquartile range are shown. (C) Representative IHC for DMBT1 expression in normal, NAG, and IM.
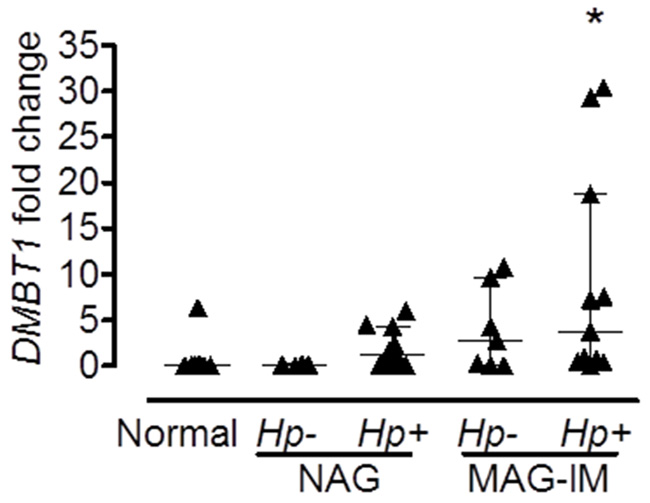
Figure 2: Expression of DMBT1 is associated with H. pylori infection. Expression of DMBT1 from gastric tissue RNA from African American individuals with different stages of gastric lesions was analyzed by real-time PCR. Infection with H. pylori was determined by Steiner silver stain as described in Materials and Methods. All subjects with normal gastric mucosa were uninfected. *p=0.007 when compared to Normal; Hp, H. pylori. Median and interquartile range are shown.
Table 2: DMBT1 epithelial immunostaining in gastric mucosa of African American and Caucasian individuals by diagnostic groups
% of epithelium stained |
African Americans |
Caucasians |
||||
|---|---|---|---|---|---|---|
Normal |
NAG |
MAG/IM |
Normal |
NAG |
MAG/IM |
|
0-5 |
25 (96) |
11 (39.3) |
7 (50) |
24 (96) |
19 (67.9) |
9 (75) |
6-20 |
1 (4) |
11 (39.3) |
7 (50) |
1 (4) |
9 (32.1) |
3 (25) |
>20 |
0 |
6 (21.4) |
0 |
0 |
0 |
0 |
Chi-square 47.83; df=10; p<0.0001. The cut-off point for this analysis was set at 5%.
Increased inflammatory response in Dmbt1-/- mice after H. pylori infection
We infected WT and Dmbt1-/- mice as described in Materials and Methods and determined the level of inflammation and damage in the gastric mucosa. Interestingly, at four months after H. pylori infection, the degree of inflammation was already evident and significantly higher in the H. pylori-infected Dmbt1-/- mice when compared to their WT counterparts (Figure 3A; p=0.048). As previously observed [45], the inflammatory response was higher in the corpus than in the antrum of both infected WT and Dmbt1-/- mice. Compared to WT mice, the Dmbt1-/- animals also exhibited significantly more atrophy of the oxyntic mucosa with presence of mucous metaplasia (Figure 3B and 3C; p=0.0065). The presence of mucous metaplasia was confirmed by Alcian Blue-Periodic acid-Schiff (AB-PAS) staining, which showed the replacement of fundic parietal and chief cells by foamy cells containing neutral and acid mucins (Figure 3D). As in WT mice, no changes were observed in the gastric mucosa of uninfected Dmbt1-/- mice (Figure 3C). Infection of WT mice with H. pylori resulted in increased Dmbt1 mRNA expression; however, H. pylori load was not different between Dmbt1-/- and WT (Supplementary Figure 2).
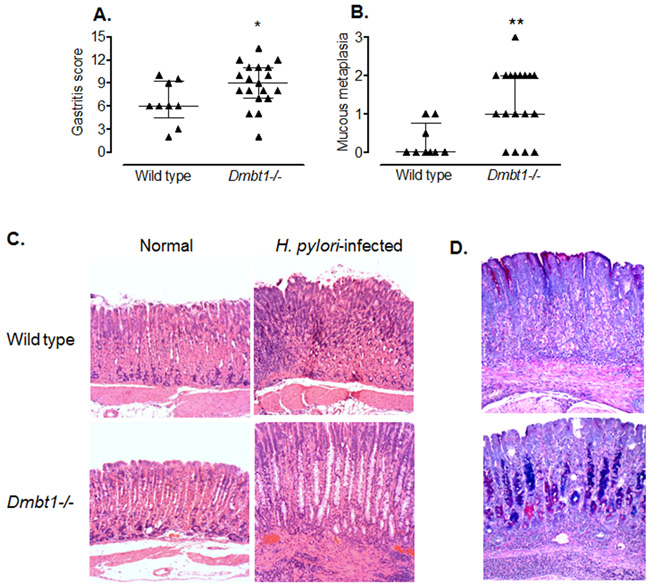
Figure 3: H. pylori infection induces premalignant lesions and more severe inflammation in Dmbt1-/- mice. (A) Semiquantitative scores of the inflammatory infiltrate in the gastric mucosae of Dmbt1-/- and WT mice 4 months after inoculation with H. pylori SS1; *p=0.048. (B) Metaplastic changes in the mucosae of the gastric corpus of H. pylori-infected Dmbt1-/- and WT mice were scored from 0 to 3 as described in Materials and Methods; **p=0.0065. (C) Representative images of sections of the gastric corpus of non-infected and H. pylori-infected Dmbt1-/- and WT mice showing more severe pseudo-pyloric metaplasia in H. pylori-infected Dmbt1-/- mice compared to H. pylori-infected WT mice. H&E stains at 100X magnification. (D) Representative images of the AB-PAS stains of gastric mucosa sections of H. pylori-infected WT and Dmbt1-/- mice showing the presence of mucous metaplasia in Dmbt1-/- mice (100X magnification). Median and interquartile range are shown.
Persistent hyper-proliferation has been suggested to contribute to gastric carcinogenesis [46]. To evaluate gastric epithelial proliferation, we stained sections of the fundic mucosa with Ki67. Although no significant difference in the number of Ki67+ cells was found between H. pylori-infected WT and Dmbt1-/- mice (p=0.28; Figure 4A and 4B), the infection increased the number of proliferating cells (Ki67+) in both groups of animals. Interestingly, this increase was greater in Dmbt1-/- mice (13.6±0.42 in non-infected vs. 33.4±7.9 in H. pylori-infected, p=0.004) compared to WT (17.1±1.1 in non-infected vs. 22.2±1.5 in infected animals, p=0.042) (Figure 4B). These results represent a 30% increase in WT compared to 150% in Dmbt1-/-.
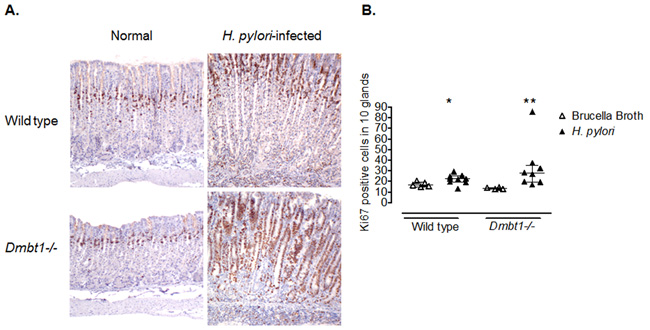
Figure 4: Enhanced proliferation of gastric epithelial cells in Dmbt1-/- mice following H. pylori infection. (A) Representative images of Ki67 labelling of gastric mucosa sections of non-infected and H. pylori-infected WT and Dmbt1-/- mice. (B) Ki67-labeledproliferatingcells were counted in 10 well-oriented and randomly selected glands of the gastric corpus; *p=0.042, **p=0.004, compared to the respective non-infected counterpart. Median and interquartile range are shown.
To determine whether differences in gene expression could explain the dissimilarities in the inflammatory changes and extension of mucous metaplasia in WT and Dmbt11-/- mice in response to H. pylori infection, versus their respective controls, we performed microarray analysis looking for genes differentially expressed between infected WT and Dmbt1-/- mice. Several genes, including Il33, Reg1, Muc13 and Fos, were downregulated in H. pylori-infected Dmbt1-/- mice when compared to infected WT mice (Table 3). Interestingly, the gene with the greatest difference in expression between H. pylori-infected WT and Dmbt1-/- mice was the Il33 gene. Since IL-33 has recently been shown to have a major role in the immune response following H. pylori infection [39], we performed real time PCR to confirm our microarray results. We found that WT mice upregulated Il33 expression in response to H. pylori infection up to 350%, while upregulation in the Dmbt1-/- reached 52% in response to the infection (Figure 5A). The levels of Il33 mRNA were significantly higher in H. pylori-infected WT than in infected Dmbt1-/- mice (p=0.049). As IL-33 signals through ERK, we determined the level of phosphorylation of ERK by immunohistochemistry in WT and Dmbt1-/- mice infected with H. pylori and found increased levels of of pERK in WT (Figure 5B and 5C).
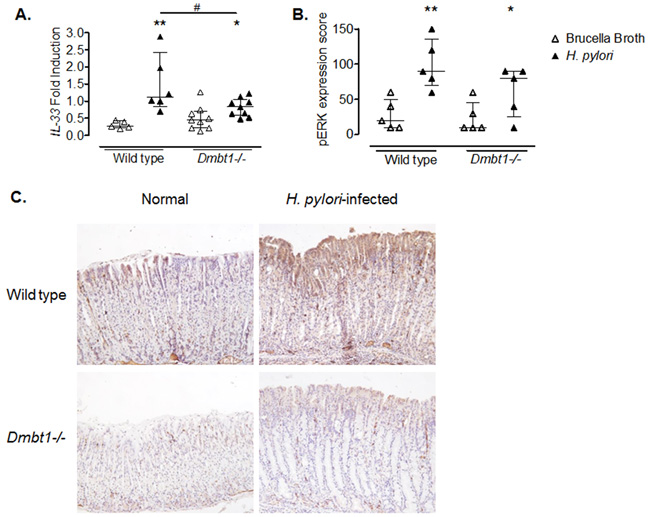
Figure 5: Decreased expression of IL-33 in H. pylori infected Dmbt1-/- mice. (A) Comparison of mRNA levels of Il33 in the gastric mucosae of Dmbt1-/- and WT mice following 4 months of infection with H. pylori. Results are shown as fold induction relative to Gapdh expression. * p=0.046; **p=0.0043; #p=0.0496. (B) pERK expression score was assessed using immunohistochemistry in the gastric mucosae of Dmbt1-/- and WT mice infected with H. pylori for 4 months as described in Material and Methods. *p=0.095; **p=0.0079. Median and interquartile range are shown. (C) Representative images of anti-pERK immunohistochemistry on gastric mucosa sections of non-infected and H. pylori-infected WT and Dmbt1-/- mice.
Table 3: Differential gene expression between WT and Dmbt1-/- mice in response to H. pylori infection
TargetID |
WT Signal* |
Dmbt1-/- Signal |
Ratio |
|---|---|---|---|
PNLIPRP2 |
0.32 |
0.66 |
0.48 |
H2-EB1 |
1.59 |
1.76 |
0.90 |
CD52 |
1.62 |
1.79 |
0.91 |
C3 |
1.76 |
1.78 |
0.99 |
H2-AB1 |
2.01 |
2.03 |
0.99 |
ZC3H12A |
1.76 |
1.75 |
1.01 |
MCPT1 |
1.61 |
1.54 |
1.05 |
H2-DMA |
1.98 |
1.82 |
1.09 |
CDKN1C |
0.46 |
0.42 |
1.10 |
CD74 |
2.44 |
2.22 |
1.10 |
RPP25 |
0.65 |
0.58 |
1.12 |
H2-AA |
1.98 |
1.76 |
1.13 |
SLC2A6 |
1.92 |
1.51 |
1.27 |
2010001M09RIK |
4.46 |
3.34 |
1.34 |
SPRR2F |
3.68 |
2.66 |
1.38 |
H2-DMB1 |
2.60 |
1.81 |
1.44 |
NFKBIZ |
3.20 |
2.08 |
1.54 |
CXCL9 |
3.18 |
1.95 |
1.63 |
IIGP2 |
3.04 |
1.78 |
1.71 |
CASP1 |
3.68 |
2.13 |
1.73 |
SMPDL3B |
3.66 |
2.05 |
1.79 |
H2-K1 |
3.25 |
1.70 |
1.91 |
TYKI |
3.15 |
1.59 |
1.98 |
DUOXA2 |
3.97 |
2.00 |
1.99 |
VIL1 |
3.57 |
1.54 |
2.32 |
LY6D |
3.61 |
1.54 |
2.34 |
CD177 |
7.55 |
3.19 |
2.37 |
INDO |
12.29 |
4.44 |
2.77 |
PSMB9 |
5.00 |
1.72 |
2.91 |
IFIT3 |
4.84 |
1.64 |
2.95 |
SLPI |
4.99 |
1.67 |
2.99 |
FOS |
4.64 |
1.53 |
3.03 |
OTTMUSG00000016644 |
6.61 |
2.01 |
3.29 |
MUC13 |
14.04 |
4.16 |
3.38 |
CLCA3 |
5.85 |
1.51 |
3.87 |
REG1 |
7.82 |
1.56 |
5.01 |
IL33 |
9.18 |
1.53 |
6.00 |
*Signal was calculated by the ratio between the signal under H. pylori infection and non-stimulated conditions.
DISCUSSION
DMBT1 binds to and aggregates multiple bacterial pathogens including H. pylori [18-22]. In some instances, such binding can prevent infection [38, 47]. In addition, a role of DMBT1 in promoting cell differentiation has also been proposed [32, 34, 48].
In the present study, we showed that the expression of DMBT1 is upregulated along the gastric precancerous cascade at both the mRNA and protein level in three different human populations. Furthermore, DMBT1 upregulation occurs as early as the NAG stage in the precancerous cascade, especially in H. pylori infected subjects, a finding that suggests a potential association of this gene with H. pylori infection in humans. From NAG, the expression of DMBT1 is gradually increased through the consecutive stages of gastric carcinogenesis: MAG, IM and dysplasia. Our results agree with a previous report showing that the expression of DMBT1 is upregulated in intestinal metaplasia compared to normal gastric mucosa [36]. Notably, our work is the first to report a consistent association of DMBT1 with precancerous gastric lesions in 3 different ethnic groups.
To the best of our knowledge, this is the first work that shows the effect of the lack of the gene in a mouse model of H. pylori infection. Deletion of Dmbt1 in mice increased inflammation and exacerbated the gastric H. pylori-driven premalignant lesions, as evidenced by greater extent of mucous metaplasia. An increase in proliferative cells was also observed in Dmbt1-/- mice. Therefore, taken together, these data suggest that DMBT1 may modulate gastric epithelial damage induced by infection and the inflammatory process and that its deletion promotes disease progression during H. pylori infection.
The role of DMBT1 as an innate defense factor has been previously shown in inflammatory diseases. Dmbt1-deficient mice for example, have enhanced susceptibility to dextran sulfate sodium (DSS)-induced colitis [31]. Rosenstiel et al. reported that DMBT1 inhibits TLR4-mediated NF-κB activation [38]. DMBT1 also has been shown to stimulate the migration of alveolar macrophages [23]. In our study, Dmbt1-/- mice had impaired Il33 response to H. pylori infection compared to WT mice. IL-33 is an IL-1 family cytokine that is expressed in barrier tissues, possesses a protective role during infections and promotes a Th2 immune response [39, 49, 50]. Interestingly, IL-33 has recently been reported to be a key player in the immune response to H. pylori. Loss of IL-33 after chronic H. pylori infection may contribute to the Th1-predominant immune response and epithelial injury that follows [39]. Furthermore, IL-33 has been shown to stimulate regulatory T cells (Tregs) during intestinal inflammation [51]. Tregs are important inflammation suppressors; therefore promoting Treg function is an additional mechanism by which the upregulation of IL-33 may help to limit gastric inflammation and damage to the epithelium. The impaired ability of Dmbt1-deficient mice to induce expression of Il33 in the gastric mucosa may in part explain why infected Dmbt1-/- mice had more severe gastric lesions. IL-33 is regulated by trefoil factor 2 (TFF2), a protein involved in mucosal repair [39]. TFF2 is expressed in neck cells of gastric glands and similar to DMBT1, H. pylori upregulates TFF2 in gastric tissues. Loss of TFF2 accelerates H. pylori-induced gastric premalignant lesions [52]. It has previously been shown that porcine DMBT1 interacts with TFF2 in the stomach, and DMBT1 is therefore considered to be a putative receptor for TFF2 [53]. Even though more studies are required to fully understand the mechanisms involved, our study suggests that DMBT1 is also involved in the regulation of IL-33. IL-33 activates various cell-specific signaling pathways [49, 53]. In vitro studies indicate that in human gastric cancer cells, IL-33 signaling occurs through pERK [39]. In agreement with that report, we found significantly higher pERK levels in H. pylori-infected WT mice compared to controls. In contrast, Dmbt1-/- mice failed to increase pERK after H. pylori infection. The latter results suggest that DMBT1 may be involved in the induction of Il33 expression, through phosphorylation of ERK, possibly leading to protective IL-4, IL-5 and IL-10 responses to counteract the inflammatory process in the stomach [39, 49, 54]. It is important to note that we do not know how the mutual expression of IL33 and DMBT1 is modulated, if in any way. In addition, even though we found that level of pERK is reduced under Dmbt1-/- conditions and seem to be associated with the expression of IL33, we acknowledge that this is not be the only way through which H. pylori induces the phosphorylation of ERK. The protective effect of DMBT1 in our model of H. pylori infection may also be due to the ability of DMBT1 to prevent H. pylori from gaining access to the gastric mucosa, as suggested by the fact that degradation of DMBT1 by E. coli, for example, increases attachment of the bacteria to epithelial cells [55].
Taken together, our data from the animal model suggest that in response to H. pylori infection, Dmbt1 may be associated with gastric mucosal protection that may reduce the risk of developing advanced gastric precancerous lesions. A major limitation of the mouse model is that wild type mice infected with mouse adapted H. pylori strains develop gastritis but do not progress to gastric adenocarcinoma. A possible explanation for that is the fact that the mouse-adapted Sydney strain (SS1), the most widely used mouse adapted H. pylori strain and the one used in our study, lacks a functional cag pathogenicity island (PAI) [56], a known virulence factor associated with gastric carcinogenesis in humans [57]. In fact, 60% of H. pylori strains in the U. S. harbor the cagA PAI, and more than 80% in the Colombian region where part our study was conducted [58]. Unfortunately, infection of mice with cag+ H. pylori strains frequently results in deletions within the cag PAI [59, 60]. On the other hand, the role of DMBT1 as a protective agent may seem contradictory with our observation of increasing expression of DMBT1 along the human gastric precancerous cascade. Sustained elevation of mucins, glycoproteins closely related to DMBT1, has been shown to promote the transition from chronic inflammation to cancer [61]. DMBT1 may initially increase as a response to H. pylori infection and/or the inflammatory response and once upregulated contribute to gastric carcinogenesis. However, the mechanisms by which DMBT1 may contribute to human gastric carcinogenesis are still not fully understood.
MATERIALS AND METHODS
Patient populations
For the mucosal expression analysis, we evaluated previously collected formalin-fixed, paraffin-embedded gastric biopsies from Hispanic individuals with preneoplastic lesions who participated in an anti-H. pylori eradication trial conducted in Colombia [45, 62, 63]. In particular, a subset of gastric mucosa biopsies at baseline was randomly selected, including 37 with MAG, 25 with IM, and 12 with dysplasia. As a replication set, we evaluated archived gastric biopsies from African American (26 normal, 28 NAG, 21 MAG with or without IM) and Caucasian (28 normal, 29 NAG, 13 MAG with or without IM) individuals attending the gastroenterology services at the Medical Center of Louisiana and the Ochsner Baptist Medical Center (formerly Memorial Medical Center), both in New Orleans, Louisiana, as previously described [8, 10]. All data and specimens were collected under IRB-approved protocols. All subjects provided written informed consent. Gastric mucosa sections were stained with modified Steiner silver stain for evaluation of H. pylori infection, as previously reported [64].
Mice infection
Six-to-eight-week old specific pathogen-free mice with deletion of the Dmbt1 gene [65] and the corresponding C57Bl/6 controls (The Jackson Laboratories, Bar Harbor, ME) were inoculated by orogastric gavage with 200 µl of Brucella broth containing 108 colony forming units (c.f.u.) of the mouse-adapted Sydney strain (SS1) of H. pylori, for 3 consecutive days. Controls were inoculated with Brucella broth alone. Bacteria were grown for 3 days in CDC anaerobic agar plates supplemented with 5% sheep blood (BD Diagnostics, Sparks, MD) under microaerobic conditions using a Campy Pouch system (BD Diagnostics) prior to inoculation. Four or seven months after inoculation, mice were euthanized by CO2 inhalation and their stomachs removed, opened along the greater curvature, cut longitudinally, and fixed in 10% neutral-buffered formalin. Mice were housed in specific pathogen-free conditions in the Animal Care Facility at Louisiana State University Health Sciences Center (LSUHSC). Animal handling and experimentation were performed in accordance with the guidelines of LSUHSC- Institutional Animal Care and Use Committee protocols.
Gene microarray
Analyses of gene expression arrays in humans and mice were done as previously described [45, 66]. Briefly, RNA was extracted from either human gastric tissues fixed in formalin and embedded in paraffin (FFPE) or from fresh frozen gastric tissue from H. pylori-infected and uninfected mice using Allprep DNA/RNA FFPE or RNEasy kits, respectively (Qiagen, Valencia, CA). Four hundred ng (400ng) of RNA were used to prepare biotin-labeled cDNA (human) or cRNA (mouse), which was later hybridized to the DASL Gene Expression Assay (human, 502 transcripts) or the mouse WG-6-v2 (30,874 transcripts) chips, respectively, according to the manufacturer’s instructions (Illumina Inc, San Diego, CA). The chips were washed and scanned to record the intensity of fluorescence emitted. For data analysis in the GenomeStudio software (Illumina Inc., v2011.1), the samples were normalized as described previously [40] using the detection p value algorithm. Analysis of the human screening array included 37 subjects with MAG, 25 with IM, and 12 with dysplasia. To identify genes associated with advanced gastric precancerous lesions, we compared MAG and dysplasia using MAG as reference, t-test to determine differential expression and scatter plots to identify genes with at least 30% change between the two extreme stages (MAG and dysplasia). Differential expression in mouse microarrays was analyzed separately for wild type (WT) (n=3; non-infected=2) or Dmbt1-/- (n=6; non-infected=3) using the uninfected tissue samples as reference and the Illumina Custom algorithm as the error model. Multiple testing corrections using the Benjamini and Hochberg false discovery rate (FDR 0.05) were applied to both analyses. MetaCore software (Thompson Reuters) was used to determine the set of common genes induced in the gastric mucosa after H. pylori infection in WT and Dmbt1-/- mice (ratio between H. pylori-infected and non-infected mice for each strain). Microarray data included in this manuscript have been deposited to the Gene Expression Omnibus (GEO) under accession number GSE83389.
Real-time PCR
RNA was converted into cDNA using SuperScript III as recommended by the vendor (Life Technologies, Foster City, CA) and subjected to real-time PCR using Assays-on-Demand Taqman probes (Life Technologies, DBMT1 assay Hs01069306_m1, Mm00455996_m1; Il33 assay Mm00505403_m1). The fold induction of the genes was determined by the 2-ΔΔCt method using GAPDH (Assay Hs03929097_g1; Mm03302249_g1) as a housekeeping gene and an internal control for normalization. Fifteen samples with MAG, 18 with IM and 11 with dysplasia were tested from the Colombian cohort. In addition, samples from African Americans included 26 cases with normal gastric histology, 25 NAG and 21 MAG with and without IM, while tissues from Caucasians comprised 28 normal, 29 NAG and 13 MAG with and without IM.
Histologic evaluation of mouse gastric mucosa
Paraffin-embedded tissues were cut into 5 µm sections and stained with hematoxylin and eosin for histological evaluation and with modified Steiner silver stain for detection of H. pylori. Gastric inflammation was assessed by the addition of the scores for mononuclear and polymorphonuclear infiltrate (0, normal; 1, mild; 2, moderate; 3, severe) and the depth of inflammation (0, normal; 1, limited to mucosa; 2, infiltrating the submucosa) in the antrum and the corpus and then combined to get an overall inflammation score [67, 68]. Mucous metaplasia was scored from 0 to 3, based on the extension of the foamy mucus-containing cells observed in the corpus as previously described [45]. Briefly, the presence of moderate foci of foamy cells affecting less than 1/3 of the parietal cells was scored 1, large foci affecting between 1/3 and 2/3 of the parietal cells was scored 2, and change affecting more than 2/3 of the parietal cells was scored 3. H. pylori density was graded from mild to severe (1 to 3).
Immunohistochemistry
DMBT1 was detected in FFPE human gastric biopsies by using a 1:100 dilution of a mouse monoclonal antibody against human DMBT1 from Santa Cruz Biotechnology (Dallas, TX) and a Mach 2 mouse polymer labeled with horseradish peroxidase (HRP) from Biocare Medical Co. (Concord, CA) as secondary antibody solution (overnight at 4°C and 30 mins at room temperature, respectively). Antigen retrieval was done in citrate buffer in a pressure cooker for 20 minutes and the staining done with DAB for 5 mins at room temperature. The percentage of gastric epithelial cells positive for DMBT1 was determined by light microscopy studying the entire histological sections. Ki67 and pERK detection was performed on mouse gastric tissue in FFPE. Immunohistochemistry (IHC) for Ki67 was conducted using the Mach 2 detection system as described above. A primary monoclonal antibody against Ki67 was used in a 1:100 dilution (BD Pharmingen, San Jose, CA) with overnight incubation at 4oC. Cells positive for Ki67 were counted in 5 to 10 well-oriented glands, and an average positive cell number per gland was calculated. Phosphorylated ERK (pERK) detection was performed using the Avidin-Biotin-Peroxidase complex system, according to the manufacturer’s instructions (Vectastain Elite ABC Peroxidase Kit; Vector Laboratories). Antigen retrieval was performed by heating slides in 0.01 M sodium citrate buffer (pH 6.0) to 95ºC under vacuum for 40 minutes. Slides were incubated with a monoclonal antibody anti-pERK (SantaCruz, 1:500 dilution) overnight, followed by incubation with biotinylated secondary antibody and then by avidin-biotin peroxidase complex for 1 h at room temperature. pERK immunostaining was evaluated semi-quantitatively assessing the average signal intensity (on a scale of 0 to 3) and the percentage of cells showing a positive stain. Intensity score and percentage of cells stained were then multiplied to give the pERK expression score.
Statistical analysis
GraphPad PRISM software v4.03 was used to compare all groups by One Way ANOVA and non-parametric tests (Mann-Whitney). The same software was used to determine differences in the percentage of DMBT1+ cells in the gastric mucosa of African American and Caucasian individuals by Chi-square using contingency tables. Heatmaps were created using R software version 3.1.3.
ACKNOWLEDGMENTS
The genomic analysis was done in the Stanley S. Scott Cancer Center’s Translational Genomics Core, LSUHSC. The IHC staining was done at VUMC and the LCRC Molecular Histopathology and Analytic Microscopy Core. JZ was supported in part by the Louisiana Cancer Research Consortium and grants from the National Institute of General Medical Sciences (NIGMS P20GM103501 subproject # 2, P30GM114732, U54GM104940-01), and the National Institute on Minority Health Disparities (NIMHD P20MD004817, U54MD008176-01). This work was also supported in part by NIH grant P01CA028842 (to PC and KTW).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136:E359–86.
2. IARC. 1994.IARC monograph on the evaluation of carcinogenic risks to humans:Schistosomes, liver flukes and Helicobacter pylori. Edition 61:177–240.
3. Correa P, Haenszel W, Cuello C, Tannenbaum S, Archer M. A model for gastric cancer epidemiology. Lancet. 1975; 2:58–60.
4. Correa P, Houghton J. Carcinogenesis of Helicobacter pylori. Gastroenterology. 2007; 133:659–72.
5. Correa P, Piazuelo MB. The gastric precancerous cascade. J Dig Dis. 2012; 13:2–9.
6. Cover TL, Peek RM Jr. Diet, microbial virulence, and Helicobacter pylori-induced gastric cancer. Gut Microbes. 2013; 4:482–93.
7. de Martel C, Forman D, Plummer M. Gastric cancer: epidemiology and risk factors. Gastroenterol Clin North Am. 2013; 42:219–40.
8. Zabaleta J, Camargo MC, Piazuelo MB, Fontham E, Schneider BG, Sicinschi LA, Ferrante W, Balart L, Correa P, Ochoa AC. Association of interleukin-1beta gene polymorphisms with precancerous gastric lesions in African Americans and Caucasians. Am J Gastroenterol. 2006; 101:163–71.
9. Zabaleta J, Schneider BG, Ryckman K, Hooper PF, Camargo MC, Piazuelo MB, Sierra RA, Fontham ET, Correa P, Williams SM, Ochoa AC. Ethnic differences in cytokine gene polymorphisms: potential implications for cancer development. Cancer Immunol Immunother. 2008; 57:107–14.
10. Zabaleta J, Camargo MC, Ritchie MD, Piazuelo MB, Sierra RA, Turner SD, Delgado A, Fontham ET, Schneider BG, Correa P, Ochoa AC. Association of haplotypes of inflammation-related genes with gastric preneoplastic lesions in African Americans and Caucasians. Int J Cancer. 2011; 128:668–75.
11. El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, Fraumeni JF Jr, Rabkin CS. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000; 404:398–402.
12. El-Omar EM, Rabkin CS, Gammon MD, Vaughan TL, Risch HA, Schoenberg JB, Stanford JL, Mayne ST, Goedert J, Blot WJ, Fraumeni JF Jr, Chow WH. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. 2003; 124:1193–201.
13. Machado JC, Figueiredo C, Canedo P, Pharoah P, Carvalho R, Nabais S, Castro Alves C, Campos ML, Van Doorn LJ, Caldas C, Seruca R, Carneiro F, Sobrinho-Simões M. A proinflammatory genetic profile increases the risk for chronic atrophic gastritis and gastric carcinoma. Gastroenterology. 2003; 125:364–71.
14. Sicinschi LA, Lopez-Carrillo L, Camargo MC, Correa P, Sierra RA, Henry RR, Chen J, Zabaleta J, Piazuelo MB, Schneider BG. Gastric cancer risk in a Mexican population: role of Helicobacter pylori CagA positive infection and polymorphisms in interleukin-1 and -10 genes. Int J Cancer. 2006; 118:649–57.
15. Mollenhauer J, Wiemann S, Scheurlen W, Korn B, Hayashi Y, Wilgenbus KK, von Deimling A, Poustka A. DMBT1, a new member of the SRCR superfamily, on chromosome 10q25.3-26.1 is deleted in malignant brain tumours. Nat Genet. 1997; 17:32–39.
16. Holmskov U, Lawson P, Teisner B, Tornoe I, Willis AC, Morgan C, Koch C, Reid KB. Isolation and characterization of a new member of the scavenger receptor superfamily, glycoprotein-340 (gp-340), as a lung surfactant protein-D binding molecule. J Biol Chem. 1997; 272:13743–49.
17. Mollenhauer J, Holmskov U, Wiemann S, Krebs I, Herbertz S, Madsen J, Kioschis P, Coy JF, Poustka A. The genomic structure of the DMBT1 gene: evidence for a region with susceptibility to genomic instability. Oncogene. 1999; 18:6233–40.
18. Ericson T, Rundegren J. Characterization of a salivary agglutinin reacting with a serotype c strain of Streptococcus mutans. Eur J Biochem. 1983; 133:255–61.
19. Hartshorn KL, White MR, Tecle T, Holmskov U, Crouch EC. Innate defense against influenza A virus: activity of human neutrophil defensins and interactions of defensins with surfactant protein D. J Immunol. 2006; 176:6962–72.
20. Nagashunmugam T, Malamud D, Davis C, Abrams WR, Friedman HM. Human submandibular saliva inhibits human immunodeficiency virus type 1 infection by displacing envelope glycoprotein gp120 from the virus. J Infect Dis. 1998; 178:1635–41.
21. Prakobphol A, Xu F, Hoang VM, Larsson T, Bergstrom J, Johansson I, Frängsmyr L, Holmskov U, Leffler H, Nilsson C, Borén T, Wright JR, Strömberg N, Fisher SJ. Salivary agglutinin, which binds Streptococcus mutans and Helicobacter pylori, is the lung scavenger receptor cysteine-rich protein gp-340. J Biol Chem. 2000; 275:39860–66.
22. Stoddard E, Cannon G, Ni H, Karikó K, Capodici J, Malamud D, Weissman D. gp340 expressed on human genital epithelia binds HIV-1 envelope protein and facilitates viral transmission. J Immunol. 2007; 179:3126–32.
23. Tino MJ, Wright JR. Glycoprotein-340 binds surfactant protein-A (SP-A) and stimulates alveolar macrophage migration in an SP-A-independent manner. Am J Respir Cell Mol Biol. 1999; 20:759–68.
24. Holmskov U, Mollenhauer J, Madsen J, Vitved L, Gronlund J, Tornoe I, Kliem A, Reid KB, Poustka A, Skjodt K. Cloning of gp-340, a putative opsonin receptor for lung surfactant protein D. Proc Natl Acad Sci USA. 1999; 96:10794–99.
25. Kang W, Nielsen O, Fenger C, Madsen J, Hansen S, Tornoe I, Eggleton P, Reid KB, Holmskov U. The scavenger receptor, cysteine-rich domain-containing molecule gp-340 is differentially regulated in epithelial cell lines by phorbol ester. Clin Exp Immunol. 2002; 130:449–58.
26. Mollenhauer J, Herbertz S, Holmskov U, Tolnay M, Krebs I, Merlo A, Schrøder HD, Maier D, Breitling F, Wiemann S, Gröne HJ, Poustka A. DMBT1 encodes a protein involved in the immune defense and in epithelial differentiation and is highly unstable in cancer. Cancer Res. 2000; 60:1704–10.
27. Kaemmerer E, Schneider U, Klaus C, Plum P, Reinartz A, Adolf M, Renner M, Wolfs TG, Kramer BW, Wagner N, Mollenhauer J, Gassler N. Increased levels of deleted in malignant brain tumours 1 (DMBT1) in active bacteria-related appendicitis. Histopathology. 2012; 60:561–69.
28. Mollenhauer J, Müller H, Kollender G, Lyer S, Diedrichs L, Helmke B, Holmskov U, Ligtenberg T, Herbertz S, Krebs I, Madsen J, Bikker F, Schmitt L, et al. The SRCR/SID region of DMBT1 defines a complex multi-allele system representing the major basis for its variability in cancer. Genes Chromosomes Cancer. 2002; 35:242–55.
29. Müller H, End C, Weiss C, Renner M, Bhandiwad A, Helmke BM, Gassler N, Hafner M, Poustka A, Mollenhauer J, Poeschl J. Respiratory Deleted in Malignant Brain Tumours 1 (DMBT1) levels increase during lung maturation and infection. Clin Exp Immunol. 2008; 151:123–29.
30. Müller H, Renner M, Helmke BM, End C, Weiss C, Poeschl J, Mollenhauer J. Deleted in Malignant Brain Tumors 1 is up-regulated in bacterial endocarditis and binds to components of vegetations. J Thorac Cardiovasc Surg. 2009; 138:725–32.
31. Renner M, Bergmann G, Krebs I, End C, Lyer S, Hilberg F, Helmke B, Gassler N, Autschbach F, Bikker F, Strobel-Freidekind O, Gronert-Sum S, Benner A, et al. DMBT1 confers mucosal protection in vivo and a deletion variant is associated with Crohn’s disease. Gastroenterology. 2007; 133:1499–509.
32. Kang W, Nielsen O, Fenger C, Leslie G, Holmskov U, Reid KB. Induction of DMBT1 expression by reduced ERK activity during a gastric mucosa differentiation-like process and its association with human gastric cancer. Carcinogenesis. 2005; 26:1129–37.
33. Mollenhauer J, Herbertz S, Helmke B, Kollender G, Krebs I, Madsen J, Holmskov U, Sorger K, Schmitt L, Wiemann S, Otto HF, Gröne HJ, Poustka A. Deleted in Malignant Brain Tumors 1 is a versatile mucin-like molecule likely to play a differential role in digestive tract cancer. Cancer Res. 2001; 61:8880–86.
34. Takito J, Hikita C, Al-Awqati Q. Hensin, a new collecting duct protein involved in the in vitro plasticity of intercalated cell polarity. J Clin Invest. 1996; 98:2324–31.
35. Conde AR, Martins AP, Brito M, Manuel A, Ramos S, Malta-Vacas J, Renner M, Poustka A, Mollenhauer J, Monteiro C. DMBT1 is frequently downregulated in well-differentiated gastric carcinoma but more frequently upregulated across various gastric cancer types. Int J Oncol. 2007; 30:1441–46.
36. Sousa JF, Ham AJ, Whitwell C, Nam KT, Lee HJ, Yang HK, Kim WH, Zhang B, Li M, LaFleur B, Liebler DC, Goldenring JR. Proteomic profiling of paraffin-embedded samples identifies metaplasia-specific and early-stage gastric cancer biomarkers. Am J Pathol. 2012; 181:1560–72.
37. Müller H, Nagel C, Weiss C, Mollenhauer J, Poeschl J. Deleted in malignant brain tumors 1 (DMBT1) elicits increased VEGF and decreased IL-6 production in type II lung epithelial cells. BMC Pulm Med. 2015; 15:32–41.
38. Rosenstiel P, Sina C, End C, Renner M, Lyer S, Till A, Hellmig S, Nikolaus S, Fölsch UR, Helmke B, Autschbach F, Schirmacher P, Kioschis P, et al. Regulation of DMBT1 via NOD2 and TLR4 in intestinal epithelial cells modulates bacterial recognition and invasion. J Immunol. 2007; 178:8203–11.
39. Buzzelli JN, Chalinor HV, Pavlic DI, Sutton P, Menheniott TR, Giraud AS, Judd LM. IL33 Is a Stomach Alarmin That Initiates a Skewed Th2 Response to Injury and Infection. Cell Mol Gastroenterol Hepatol. 2015; 1:203–221.e3.
40. Shang J, Zhao J, Wu X, Xu Y, Xie J, Zhao J. Interleukin-33 promotes inflammatory cytokine production in chronic airway inflammation. Biochem Cell Biol. 2015; 93:359–66.
41. Mine Y, Makihira S, Yamaguchi Y, Tanaka H, Nikawa H. Involvement of ERK and p38 MAPK pathways on Interleukin-33-induced RANKL expression in osteoblastic cells. Cell Biol Int. 2014; 38:655–62.
42. Armengol C, Bartolí R, Sanjurjo L, Serra I, Amézaga N, Sala M, Sarrias MR. Role of scavenger receptors in the pathophysiology of chronic liver diseases. Crit Rev Immunol. 2013; 33:57–96.
43. Malamud D, Abrams WR, Barber CA, Weissman D, Rehtanz M, Golub E. Antiviral activities in human saliva. Adv Dent Res. 2011; 23:34–37.
44. Wu Z, Golub E, Abrams WR, Malamud D. gp340 (SAG) binds to the V3 sequence of gp120 important for chemokine receptor interaction. AIDS Res Hum Retroviruses. 2004; 20:600–07.
45. Garay J, Piazuelo MB, Majumdar S, Li L, Trillo-Tinoco J, Del Valle L, Schneider BG, Delgado AG, Wilson KT, Correa P, Zabaleta J. The homing receptor CD44 is involved in the progression of precancerous gastric lesions in patients infected with Helicobacter pylori and in development of mucous metaplasia in mice. Cancer Lett. 2016; 371:90–98.
46. Naumann M, Crabtree JE. Helicobacter pylori-induced epithelial cell signalling in gastric carcinogenesis. Trends Microbiol. 2004; 12:29–36.
47. Wu Z, Lee S, Abrams W, Weissman D, Malamud D. The N-terminal SRCR-SID domain of gp-340 interacts with HIV type 1 gp120 sequences and inhibits viral infection. AIDS Res Hum Retroviruses. 2006; 22:508–15.
48. Bisgaard HC, Holmskov U, Santoni-Rugiu E, Nagy P, Nielsen O, Ott P, Hage E, Dalhoff K, Rasmussen LJ, Tygstrup N. Heterogeneity of ductular reactions in adult rat and human liver revealed by novel expression of deleted in malignant brain tumor 1. Am J Pathol. 2002; 161:1187–98.
49. Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol. 2010; 10:103–10.
50. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016; 16:676–89.
51. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, Pott J, Griseri T, Bollrath J, Hegazy AN, Harrison OJ, Owens BM, Löhning M, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014; 513:564–68.
52. Fox JG, Rogers AB, Whary MT, Ge Z, Ohtani M, Jones EK, Wang TC. Accelerated progression of gastritis to dysplasia in the pyloric antrum of TFF2 -/- C57BL6 x Sv129 Helicobacter pylori-infected mice. Am J Pathol. 2007; 171:1520–28.
53. Thim L, Mørtz E. Isolation and characterization of putative trefoil peptide receptors. Regul Pept. 2000; 90:61–68.
54. Yu XX, Hu Z, Shen X, Dong LY, Zhou WZ, Hu WH. IL-33 Promotes Gastric Cancer Cell Invasion and Migration Via ST2-ERK1/2 Pathway. Dig Dis Sci. 2015; 60:1265–72.
55. Grys TE, Siegel MB, Lathem WW, Welch RA. The StcE protease contributes to intimate adherence of enterohemorrhagic Escherichia coli O157:H7 to host cells. Infect Immun. 2005; 73:1295–303.
56. Crabtree JE, Ferrero RL, Kusters JG. The mouse colonizing Helicobacter pylori strain SS1 may lack a functional cag pathogenicity island. Helicobacter. 2002; 7:139–40.
57. Peek RM Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002; 2:28–37.
58. Sicinschi LA, Correa P, Peek RM, Camargo MC, Piazuelo MB, Romero-Gallo J, Hobbs SS, Krishna U, Delgado A, Mera R, Bravo LE, Schneider BG. CagA C-terminal variations in Helicobacter pylori strains from Colombian patients with gastric precancerous lesions. Clin Microbiol Infect. 2010; 16:369–78.
59. Philpott DJ, Belaid D, Troubadour P, Thiberge JM, Tankovic J, Labigne A, Ferrero RL. Reduced activation of inflammatory responses in host cells by mouse-adapted Helicobacter pylory isolates. Cell Microbiol. 2002; 4:285–96.
60. Sozzi M, Crosatti M, Kim SK, Romero J, Blaser MJ. Heterogeneity of Helicobacter pylori cag genotypes in experimentally infected mice. FEMS Microbiol Lett. 2001; 203:109–14.
61. Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009; 9:874–85.
62. Correa P, Fontham ET, Bravo JC, Bravo LE, Ruiz B, Zarama G, Realpe JL, Malcom GT, Li D, Johnson WD, Mera R. Chemoprevention of gastric dysplasia: randomized trial of antioxidant supplements and anti-helicobacter pylori therapy. J Natl Cancer Inst. 2000; 92:1881–88.
63. Mera R, Fontham ET, Bravo LE, Bravo JC, Piazuelo MB, Camargo MC, Correa P. Long term follow up of patients treated for Helicobacter pylori infection. Gut. 2005; 54:1536–40.
64. Garvey W, Fathi A, Bigelow F. Modified Steiner for the Demonstration of Spirochetes. J Histotechnol. 2016; 8:15–17.
65. De Lisle RC, Xu W, Roe BA, Ziemer D. Effects of Muclin (Dmbt1) deficiency on the gastrointestinal system. Am J Physiol Gastrointest Liver Physiol. 2008; 294:G717–27.
66. Kim SH, Sierra RA, McGee DJ, Zabaleta J. Transcriptional profiling of gastric epithelial cells infected with wild type or arginase-deficient Helicobacter pylori. BMC Microbiol. 2012; 12:175–83.
67. Rogers AB. Histologic scoring of gastritis and gastric cancer in mouse models. Methods Mol Biol. 2012; 921:189–203.
68. Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol. 1996; 20:1161–81.