
INTRODUCTION
Breast cancer (BC) has the highest incidence of cancer occurrence in the female population and is the number one cause of cancer deaths for women worldwide [1–5]. In 2017, there were 252,710 estimated new cases of BC and 40,610 expected BC deaths among women in the United States [1]. Resource-stratified guidelines provide a vehicle for designing programs to promote early detection, diagnosis, and treatment using existing infrastructure and renewable resources and methods. Existing strategies for evaluating the current state and projecting future burden have a major role in developing new strategies to improve BC outcomes at the national and international levels [6–8]. While China’s mainland currently has 470,000 BC patients, only 20 percent of them were diagnosed at early stage, whereas, in USA, 80 percent of BC patients were diagnosed at an early stage, securing best practice and best outcome for the patients (http://www.chinadaily.com.cn/china/2012-03/12/content_14814002.htm). Thus, we must increase BC risk awareness in the general public by recommending regular screening starting at 40 years of age to promote early detection and treatment.
Cancer is genetically heterogeneous regarding to molecular characteristics and pathogenic pathways. A wide spectrum of biomarkers, including DNA markers, is used for determining genomic instability, molecular subtype determination, and prognosis, and estimating sensitivity to different drugs in practice. Systematic analysis of genetic changes in colon cancer development demonstrated that multiple mutations are necessary for the evolution of normal cells into cancer cells [9]. There is growing evidence that multiple mutations are responsible for the development of cancer; cancer cells must exhibit mutator phenotypes [10, 11], which are likely to be responsible for the genomic instability found in cancer tissues. Mutator phenotype, including defective mismatch repair, is known to cause microsatellite instability, associated with multiple hereditary cancers [12, 13]. Loss of chromosomal materials would inactivate tumor suppressor genes while gain of chromosomal materials has the potential to activate tumor-promoting genes [8]. Genomic instability and chromosome copy number variations are the hallmarks of neoplastic transformation and a herald of genomic damage, leading to the conclusion that multiple mutations are the driving force in the carcinogenic processes [14–16]. Thus, the development of diagnostic markers for BC is vital for early detection and treatment of BC patients.
Since the 1990s, a number of genetic and DNA-based molecular marker techniques have been developed, including random amplified polymorphic DNA (RAPD) [17, 18]. Since then, RAPD technique either alone or in combination with other techniques is widely used for the genetic characterization of different medicinal plants, and other organisms [18, 19]. Although RAPD has few requirements of the amount of template DNA, produces no hazardous contamination, and is simple and inexpensive, the few disadvantages that poses including poor reproducibility and low production or yield. Resolution and production of RAPD are greatly increased by prolonging the RAMP time from the stage of annealing to extension in PCR [18, 19]. To improve the effectiveness of the RAPD method, we developed a highly effective DNA marker technique using improved RAPD with high-GC content primers, in which the GC content reaches 8–10 nucleotides for a 10 nucleotide primer (80~100%) [20, 21]. The Sequence Characterized Amplified Region (SCAR) markers are very stable, sensitive, and reliable. These markers are also effective for diagnostic purposes by using a simple PCR assay, which are generally derived from the molecular cloning of RAPD fragments in medicinal plants [21–28].
Different types of cancers have been associated with abnormal DNA fingerprinting; therefore, in this study we applied this effective improved RAPD technique to generate genetic fingerprints that detect genomic alterations in human breast cancer and develop diagnostic SCAR markers.
RESULTS
RAPD amplification from breast tumor tissue DNA samples using high-GC primers
To increase the efficiency of RAPD amplification and obtain more specific bands, standard primers were replaced with high-GC content primers (FY-10, FY-13 and FY-32). In addition, the RAMP time from the stage of annealing (36ºC) to extension (72ºC) was set to 5% (0.125 ºC/s). This improved RAPD was used to amplify genomic DNA from five breast cancer patients. For each patient, two tissue samples were obtained from both the tumor and the normal tissue adjacent to the tumor. The PCR products were resolved by electrophoresis on a 1.5% agarose gel and showed signal difference in intensity of RAPD bands within patients (Figure 1).
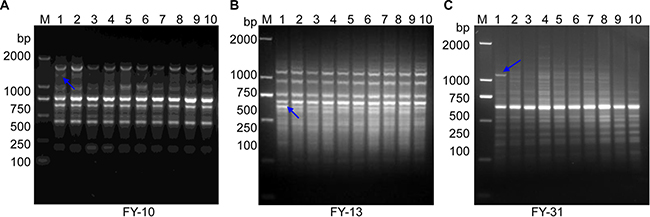
Figure 1: Improved RAPD PCR from breast cancer tissues and their adjacent tissues. (A) High-GC primer FY-10. (B) High-GC primer FY-13. (C) High -GC primer FY-31. Five pairs of genomic DNA from breast cancer tissues and their adjacent or surrounding normal tissues were subjected to improved RAPD amplification (high-GC RAMP-PCR). Lanes 1, 3, 5, 7 and 9 are DNA from breast cancer tissues (see Table 1). Lanes 2, 4, 6, 8 and 10 are their matched DNA from adjacent tissue. The blue arrows indicate bands that were excised for DNA cloning. Lane “M” shows the DL2000 DNA molecular weight marker (bp).
Table 1: The histopathologic profiles of the 5 breast carcinomas specimens
1 (TG53) |
3 (TG55) |
5 (TG57) |
7 (TG59) |
9 (TG62) |
|
|---|---|---|---|---|---|
Histology |
Ductal |
Ductal |
Ductal |
Ductal |
Ductal |
Diagnostic age |
49 |
38 |
68 |
45 |
48 |
Stage |
T3 |
T4 |
T2 |
T2 |
T3 |
Grade |
II |
III |
II |
III |
III |
Estrogen receptor |
+,90% |
− |
− |
− |
− |
Progesterone receptor |
+,10% |
− |
− |
− |
− |
HER-2 |
1+ |
1+ |
0 |
3+ |
0 |
E-Cadherin |
+ |
+ |
+ |
+ |
+ |
p53 |
− |
+, 90% |
+, 40% |
+, 30% |
+, 60% |
Ki-67 |
+, 40% |
+, 75% |
+, 70% |
+, 15% |
+, 50% |
Table 2: The sequences of high-GC primers
Name |
Sequence (5′-3′) |
|---|---|
FY-10 |
GCTCCCGCCG |
FY-13 |
GCGTCCCGCC |
FY-31 |
CGTGGCCCCG |
Cloning and sequencing of DNA fragments
Three different bright bands with over-amplification in Figure 1 were excised and purified from the agarose gel for cloning into the pGM-T vector (Figure 2A). The positive cloned DNA fragments, 10-1, 13-4 and 31-2 (Figure 2B–2D) were selected based on insert sizes by EcoRI digestion matching those of the RAPD fragments in Figure 1 and Figure 2A and then sequenced by the Sanger method. Sequence homology was determined using the online program BLAST (http://www.ncbi.nlm.nih.gov/BLAST/) against the GenBank database.
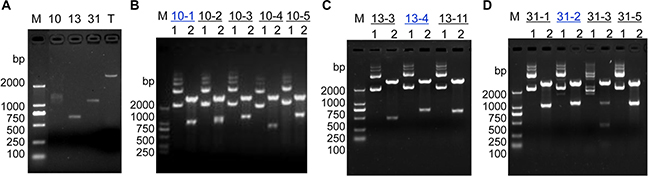
Figure 2: Molecular cloning for RAPD products. (A) Agarose gel electrophoresis for RAPD DNA fragments 10, 13 and 31 derived from high-GC primers FY-10, FY-13 and FY-31, respectively. “T” indicates “pGM-T vector”. (B) Enzymatic identification of positive clones from RAPD fragment 10. (C) Enzymatic identification of positive clones from RAPD fragment 13. (D) Enzymatic identification of positive clones from RAPD fragment 31. Lane 1 contains undigested plasmid DNA; lane 2 contains plasmid DNA after EcoRI digestion. Clones 10-1, 13-4 and 31-2 (blue text color) were selected for Sanger sequencing. Lane “M” contains the DL2000 DNA molecular weight marker (bp).
Sequencing of the aforementioned three cloned RAPD fragments from human breast cancer tissue revealed that clone 10-1 consisted of 1027 nucleotides is located in the intron of DPEP1 transcript variant 2, and overlapped DPEP1 (dipeptidase 1) transcript variant 1 in GenBank (NM_004413) (Figure 3A and Figure 4A). Clone 13-4 consisted of 663 nucleotides (Figure 3B), located only 536 bp away from the PHKG2 gene (Homo sapiens phosphorylase kinase, gamma 2, NM_000294) (Homo sapiens chromosome 16, GRCh38 Primary Assembly. Sequence ID: ref|NC_000016.10|Length: 90338345, Range of clone13-4: 30761532 to 30762194 vs Range of PHKG2: 30748299 to 30761176) (Data not shown), and is also mainly located within or overlapping the first exon of RNF40 (Homo sapiens ring finger protein 40, E3 ubiquitin protein ligase, NM_014771.3) (Figure 4B and Figure 5) in GenBank. Clone 31-2 consisted of 1104 nucleotides that did not match any known genes in GenBank (Figure 3C). Clone 10-1 is mapped to chromosome 16p24.3, clone 13-4 is mapped to chromosome 16p11.2, and clone 31-2 is mapped to chromosome 11q13.5.

Figure 3: Results of Sanger-sequencing of the cloned DNA fragments. (A) The sequence of clone 10-1. (B) The sequence of clone 13-4. (C) The sequences of clone 31-2.
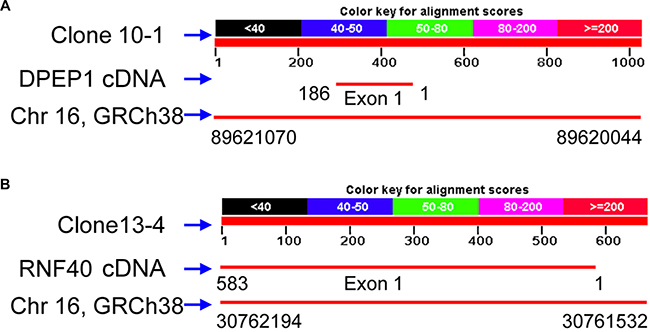
Figure 4: The Human genome locations of clones 10-1 and 13-4 with their partial cDNAs of DEPEP1 and RNF40, respectively.

Figure 5: The sequence of clone 13-4 aligns with RNF40 cDNA. Depicted is the BLAST output showing alignment of the clone 13-4 and RNF40 cDNA sequences. The sequence of clone 13-4 showed 583bp (Plus strand) identity with the cDNA of RNF40 (minus strand).
Development of diagnostic SCAR markers
To generate stable diagnostic SCAR markers from our cloned RAPD fragments, three pairs of primers for semi-quantitative PCR (Table 3) and three pairs of primers for real-time PCR (Table 4) were designed and synthesized based on the cloned sequences. The semi-quantitative SCAR primer pairs were used to amplify ten samples of genomic DNA collected from the breast tumors of five breast cancer patients. Genomic DNA from non-tumor adjacent tissue was used to test for amplification marker-specificity and to verify that the genomic DNA was over-amplified in the tumor. The PCR results indicated that the products with expected size were observed in all samples by three SCAR markers (Figure 6A, 6B, and data not shown). SCAR markers BC10-1 and BC31-2 showed higher signals indicating that these SCAR markers have genomic DNA over-amplified in the tumor tissues (Figure 6).
Table 3: Sequences of SCAR primers, PCR product size (bp)
SCAR |
5′-primer |
Sequence (5′-3′) |
3′-primer |
Sequence (5′-3′) |
Size |
Tm (°C) |
|---|---|---|---|---|---|---|
BC10-1 |
BC10-1L |
GATCACAGGCAGTCAGAGAGG |
BC10-1R |
CAGGCCACTTGAGACTCTCTG |
590 |
60 |
BC31-2 |
BC31-2L |
ACAAACGGCCCCATTTCTTTG |
BC31-2R |
TTGGTGGTTGTGGTAGATGGG |
512 |
60 |
GAPDH |
GAPDH5G |
ACCCAGAAGACTGTGGATGG |
GAPDH3G |
TGACAAAGTGGTCGTTGAGG |
376 |
60 |
Abbreviations: SACR, sequence-characterized amplified region; bp, base pair.
Table 4: Sequences of real time PCR primers for SACR markers, probes
SCAR |
5′-primer |
Sequence (5′-3′) |
3′-primer |
Sequence (5′-3′) |
Probe |
|---|---|---|---|---|---|
BC10-1 |
BC10-1L87 |
cccaggcagtagtgtagggta |
BC10-1R87 |
ccctcctctccatttctcaa |
87 |
BC13-4 |
BC13-4L34 |
tctggcttcttcccaaacac |
BC13-4R34 |
cgcggtgtcagttactatgg |
34 |
beta-actin |
Q-b-actin55LG |
aagtcccttgccatcctaaaa |
Q-b-actin55RG |
atgctatcacctcccctgtg |
55 |
Abbreviations: SACR, sequence-characterized amplified region.
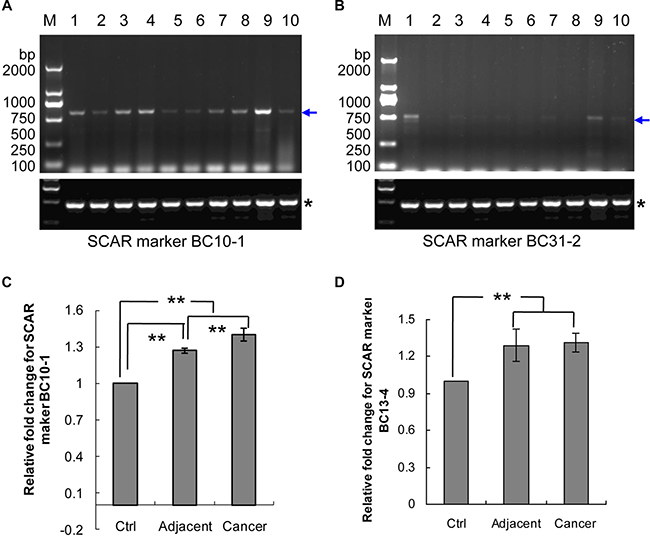
Figure 6: Genomic DNA amplification of SCAR markers BC10-1, BC13-4 and BC31-2 in breast cancer patients. (A) SCAR marker BC10-1 in five pairs of genomic DNA from breast cancer tissues and their adjacent tissues. (B) SCAR marker BC31-2 in five pairs of genomic DNA from breast cancer tissues and their adjacent tissues. Lanes 1, 3, 5, 7 and 9 contain DNA from breast cancer tissues (see Table 1). Lanes 2, 4, 6, 8 and 10 contain their matched adjacent tissue DNA. Blue arrows indicate the amplified band, whereas the stars “*” indicate the internal control. (C) Real-time PCR for SCAR marker BC10-1. (D) Real-time PCR for SCAR marker BC13-4. “Cancer”, breast cancer tissues; “Adjacent”, normal tissues adjacent to or surrounding the breast tumor; “Ctrl”, normal women blood DNA; “**”p value ≤ 0.05.
Real-time PCR was performed, including ten samples of women’s blood DNA, 22 samples of adjacent normal tissue DNA and 30 samples of breast tumor tissue DNA. The results of SCAR marker BC10-1 showed gradually increased levels from blood DNA to tumor DNA (p-value ≤ 0.05) (Figure 6C). SCAR marker BC13-4 is present in increased levels in DNA from adjacent and tumor samples compared to blood DNA (p-value ≤ 0.05), but no difference in levels between adjacent DNA and tumor DNA (Figure 6D). SCAR marker BC31-2 did not show increased levels between blood DNA and tumor DNA (data not shown)
DPEP1 mRNA expression in BC cells and invasive ductal carcinomas
The mRNA expression from the DPEP1 gene was performed by real-time PCR using RNA extracted from BC cell lines BT549, MDA-MB-231 and MDA-MB-435, and non-tumorigenic mammary epithelial cell line MCF10A. We found that the level of DPEP1 mRNA expression was lower in non-tumorigenic cell line MCF10A, but elevated in other BC cell lines (Figure 7A). To determine the DPEP1 levels in BC development, we collected invasive ductal carcinoma specimens from 33 human BC patients and 11 adjacent normal tissues with informed consent. Total RNAs were purified from each tumor tissue and the surrounding normal tissues followed by real-time PCR. Figure 7B shows that the mRNA levels of DPEP1 in BC tissues were significantly higher (11.6-fold) than that of the adjacent normal tissues.
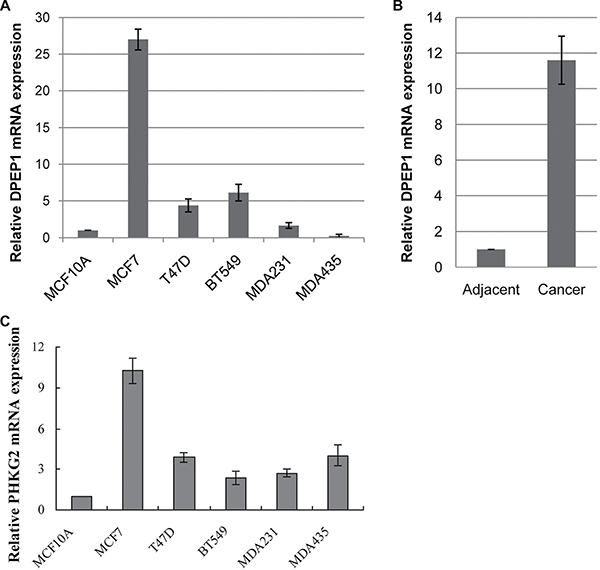
Figure 7: Analysis of DPEP1 and PHKG2 expression in breast cancer cell lines and tissues. (A) DPEP1 gene expression in breast cancer cell lines. (B) DPEP1 gene expression in invasive ductal carcinoma specimens. (C) PHKG2 gene expression in breast cancer cell lines. “MDA231”, MDA-MB-231 cells; “MDA435”, MDA-MB-435 cells.
PHKG2 mRNA expression in BC cells
The mRNA expression from the PHKG2 gene was performed by real-time PCR using RNA extracted from breast cancer cell lines; we found that the level of PHKG2 mRNA expression was lowest in the non-tumorigenic cell line MCF10A, but elevated in the other BC cell lines (Figure 7C).
DISCUSSION
The RAPD technique either alone or in combination with other technique is widely used for the genetic characterization of different plants, animals or other organisms, particularly in medicinal plants [17–19]. In a recent cancer study, RAPD was also used for screening genome-wide changes in DNA methylation, which was called Methylation Sensitive-Random Amplified Polymorphic DNA-Polymerase Chain Reaction (MS-RAPD-PCR) [29]. Cancer patients are predisposed to fungal infections caused by Candida albicans, especially oral or respiratory tract candidiasis. Biernasiuk and colleagues [30] applied RAPD-PCR to successfully estimate genetic diversity of C. albicans isolated from the upper respiratory tract of patients with non-small cell lung cancer. However, there are some disadvantages to RAPD, like poor reproducibility and low yield of PCR products (low production). The resolution and production of RAPD are greatly increased by prolonging the RAMP time from the stage of annealing to extension in PCR [18, 19]. Recently, the technique was modified using high-GC content primers to improve the effectiveness of the RAPD method (high-GC RAMP-PCR technique) [20]. This improved RAPD successfully increased the number of RAPD bands produced from a given DNA sample. Using improved RAPD with high-GC content primers, we found that the average number of bands per primer increased from 6.41 to 15.04 in comparison to regular RAPD with standard primers. The average number of polymorphisms per primer was also increased from 4.647 to 10.22 [20]. Thus, we can produce more polymorphic bands using less RAPD primers. With these successes, we decided to apply this simple yet effective improved RAPD technique with high-GC primers to generate more fingerprints for detecting genomic alterations in human BC and developing diagnostic SCAR markers. High-GC primers FY-10, FY-13 and FY-32 were used to amplify genomic DNA successfully from five pairs of BC tissues and their adjacent tissues. Three different over-amplified bands were purified from the agarose gel, cloned and sequenced, and then used to successfully generate stable diagnostic SCAR markers BC10-1, BC13-4 and BC31-2.
Real-time PCR was performed using DNAs from bloods, tumor tissues and normal tissue adjacent to the tumor. SCAR marker BC10-1 results showed gradually increased levels from blood DNA to tumor DNA. SCAR marker BC13-4 are shown to have increased levels in both tumor tissues and normal tissues adjacent to the tumors compared to DNA from blood; however, there was no difference between DNA from tumor samples and adjacent tissues. SCAR marker BC31-2 did not show increased levels of tumor DNA compared to blood DNA. These results indicate that SCAR markers BC10-1 and BC13-4 could be useful diagnostic markers of genomic instability or chromosome copy number variation in BC [14–16].
To note, the PHKG2 (Phosphorylase Kinase Subunit Gamma-2) gene, which encodes a subunit of phosphorylase kinase (Phk), is located close to SCAR marker BC13-4. Heritable deficiency of Phk, a regulatory enzyme of glycogen metabolism, is responsible for 25% of all cases of glycogen storage disease and occurs in 1 in 100,000 births. Liver phosphorylase b kinase (PhK) deficiency (glycogen storage disease type IX), one of the most common causes of glycogen storage disease, is brought by mutations in the PHKA2, PHKB and PHKG2 genes. Mutations in the testis/liver isoform of PHKG2, which leads to approximately 10–15% of cases, have been associated with autosomal liver glycogenosis in gsd rats and humans [31–33]. However, the role of PHKG2 in BC is unknown. Real-time PCR indicated that the level of mRNA expression for the PHKG2 gene was lowest in non-tumorigenic BC cell line MCF10A, compared to elevated levels in all other tumorigenic BC cells tested. Of course, the levels of protein should be tested to further confirm their significance. Clone 10-1 was located and shown to overlap with DPEP1 gene which promoted metastasis in colon cancer [34–36] but inhibited tumor cell invasiveness in pancreatic ductal adenocarcinoma [37]. The role of DPEP1 in different types of cancer is controversial [34–38]. Our results, for the first time, showed that the mRNA levels of DPEP1 in BC tissues were significantly higher (11.6-fold) than that of the adjacent normal tissues, which may play a role in promoting BC metastasis and invasiveness. Taken together, the DPEP1 and PHKG2 gene may be prognostic targets, which would play a vital role in BC carcinogenesis. Further studies of how the DPEP1, PHKG2 and RNF40 function in BC progression and how they might be used for risk prediction and therapy are underway.
MATERIALS AND METHODS
Breast carcinoma DNA and RNA preparation
Human breast invasive ductal carcinoma specimens were collected from surgically removed tumor tissues at Southwest Medical University Affiliated Hospital in China with informed consent [39–42]. The protocol for human samples was approved by the Ethics Committee of Southwest Medical University. The patients were Chinese women, between 26 to 68 years old and did not receive preoperative radiotherapy or chemotherapy. The histopathologic profiles of the 5 breast carcinomas specimens for RAPD amplification are shown in Table 1. The cancer tissues and adjacent tissues or surrounding normal tissues were immediately frozen in liquid nitrogen and stored at −80ºC. DNA extraction from tissues was performed using the Proteinase K and phenol/chloroform method [40, 43–45]. Total RNA was isolated from cancer cells and tissues using the RNeasy Mini Kit (50) (Cat No.74104, QIAGEN) and stored at −80ºC. The RNA and DNA quality was determined by a 1% agarose gel electrophoresis and spectrophotometry. Genomic DNA from healthy women was also isolated from peripheral leukocytes or blood using the previously described method [43]. The final concentration of all DNA samples was adjusted to 20 ng/μl and stored at −20 ºC until use.
High-GC primers for RAPD
The 10-bp length oligos with high-GC contents (90% of G+C) were designed and synthesized at Beijing DNA chem. Biotechnology Co., Ltd (Beijing, China) and were as previously described [20]. The sequences of high-GC primers FY-10, FY-13 and FY-31 are presented in Table 2.
Amplification of DNA by high-GC RAPD PCR
All the contents of the PCR (10 μl total) were as follows: 1 μl of 2.5 μmol/L primers, 0.5 μl of DNA template (10 ng), 5 μl of 2×PCR Taq MasterMix (TianGen Biotech Co. Ltd., Beijing, China), 3.5 μl of deionized water. PCR conditions were as follows: initial denaturation at 95ºC for 90 s, followed by 40 cycles of 40 s at 94ºC, 60 s at 36ºC, 90 s at 72 ºC, and final extension of 5 min at 72ºC. PCR amplification was executed in an “Applied Biosystems Veriti® 96-Well Thermal Cycler” (Life Technology, USA), and the RAMP time from annealing to extension with a RAMP rate for 5% (0.125 ºC/s) was used [20, 21].
Agarose gel electrophoresis
The amplified RAPD-PCR products were resolved by electrophoresis on a 1.5% agarose gel in 1× TAE (Tris-acetate-EDTA) buffer at 60 volts for 210 minutes at 4 ºC. To increase the resolution, the electrophoresis time was extended to 240 minutes at 60 volts at 4 ºC. Then, the gels were visualized by 0.5 μg/mL ethidium bromide (EB) staining, and the images were documented using the ChemiDoc XR (Bio-Rad, USA) [19, 43]. Bands with different intensity between BC tumors and adjacent normal tissues were selected for cloning.
Molecular cloning of RAPD fragments from amplification by high-GC primers
Cloning of RAPD fragments was described previously [21, 23, 27, 28, 44]. Bands with differential amplification were excised from agarose gels and purified with a TIANgel Mini Purification Kit (DP209, China) according to the company provided protocol. Purified DNA fragments were ligated into pGM-T vector (VT202, Tiangen reagents, Beijing, China), and transformed into DH5α E. coli complement cells. The recombinant clones were selected on LB agar plates from blue and white colonies. The presence of right insert was verified by PCR by using T7/SP6 primer pairs, and EcoRI digestion from purified plasmids. The positive clones were selected for Sanger sequencing.
Development of diagnostic SCAR markers
Homology searches within the human genome were performed using BLAST (http://www.ncbi.nlm.nih.gov/BLAST/). The sequence of each cloned RAPD fragment was used to design pairs of SCAR primers using Primer 3 software (http://frodo.wi.mit.edu/primer3). Sequences of the SCAR primers, amplification length and PCR conditions are shown in Table 3. Ten DNA samples, which we mentioned previously, were used as templates for PCR amplification for development of SCAR markers. The PCR reaction solution consisted of 5 μl 2×Taq PCR MasterMix, 1 μl of 2.5 μM each pair of SCAR primers and 1 μl of genomic DNA (20ng), with a total volume of 10μl. Amplification reactions were performed with an initial pre-denaturation of 90 s at 95°C followed by 33 cycles of denaturation at 94°C for 40 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s. The final extension step was performed at 72°C for 5 min. The amplified PCR products were resolved by electrophoresis on a 1.5% agarose gel [19, 43]. For semi-quantitative genomic DNA PCR using SCAR markers, GAPDH gene served as a loading control.
Quantitative real-time PCR amplification
Three pairs of primers for real-time PCR of SCAR markers and β-actin were designed using software from assay design center of Roch (http://lifescience.roche.com/shop/CategoryDisplay?catalogId=10001&tab=&identifier=Universal+Probe+Library). Sequences of real-time PCR primers and probes for genomic DNA are shown in Table 4. Real-time PCR was performed using the FastStartUniversal Probe Master (Rox) (Cat No. 4914058001, Roch) and the StepOnePlus Real-Time PCR system (Applied Biosystems, USA) as described previously [39, 41, 42, 46–48]. The β-actin gene served as an internal control for normalizing the relative levels of SCAR markers.
Real-time PCR analysis of PHKG2 and DPEP1 mRNA expression
The cDNA libraries were generated from 1 μg of total RNA using a reverse transcriptase kit (Invitrogen, USA). The method for real-time PCR was described previously [41, 42, 46–48]. Sequences of the primers and probes for human PHKG2 and DPEP1 mRNA are shown in Table 5. Levels of the 18S rRNA were measured in the same reaction for each sample to serve as an internal control for normalizing the relative mRNA levels of PHKG2 and DPEP1.
Table 5: Sequences of real time PCR primers for PHKG2 and DPEP1 mRNA expression, probes
Gene |
5′-primer |
Sequence (5′-3′) |
3′-primer |
Sequence (5′-3′) |
Probe |
|---|---|---|---|---|---|
PHKG2 |
Q-PHKG2-L2 |
caatatgcagatccgactttca |
Q-PHKG2-R2 |
ggggtcccacacaactctc |
2 |
DPEP1 |
Q-DPEP1-13L |
tgcactgcagacttctttcg |
Q-DPEP1-13R |
gccaggggaggtcattgt |
13 |
18S |
18S48R |
gcaattattccccatgaacg |
18S48L |
gggacttaatcaacgcaagc |
48 |
Statistical analysis
Student’s t-test was used to determine any significant differences. A p-value of less than 0.05 was considered to be significant for all tests. All the experiments were repeated thrice.
ACKNOWLEDGMENTS AND FUNDING
This work was supported in part by the National Natural Science Foundation of China (81672887, 81172049), the Science and Technology Innovation Team of Colleges and Universities of Sichuan Province (13TD0032), the Applied Basic Research Program of Science and Technology Department of Sichuan Province (14JC0797, 2015JY0038), and the Research Foundation of the Education Department of Sichuan Province (17ZA0427, 17ZB0467).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67:7–30. doi: 10.3322/caac.21387.
2. Wang Y, Gao Y, Battsend M, Chen K, Lu W, Wang Y. Development of a risk assessment tool for projecting individualized probabilities of developing breast cancer for Chinese women. Tumor Biol. 2014; 35:10861–10869.
3. Imani S, Zhang X, Hosseinifard H, Fu S, Fu J. The diagnostic role of microRNA-34a in breast cancer: a systematic review and meta-analysis. Oncotarget. 2017; 8:23177–23187. doi: 10.18632/oncotarget.15520.
4. Moore SP, Antoni S, Colquhoun A, Healy B, Ellison-Loschmann L, Potter JD, Garvey G, Bray F. Cancer incidence in indigenous people in Australia, New Zealand, Canada, and the USA: a comparative population-based study. Lancet Oncol. 2015; 16:1483–1492.
5. Imani S, Hosseinifard H, Cheng J, Wei C, Fu J. Prognostic Value of EMT-inducing Transcription Factors (EMT-TFs) in Metastatic Breast Cancer: A Systematic Review and Meta-analysis. Sci Rep. 2016; 6:28587. doi: 10.1038/srep28587.
6. Anderson BO, Ilbawi AM, El Saghir NS. Breast Cancer in Low and Middle Income Countries (LMICs): A Shifting Tide in Global Health. Breast J. 2015; 21:111–118.
7. Eberth JM, Xu Y, Smith GL, Shen Y, Jiang J, Buchholz TA, Hunt KK, Black DM, Giordano SH, Whitman GJ, Yang W, Shen C, Elting L, et al. Surgeon influence on use of needle biopsy in patients with breast cancer: a national medicare study. J Clin Oncol. 2014; 32:2206–2216.
8. Calhoun KE, Anderson BO. Needle biopsy for breast cancer diagnosis: a quality metric for breast surgical practice. J Clin Oncol. 2014; 32:2191–2192.
9. Fearon ER, Vogelstein B. A enetic model for colorectal tumorigenesis. Cell. 1990; 61: 759–767.
10. Loeb LA. Microsatellite instability: Marker of a mutator phenotype in cancer. Cancer Res. 1994; 54:5059–5063.
11. Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991; 51:3075–3079.
12. Eshleman JR, Markowitz SD. Microsatellite instability in inherited and sporadic neoplasms. Curr Opin Oncol. 1995; 7:83–89.
13. Ong TM, Song B, Qian HW, Wu ZL, Whong WZ. Detection of genomic instability in lung cancer tissues by random amplified polymorphic DNA analysis. Carcinogenesis. 1998; 19:233–235.
14. Ueno T, Emi M, Sato H, Ito N, Muta M, Kuroi K, Toi M. Genome-wide copy number analysis in primary breast cancer. Expert Opin Ther Targets. 2012; 16:S31–5. doi: 10.1517/14728222.2011.636739.
15. Wu C, Yi Q, Wang F, Liu Q, Liu L, Lin A, Chen Q. Genetic instability in patients with pancreatic cancer analyzed by SCARs and electrochemical sensors. Clin Lab. 2014; 60:1169–1175.
16. Tchatchou S, Burwinkel B. Chromosome copy number variation and breast cancer risk. Cytogenet Genome Res. 2008; 123:183–187.
17. Williams JG, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990; 18:6531–6535.
18. Fu J, Li L, Xu X, Wang Z, Tang G, Yin C, Lu G. An Improved Method for Increasing the Efficiency of the Technique of Random Amplified Polymorphic DNA (RAPD). Hereditas. 2000; 22:251–252.
19. Fu J, Yang L, Khan MA, Mei Z. Genetic characterization and authentication of Lonicera japonica Thunb. by using improved RAPD analysis. Mol Biol Rep. 2013; 40: 5993–5999.
20. Wei CL, Cheng JL, Khan MA, Yang LQ, Imani S, Chen HC, Fu JJ. An improved DNA marker technique for genetic characterization by using RAMP-PCR with high GC primers. Genet Mol Res. 2016; 15. doi: 10.4238/gmr.15038721.
21. Cheng JL, Li J, Qiu YM, Wei CL, Yang LQ, Fu JJ. Development of four novel SCAR markers for genetic characterization of Lonicera japonica from high GC-RAMP-PCR and DNA cloning. Genet Mol Res. 2016; 15. doi: 10.4238/gmr.15027737.
22. Cheng J, Long Y, Khan MA, Wei C, Fu S, Fu J. Development and significance of RAPD-SCAR markers for Litchi chinensis Sonn. variety authentication by improved RAPD amplification and molecular cloning. Electronic J Biotechnol. 2015; 18:35–39.
23. Fu JJ, Khan MA, Mei ZQ, Tania M, Yang LQ, Cheng JL. Development of RAPD-SCAR markers for different Ganoderma species authentication by improved RAPD amplification and molecular cloning. Genet Mol Res. 2015; 14: 5667–5676.
24. Zhang C, Mei Z, Cheng J, He Y, Khan MA, Luo P, Imani S, Fu J. Development of SCAR markers based on improved RAPD amplification fragments and molecular cloning for authentication of herbal medicines Angelica sinensis, Angelica acutiloba and Levisticum officinale. Nat Prod Commun. 2015; 10:1743–1747.
25. Mei Z, Zhou B, Wei C, Cheng J, Imani S, Chen H, Fu J. Genetic Authentication of Gardenia jasminoides Ellis var. grandiflora Nakai by Improved RAPD-Derived DNA Markers. Molecules. 2015; 20:20219–29. doi: 10.3390/molecules201119687.
26. Lam KY, Chan GK, Xin GZ, Xu H, Ku CF, Chen JP, Yao P, Lin HQ, Dong TT, Tsim KW. Authentication of Cordyceps sinensis by DNA Analyses: Comparison of ITS Sequence Analysis and RAPD-Derived Molecular Markers. Molecules. 2015; 20:22454–22462.
27. Khan MA, Cheng JL, Mei ZQ, Wei CL, Fu JJ. Development of two novel specific SCAR markers by cloning improved RAPD fragments from the medicinal mushroom Ganoderma lucidium (Leysser: Fr) Karst. Genet Mol Res. 2016; 15. doi: 10.4238/gmr.15038536.
28. Cheng JL, Yin ZC, Mei ZQ, Wei CL, Chen HC, Wu XS, Fu JJ. Development and significance of SCAR marker QG12-5 for Canarium album (Lour.) Raeusch by molecular cloning from improved RAPD amplification. Genet Mol Res. 2016. doi: 10.4238/gmr.15038347.
29. Singh KP. Screening of DNA methylation changes by methylation-sensitive random amplified polymorphic DNA-polymerase chain reaction (MS-RAPD-PCR). Methods Mol Biol. 2014;1105:71–81.
30. Biernasiuk A, Korona-Głowniak I, Grzegorczyk A, Malm A. Differentiation by random amplified polymorphic DNA-polymerase chain reaction (RAPD-PCR) of Candida albicans isolated from upper respiratory tract in patients with non-small cell lung cancer. Acta Biochim Pol. 2014; 61:727–729.
31. Bali DS, Goldstein JL, Fredrickson K, Rehder C, Boney A, Austin S, Weinstein DA, Lutz R, Boneh A, Kishnani PS. Variability of disease spectrum in children with liver phosphorylase kinase deficiency caused by mutations in the PHKG2 gene. Mol Genet Metab. 2014; 111:309–13. doi: 10.1016/j.ymgme.2013.12.008.
32. Maichele AJ, Burwinkel B, Maire I, Søvik O, Kilimann MW. Mutations in the testis/liver isoform of the phosphorylase kinase gamma subunit (PHKG2) cause autosomal liver glycogenosis in the gsd rat and in humans. Nat Genet. 1996; 14:337–340.
33. Albash B, Imtiaz F, Al-Zaidan H, Al-Manea H, Banemai M, Allam R, Al-Suheel A, Al-Owain M. Novel PHKG2 mutation causing GSD IX with prominent liver disease: report of three cases and review of literature. Eur J Pediatr. 2014; 173:647–53. doi: 10.1007/s00431-013-2223-0.
34. Park SY, Lee SJ, Cho HJ, Kim TW, Kim JT, Kim JW, Lee CH, Kim BY, Yeom YI, Lim JS, Lee Y, Lee HG. Dehydropeptidase 1 promotes metastasis through regulation of E-cadherin expression in colon cancer. Oncotarget. 2016; 7:9501–12. doi: 10.18632/oncotarget.7033.
35. Eisenach PA, Soeth E, Röder C, Klöppel G, Tepel J, Kalthoff H, Sipos B. Dipeptidase 1 (DPEP1) is a marker for the transition from low-grade to high-grade intraepithelial neoplasia and an adverse prognostic factor in colorectal cancer. Br J Cancer. 2013; 109:694–703. doi: 10.1038/bjc.2013.363.
36. McIver CM, Lloyd JM, Hewett PJ, Hardingham JE. Dipeptidase 1: a candidate tumor-specific molecular marker in colorectal carcinoma. Cancer Lett. 2004; 209:67–74.
37. Zhang G, Schetter A, He P, Funamizu N, Gaedcke J, Ghadimi BM, Ried T, Hassan R, Yfantis HG, Lee DH, Lacy C, Maitra A, Hanna N, et al. DPEP1 inhibits tumor cell invasiveness, enhances chemosensitivity and predicts clinical outcome in pancreatic ductal adenocarcinoma. PLoS One. 2012;7:e31507.
38. Green AR, Krivinskas S, Young P, Rakha EA, Paish EC, Powe DG, Ellis IO. Loss of expression of chromosome 16q genes DPEP1 and CTCF in lobular carcinoma in situ of the breast. Breast Cancer Res Treat. 2009;113:59–66.
39. Fu J, Zhang L, He T, Xiao X, Liu X, Wang L, Yang L, Yang M, Zhang T, Chen R, Xu J. TWIST represses estrogen receptor-alpha expression by recruiting the NuRD protein complex in breast cancer cells. Int J Biol Sci. 2012; 8:522–32.
40. Khan MA, Zhu L, Tania M, Xiao X, Fu J. Relationship between SPOP mutation and breast cancer in Chinese population. Genet Mol Res. 2015; 14:12362–12366.
41. Imani S, Wei C, Cheng J, Khan MA, Fu S, Yang L, Tania M, Zhang X, Xiao X, Zhang X, Fu J. MicroRNA-34a targets epithelial to mesenchymal transition-inducing transcription factors (EMT-TFs) and inhibits breast cancer cell migration and invasion. Oncotarget. 2017; 8:21362–21379. doi: 10.18632/oncotarget.15214.
42. Wei C, Cheng J, Zhou B, Zhu L, Khan MA, He T, Chen H, Zhang D, Zhao Y, Fu J. Tripartite motif containing 28 (TRIM28) promotes breast cancer metastasis by stabilizing TWIST1 protein. Sci Rep. 2016; 6:29822. doi: 10.1038/srep29822.
43. Fu JJ, Li LY, Lu GX. Relationship between microdeletion on Y chromosome and patients with idiopathic azoospermia and severe oligozoospermia in the Chinese. Chin Med J. 2002; 115:72–75.
44. Fu JJ. Short protocols in medical molecular biology. China Medical Science Press, Beijing. 2012.
45. Cheng J, Fu S, Wei C, Tania M, Khan MA, Imani S, Zhou B, Chen H, Xiao X, Wu J, Fu J. Evaluation of PIK3CA mutations as a biomarker in Chinese breast carcinomas from Western China. Cancer Biomark. 2017; 19:85–92. doi: 10.3233/CBM-160380.
46. Yang L, Khan MA, Mei Z, Yang M, Zhang T, Wei C, Yang W, Zhu L, Long Y, Fu J. Development of RAPD-SCAR markers for Lonicera japonica Thunb. (Caprifolicaceae) variety authentication by improved RAPD and DNA cloning. Rev Biol Trop. 2014; 62:1649–1657.
47. Khan MA, Tania M, Wei C, Mei M, Fu S, Cheng J, Xu J, Fu J. Thymoquinone inhibits cancer metastasis by downregulating TWIST1 expression to reduce epithelial to mesenchymal transition. Oncotarget. 2015; 6:19580–19591. doi: 10.18632/oncotarget.3973.
48. Fu J, Qin L, He T, Qin J, Hong J, Wong J, Liao L, Xu J. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011;21:275–289.