
INTRODUCTION
The deafness, autosomal dominant 5 (DFNA5) gene was identified in our lab in 1998, as a gene causing autosomal dominant non-syndromic hearing loss [1]. Experiments in our laboratory have demonstrated that the DFNA5 protein has the capacity to induce regulated cell death [2–4]. In addition, we recently showed that the mitochondria and especially the MAPK-related pathways play a role in DFNA5-induced regulated cell death [2, 3]. A number of papers on DFNA5 have been published, pointing towards a possible involvement in cancer [2–14]. Furthermore, DFNA5 has been identified in several tumor suppressor genomic methylation screens [8, 9, 11].
Methylation of promoter CpG islands, frequently associated with transcriptional silencing, may serve as a mechanism to inactivate tumor suppressor genes in cancer [15–19]. This is also true for breast cancer [16–19]. Thus, the identification of methylation markers that are sensitive and specific for breast cancer may improve early detection, which is of tremendous importance in achieving a better prognosis [20, 21].
Epigenetic silencing through DFNA5 methylation was previously shown in 52% of primary gastric tumors [11] and in respectively 65% [9] and 34% [6] of colorectal cancers. In 2008, Kim et al. performed a DFNA5 methylation analysis in breast cancer on a limited number of samples (N = 34) using a PCR-based methylation assay, analyzing only a single CpG site [10]. They showed DFNA5 methylation in 53% of primary breast cancer samples and 15.3% of 13 histological normal breast tissues at a distance of the tumor. In seven breast samples of healthy women, DFNA5 methylation was completely absent. The DFNA5 methylation status correlated positively with lymph node metastasis [10]. However, the number of samples and the associations with clinicopathological and survival parameters in the latter study were limited.
In this study, we aimed to analyze the methylation status of DFNA5 in breast cancer in more detail, potentially resulting in the identification of a new detection and/or prognostic marker. We hypothesize that DFNA5 promoter methylation can be a valuable epigenetic biomarker, based upon strong indications for its role as tumor suppressor gene and its function in regulated cell death. We analyzed DFNA5 promoter methylation in breast adenocarcinomas, normal breast tissues (matched tumor) and healthy breast reduction tissues in a large number of samples (N = 123). We also looked for associations between DFNA5 methylation and clinicopathological and survival parameters.
RESULTS
DFNA5 promoter methylation in primary untreated breast adenocarcinomas compared to healthy controls
DFNA5 promoter methylation was investigated in 123 primary breast adenocarcinoma samples and 24 healthy breast reduction samples. All analyses were corrected for age because of the age difference between both groups (age range: 28–89 years for the breast adenocarcinomas and 17–69 years for the healthy breast reductions). Our analysis showed a statistically significant difference in the average DFNA5 promoter methylation percentage between the primary breast adenocarcinomas and the healthy breast reductions (p = 1.6*10−3; R² adjusted = 0.160). The median DFNA5 promoter methylation was 9.00% [range: 1.25%–86.75%] for the breast adenocarcinomas, compared to 3.75% [range: 1.50%–7.00%] for the healthy breast reductions. To assess the reproducibility of our results, we calculated the intraclass correlation coefficient (ICC) for each of the two techniques used. When repeating the analysis starting from pyrosequencing, an ICC of 0.998 [95% CI: 0.997–0.999] was obtained, whereas an ICC of 0.924 [95% CI: 0.858–0.960] was observed when repeating the analysis starting from PCR.
Based on the R²-value of the linear regression model, we determined which of the 4 individual CpGs differentiated best between breast adenocarcinoma samples and healthy breast reduction samples. A statistically significant difference in methylation between both groups was observed when looking at CpG1 (p = 0.017; R² adjusted = 0.096), CpG3 (p = 7.5*10−3; R² adjusted = 0.148) or CpG4 alone (p = 6.1*10−4; R² adjusted = 0.168; Figure 1) alone. Based on the p-value and the R²-value, the methylation percentage of CpG4 was better suited to differentiate between breast adenocarcinomas and healthy controls than the average DFNA5 methylation percentage. Therefore, DFNA5 CpG4 methylation percentage was used in the subsequent analyses. All analyses were also performed with the average DFNA5 methylation over all four CpGs and this did not affect the overall conclusions (data not shown in this manuscript). The median DFNA5 CpG4 methylation percentage for the breast adenocarcinomas was 12% [range: 0%–96%], compared to 4% [range: 1%–7%] for the healthy breast reductions (Figure 1). Methylation values of CpG2 were least suited to differentiate between breast cancer and healthy control samples (p = 0.053; R² adjusted = 0.073).
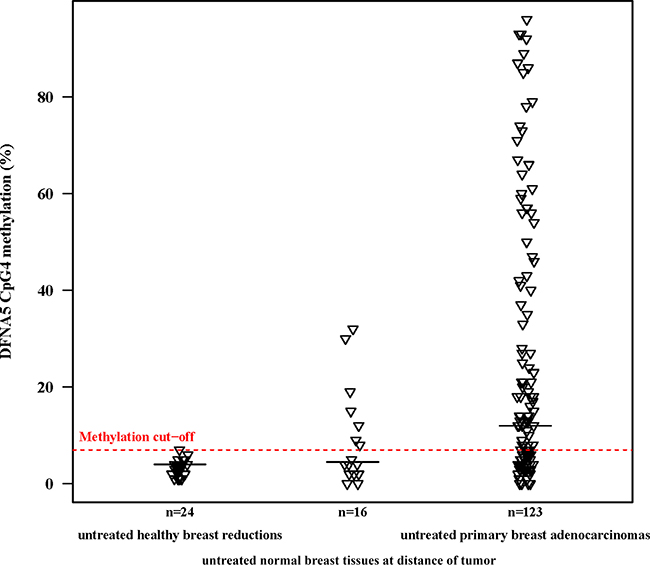
Figure 1: DFNA5 CpG4 methylation percentages of primary breast adenocarcinomas compared to normal breast tissues at a distance of the tumor and healthy breast reductions. The plot shows DFNA5 CpG4 promoter methylation percentages in 123 untreated, primary breast adenocarcinomas; 16 untreated, histological normal breast tissues at a distance of the tumor and 24 untreated, healthy breast reduction samples of non-cancerous patients. The optimal methylation cutoff (7.0%), determined by a ROC analysis, showed DFNA5 CpG4 methylation in 61.8% of 123 breast adenocarcinomas, in 43.8% of 16 histological normal breast tissues at a distance of the tumor and in none of 24 healthy breast reduction samples. The horizontal black lines indicate the median DFNA5 CpG4 methylation percentage per group.
Based upon the DFNA5 CpG4 methylation percentages of breast adenocarcinomas and healthy breast reductions, we constructed a receiver operating characteristic (ROC) curve. The area under the curve (AUC) was 0.830 [95% CI: 0.765–0.896] (Figure 2). The optimal sensitivity and specificity for the detection of breast cancer was obtained with a methylation cutoff of 7.0%. DFNA5 CpG4 methylation was considered positive if the methylation percentage was higher than the cutoff value. We detected DFNA5 CpG4 methylation in 76 out of 123 breast cancer patients, yielding a sensitivity of 61.8% for the detection of breast cancer. Moreover, none of the 24 healthy controls tested positive for DFNA5 CpG4 methylation, which resulted in a specificity of the assay of 100% in our dataset.
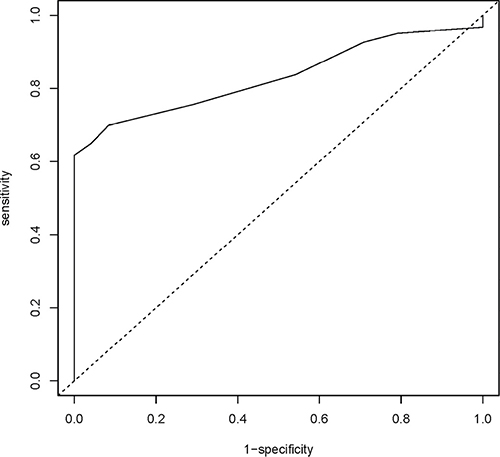
Figure 2: DFNA5 CpG4 methylation percentage as a biomarker for breast adenocarcinomas. Sensitivity and specificity at various cutoff values for our dataset (123 breast adenocarcinomas and 24 healthy breast reductions) are shown in the ROC curve. The full line represents the ROC curve. The dotted line represents the line of no discrimination between tumor and healthy breast samples. The determined optimal cutoff value for DFNA5 CpG4 methylation is 7.0%.
DFNA5 promoter methylation in primary untreated breast adenocarcinomas and histologically normal breast tissues at a distance of the tumor
We had access to tissues of 16 breast cancer patients from whom both breast adenocarcinoma tissue and histologically normal breast tissue at a distance of the tumor were available. The DFNA5 methylation values obtained after pyrosequencing were analyzed using the paired samples t-test (Figure 1). We were not able to find a significant mean difference in DFNA5 CpG4 methylation between breast cancer and matched normal samples (p = 0.10; Figure 3). We observed a median DFNA5 CpG4 methylation difference of 3.5% [range: -29%–73%] between breast cancer and matched normal samples. Figure 3 reveals that in 75% (12/16) of the patients the DFNA5 CpG4 methylation percentage was higher in the tumor sample compared to the normal sample. Interestingly, in 25% (4/16) of the patients, the DFNA5 CpG4 methylation percentage in the matched normal sample was higher compared to the tumor sample.
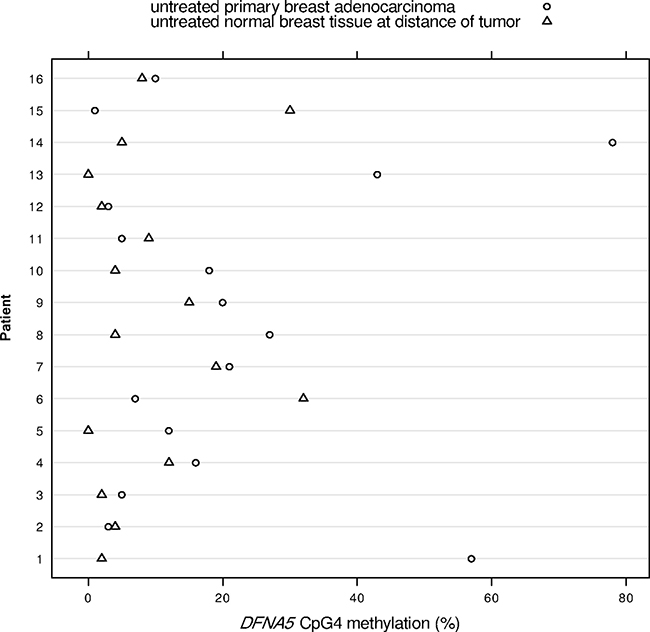
Figure 3: DFNA5 CpG4 methylation percentages in 16 paired breast adenocarcinomas and histologically normal breast tissues at a distance of the tumor. The x-axis shows the DFNA5 CpG4 methylation percentage and each number on the y-axis depicts a patient, from whom both breast adenocarcinoma tissue and histologically normal breast tissue at a distance of the tumor were available.
Associations between DFNA5 CpG4 methylation and clinicopathological parameters
Association analyses between DFNA5 CpG4 methylation percentage and available clinicopathological parameters were performed (Table 1). Not all clinicopathological parameters were available for all patients. A statistically significant association between DFNA5 CpG4 methylation percentage and human epidermal growth factor receptor 2 (HER2) amplification in 58 breast adenocarcinoma patients (45 without HER2 amplification and 13 with HER2 amplification) was found (p = 0.030; Table 1). The median DFNA5 CpG4 methylation was 19% [Q1-Q3: 13%–43%] for the breast adenocarcinomas with HER2 amplification, compared to 8% [Q1-Q3: 4%–18%] for the breast adenocarcinomas without HER2 amplification. No associations were found between DFNA5 CpG4 methylation and ischemia time, pathological tumor-node-metastasis (pTNM) staging, estrogen receptor (ER) status, progesterone receptor (PgR) status, lymphovascular invasion, tumor grade (Nottingham grading system), mitotic activity index (MAI) or maximal tumor diameter (Table 1).
Table 1: Clinicopathological characteristics of the 123 breast adenocarcinomas
Clinicopathological parameter |
Number (%) |
P-value |
|---|---|---|
pT (pTNM) |
0.25 |
|
1 |
54 (43.9) |
|
2 |
35 (28.5) |
|
3 |
5 (4.1) |
|
4 |
3 (2.4) |
|
unknown |
26 (21.1) |
|
pN (pTNM) |
0.80 |
|
0 |
56 (45.5) |
|
1 |
34 (27.7) |
|
2 |
3 (2.4) |
|
3 |
1 (0.8) |
|
unknown |
29 (23.6) |
|
ER |
0.20 |
|
ER+ |
100 (81.3) |
|
ER- |
19 (15.4) |
|
unknown |
4 (3.3) |
|
PgR |
0.17 |
|
PgR+ |
78 (63.4) |
|
PgR- |
36 (29.3) |
|
unknown |
9 (7.3) |
|
HER2 |
0.030 |
|
HER2+ |
13 (10.6) |
|
HER2- |
45 (36.6) |
|
unknown |
65 (52.8) |
|
Lymphovascular invasion |
0.81 |
|
yes |
22 (17.9) |
|
no |
27 (22.0) |
|
unknown |
74 (60.2) |
|
Tumor grade |
0.13 |
|
low grade |
42 (34.1) |
|
intermediate grade |
29 (23.6) |
|
high grade |
30 (24.4) |
|
unknown |
22 (17.9) |
|
MAI |
0.64 |
|
Tumor diameter |
0.32 |
P-values of the associations between DFNA5 CpG4 methylation and the clinicopathological parameters are indicated in the third column. Some clinicopathological parameters were not available for all patients.
Effect of DFNA5 CpG4 methylation on survival
Overall survival (OS), progression free survival (PFS) and disease free survival (DFS) were investigated by survival analysis over a 5-year period. Kaplan Meier and Cox proportional hazard analyses were performed to determine the prognostic value of DFNA5 CpG4 methylation in breast adenocarcinoma. Follow-up data were not available for all patients.
Kaplan Meier analyses were performed, comparing 2 groups based upon the dichotomized DFNA5 CpG4 methylation values, using the cutoff based on the ROC analysis. In our dataset (N = 120), with a limited number of events (N = 11), there was no statistically significant difference in 5-year OS between methylated (N = 47) and non-methylated breast adenocarcinomas (N = 73) (p = 0.39; Figure 4). The 5-year OS rate was 89.0% (65/73) for the methylated and 93.6% (44/47) for the non-methylated breast adenocarcinoma patients. For the 5-year PFS, the number of events was also limited (N = 8) in our dataset (N = 111). The 5-year PFS rate was 91.0% (61/67) for the methylated and 95.5% (42/44) for the non-methylated breast adenocarcinoma patients (p = 0.36; Figure 4). The 5-year DFS rate was 84.5% (60/71) for the methylated and 93.3% (42/45) for the non-methylated breast adenocarcinoma patients, with a total of 14 events in 116 patients (p = 0.16; Figure 4; Supplementary Table 1).
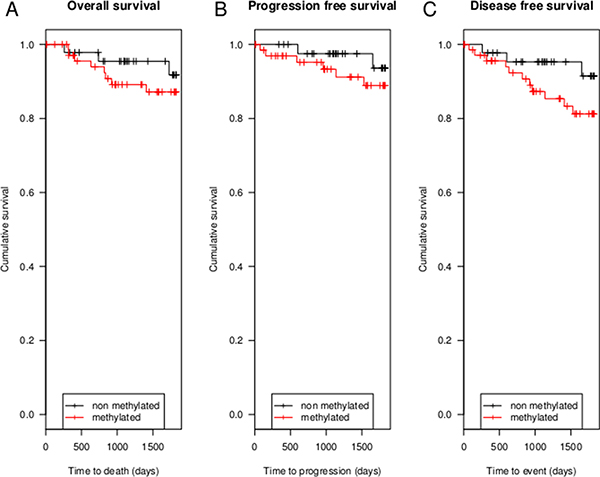
Figure 4: Kaplan Meier analyses of 5-year OS, 5-year PFS and 5-year DFS. (A) shows 5-year OS in 120 breast adenocarcinoma patients, 11 patients died in 5 years after diagnosis. In (B) 5-year PFS in 111 breast adenocarcinoma patients is shown. In only 8 patients recurrence or metastatic disease occurred. (C) shows 5-year DFS in 116 patients. In 5 years after diagnosis a total of 14 patients have had metastatic disease, recurrence or died.
Cox proportional hazard models were fit to estimate the effect of the non-dichotomized DFNA5 CpG4 methylation percentages, accounting for age. The trend towards association between DFNA5 CpG4 methylation and 5-year DFS could be confirmed (p = 0.051; Supplementary Table 1). DFNA5 CpG4 methylation percentage had no statistically significant effect on neither 5-year OS (p = 0.19), nor on 5-year PFS (p = 0.10).
The impact of other covariates, including radiotherapy, adjuvant chemotherapy, adjuvant trastuzumab and hormonal therapy on 5-year OS, PFS and DFS could not be tested due to the limited number of events in our dataset.
DISCUSSION
In this study we evaluated the potential use of DFNA5 promoter methylation as a biomarker for the detection of breast cancer. We analyzed four specific CpGs in the promoter of DFNA5 (Supplementary Figure 1). These CpG dinucleotides were of interest because Akino et al. [11] found that methylation of the region around the transcription start site of DFNA5 correlated with silencing of the gene. As we demonstrated that the methylation percentage of CpG4 alone was better suited to differentiate between breast adenocarcinomas and healthy controls than the average DFNA5 methylation percentage, we performed all our analyses using DFNA5 CpG4 methylation percentages. The median DFNA5 CpG4 methylation percentage was significantly higher (12%) in primary breast adenocarcinomas compared to healthy breast reduction samples (4%) (Figure 1). These results were used to construct a ROC curve that had an AUC of 0.830 (Figure 2). Based on this ROC curve, we were able to determine a methylation cutoff (7%), enabling us to dichotomize our study population. Using this cutoff, DFNA5 CpG4 was methylated in 61.8% of the 123 primary breast adenocarcinomas and in none of the 24 breast samples from healthy women without cancer undergoing a breast reduction. We conclude that DFNA5 CpG4 methylation can be a biomarker for the detection of breast cancer in solid biopsies. Although a different CpG in the DFNA5 promoter was analyzed and a different technique (TaqMan-MSP) was used, our results are in line with those obtained by Kim et al. in 2008. They demonstrated DFNA5 methylation in 53% (18/34) of the breast adenocarcinomas, 15.4% (2/13) of the normal breast tissues at a distance of the tumor and none (0/7) of the normal breast tissues from non-cancerous patients [10].
Analysis of DFNA5 CpG4 methylation in histologically normal breast tissues at a distance of the tumor showed a higher DFNA5 CpG4 methylation percentage in 25% (4/16) of the matched normal samples compared to the tumor samples (Figure 3). These observations can be explained by “field cancerization”, which is the occurrence of genetic, epigenetic and biochemical aberrations in structurally intact cells in histologically normal tissues adjacent to cancerous lesions. The concept of field cancerization was first described in oral squamous cell carcinoma by Slaughter et al. in 1953 [22]. Since then, field cancerization has been described in many different organ systems, including breast cancer [23–27]. In addition to genetic abnormalities (such as chromosomal anomalies and loss of heterozygosity) epigenetic alterations, in particular changes in the DNA methylation status, have been found in normal-appearing tissue surrounding the tumor site in breast cancer [24, 28–33]. Moreover, epigenetic modifications are believed to be early events in breast cancer development due to their presence in pre-invasive ductal carcinoma in situ (DCIS) [32, 34–36], which makes them very suitable as early detection biomarkers. We do not know whether DFNA5 methylation is an early event as currently DFNA5 methylation has not been analyzed in DCIS. However, our data and the data from Kim et al. [10] suggest that DFNA5 methylation might be an early biomarker, making it a suitable candidate to include in a panel of breast cancer detection markers. Further research on the role of DFNA5 in field cancerization is needed, as this phenomenon is regarded as clinically significant due to its presumed role in the local recurrence of cancer. Indeed, the region showing these molecular abnormalities is not always completely removed by surgery and therefore might lead to newly occuring neoplasms.
DFNA5 CpG4 methylation performs well compared to other gene promoter methylation markers described in literature thus far. However, combining DFNA5 CpG4 methylation with other markers could improve its sensitivity for the detection of breast cancer, as has been observed for other methylation markers. One example is the Ras-associated domain family member 1 (RASSF1A) gene, one of the most frequently hypermethylated genes in human cancer. CpG island hypermethylation of RASSF1A has been demonstrated in 49%–77% of breast cancers [33, 37–39]. In a comprehensive study of RASSF1A promoter methylation in breast tissue samples, it was demonstrated that primary tumors had significantly higher promoter methylation compared to healthy breast reduction samples, while normal breast samples at a distance of the tumor had intermediate methylation levels [24]. These results correspond to our findings in DFNA5.
A next step in the study of the potential utility of DFNA5 CpG4 methylation as a biomarker, is to analyze DFNA5 CpG4 methylation in circulating DNA from both breast cancer patients and healthy individuals. Several studies have provided proof of principle for the detection of promoter hypermethylation of breast adenocarcinoma derived DNA in blood [40–42]. Using liquid biopsies, DFNA5 CpG4 methylation has the potential to be a suitable low invasive early detection biomarker for breast cancer.
We found that DFNA5 CpG4 methylation was associated with HER2 amplification (Table 1). Currently, we do not have an explanation for this observation. Further functional studies are needed to elucidate the cause of the association with HER2 status, possibly leading to more insights into tumorigenesis, or a better molecular subclassification. Like Kim et al. [10], we could not find an association with ER status in our study population, which is in contrast to the study of Thompson and Weigel [14]. A possible explanation could be that in the study of Thompson and Weigel DFNA5 expression in cell lines and 29 primary breast tumors was analyzed instead of DFNA5 methylation [14]. Moreover, the scoring system for ER status has changed over time [43, 44]. Furthermore, we were not able to find an association with lymph node metastasis (pN), which is in contrast to the study of Kim et al. [10].
The survival models for OS, PFS and DFS were not statistically significant, possibly due to the limited number of events in our study population. However, the survival curves suggest a trend, wherein breast cancer patients without DFNA5 CpG4 methylation perform better compared to breast cancer patients with DFNA5 CpG4 methylation (Figure 4). The strongest trend is observed for DFS. Larger prospective studies are needed to investigate the prognostic role of DFNA5 methylation in breast cancer.
Finally, DFNA5 has never been identified in classic tumor suppressor gene screens that aim to identify genes with inactivating somatic mutations. Moreover, we consulted the COSMIC database and compared the number of somatic DFNA5 mutations with somatic mutations in non-cancer genes, similar results were found (data not shown in this manuscript). Taken together, we can conclude that inactivation of DFNA5 through gene mutations, in contrast to promoter hypermethylation, is not a main mechanism for DFNA5 in cancer.
In conclusion, we demonstrated that 1) DFNA5 CpG4 methylation can be a biomarker for the detection of breast cancer in solid biopsies, 2) DFNA5 is methylated in breast adenocarcinomas in contrast to healthy breast reduction samples, 3) DFNA5 methylation is often present in normal breast tissue surrounding the tumor, which may be explained by field cancerization. Firstly, this suggests that DFNA5 methylation is an early event, which offers opportunities for DFNA5 methylation as early detection maker for breast cancer. Secondly, this indicates the importance of using healthy reference tissue from non-cancerous patients in detection biomarker research. Further research is needed to investigate if DFNA5 methylation could reliably be analyzed in liquid biopsies, so that it may be developed as low invasive, early detection biomarker for breast cancer.
MATERIALS AND METHODS
Study population and tissue samples
We collected 123 well-characterized primary breast adenocarcinomas (108 ductal– 10 lobular– 2 mixed– 3 unknown) and 24 breast samples from women without cancer undergoing a breast reduction. If available, we also retrieved histologically normal breast tissue from breast cancer resection specimens (N = 16; 14 ductal– 2 mixed). All samples were formalin-fixed, paraffin-embedded (FFPE) and were retrieved from the tumorbank of the Antwerp University Hospital (tumorbank@UZA). Characteristics of the study population are shown in Table 1. This study was approved by the ethical committee of the Antwerp University Hospital and the University of Antwerp.
DNA isolation
A 5μm-section of each tissue block was stained with hematoxylin and eosin (HE) and inspected by the study pathologist (Prof. Dr. P. Pauwels). Inclusion criteria for tumor samples were primary malignancy without necrosis and tumor cell content of at least 40%. Ten sections (5 μm thick) were deparaffinized and subjected to DNA extraction using the QIAamp® DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
Bisulfite conversion, PCR and pyrosequencing
Bisulfite modification was performed using 2 μg genomic DNA as input in a MethylEasy™ Xceed Kit (Human Genetic Signatures, Sydney NSW 2113, Australia) according to the manufacturer’s instructions. For PCR, bisulfite-modified DNA was amplified by AmpliTaq Gold® DNA Polymerase (Applied Biosystems, California 94404, USA) using the following degenerate primers (5′-Biotin-RAACCCCTCCCRCAACCT-3′ and 5′-GGYGGAGAGAGGGTTYGTT-3′, Y = C/T and R = A/G; Supplementary Figure 1). The PCR program consisted of an initial enzyme activation at 95°C for 10 minutes, followed by 50 cycles of 45 seconds at 95°C, 45 seconds at 60°C and 45 seconds at 72°C; and a final extension at 72°C for 10 minutes. Subsequently, four specific CpG dinucleotides around the transcription start site of DFNA5 were sequenced using the following sequencing primer (5′-YGGGYGTTTTAGAGT-3′, Y = C/T) on the PyroMark Q24 platform (Qiagen, Hilden, Germany) according to the manufacturer’s instructions (Supplementary Figure 1). Analysis of the results was performed with the PyroMark Q24 software (Qiagen, Hilden, Germany).
Statistical analysis
All analyses were performed with log10 transformed methylation percentages to obtain normality. Due to the age difference between the healthy samples and the breast adenocarcinoma samples, age was included as a covariate throughout all analyses. For the comparison of DFNA5 methylation percentages between breast adenocarcinomas and healthy breast reductions, linear regression models were fit with the logarithm of the methylation percentage as dependent variable and disease status as independent variable. Intraclass correlation was calculated for absolute agreement. The association between DFNA5 CpG4 methylation percentage and clinicopathological parameters was tested by fitting linear regression models with the logarithm of the methylation percentage as dependent variable and the clinicopathological parameter as independent variable. The diameter, MAI and ischemia time were entered as independent variables after log10 transformation. To analyze whether ischemia time, the time between resection of the tissue and the time of fixation in formalin, had an influence on DFNA5 methylation, we corrected for disease status. For survival analyses, patients were censored at time of last follow-up, with a maximum of 5 years. OS was defined as: the time of diagnosis to death; PFS as: the time of diagnosis to recurrence or metastatic disease and DFS as: the time of diagnosis to recurrence, metastatic disease or death. All p-values are two-sided, and p-values less than or equal to 0.05 were considered statistically significant. Data analysis was performed using SPSS software (version 22; SPSS Inc., Chicago, IL).
Abbreviations
DFNA5: deafness, autosomal dominant 5; ICC: intraclass correlation coefficient; ROC: receiver operating characteristic; AUC: area under the curve; HER2: human epidermal growth factor receptor 2; pTNM: pathological tumor-node-metastasis; ER: estrogen receptor; PgR: progesterone receptor; MAI: mitotic activity index; OS: overall survival; PFS: progression free survival; DFS: disease free survival; DCIS: ductal carcinoma in situ; RASSF1A: Ras-associated domain family member 1.
Authors’ contributions
L.C. performed the experiments. L.C. and E.F. analyzed the data and L.C., K.o.D.B. and G.V.C. wrote the manuscript. P.P. analyzed all HE-stained slices. All authors designed and supervised experiments and contributed, with their critical revision, to the final version of the manuscript.
ACKNOWLEDGMENTS
The human biological material used in this publication was provided by the UZA tumorbank, Antwerp University Hospital, Belgium, which is funded by the National Cancer Plan (tumorbank@UZA). With a special thank you to Annemieke De Wilde and Sofie Goethals. Furthermore, we want to thank the datamanagers of the multidisciplinary oncological center, Antwerp University Hospital, Belgium, to provide us the clinicopathological information of our study population. Finally, we want to acknowledge the contribution of our lab technicians, Christophe Hermans and An Schepers and statistician Dr. Kristien Wouters.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
L.C. has a Ph.D. fellowship of the Research Foundation–Flanders (FWO; 11Y9815N).
REFERENCES
1. Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt PJ, van de Heyning P, McGuirt WT, Smith RJ, Willems PJ, Legan PK, Richardson GP, van Camp G. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet. 1998; 20:194–7. doi: 10.1038/2503.
2. Van Rossom S, Op de Beeck K, Franssens V, Swinnen E, Schepers A, Ghillebert R, Caldara M, van Camp G, Winderickx J. The splicing mutant of the human tumor suppressor protein DFNA5 induces programmed cell death when expressed in the yeast Saccharomyces cerevisiae. Front Oncol. 2012; 2:77. doi: 10.3389/fonc.2012.00077.
3. Van Rossom S, Op de Beeck K, Hristovska V, Winderickx J, van Camp G. The deafness gene DFNA5 induces programmed cell death through mitochondria and MAPK-related pathways. Front Cell Neurosci. 2015; 9:231. doi: 10.3389/fncel.2015.00231.
4. Op de Beeck K, van Camp G, Thys S, Cools N, Callebaut I, Vrijens K, van Nassauw L, van Tendeloo VF, Timmermans JP, van Laer L. The DFNA5 gene, responsible for hearing loss and involved in cancer, encodes a novel apoptosis-inducing protein. Eur J Hum Genet. 2011; 19:965–73. doi: 10.1038/ejhg.2011.63.
5. Wang C, Tang L, Shen D, Wang C, Yuan Q, Gao W, Wang Y, Xu R, Zhang H. The expression and regulation of DFNA5 in human hepatocellular carcinoma DFNA5 in hepatocellular carcinoma. Mol Biol Rep. 2013; 40:6525–31. doi: 10.1007/s11033-013-2581-8.
6. Yokomizo K, Harada Y, Kijima K, Shinmura K, Sakata M, Sakuraba K, Kitamura Y, Shirahata A, Goto T, Mizukami H, Saito M, Kigawa G, Nemoto H, et al. Methylation of the DFNA5 gene is frequently detected in colorectal cancer. Anticancer Res. 2012; 32:1319–22.
7. Op de Beeck K, van Laer L, van Camp G. DFNA5, a gene involved in hearing loss and cancer: a review. Ann Otol Rhinol Laryngol. 2012; 121:197–207.
8. Fujikane T, Nishikawa N, Toyota M, Suzuki H, Nojima M, Maruyama R, Ashida M, Ohe-Toyota M, Kai M, Nishidate T, Sasaki Y, Ohmura T, Hirata K, et al. Genomic screening for genes upregulated by demethylation revealed novel targets of epigenetic silencing in breast cancer. Breast Cancer Res Treat. 2010; 122:699–710. doi: 10.1007/s10549-009-0600-1.
9. Kim M, Chang X, Yamashita K, Nagpal J, Baek J, Wu G, Trink B, Ratovitski E, Mori M, Sidransky D. Aberrant promoter methylation and tumor suppressive activity of the DFNA5 gene in colorectal carcinoma. Oncogene. 2008; 27:3624–34. doi: 10.1038/sj.onc.1211021.
10. Kim M, Lebron C, Nagpal J, Chae Y, Chang X, Huang Y, Chuang T, Yamashita K, Trink B, Ratovitski E, Califano J, Sidransky D. Methylation of the DFNA5 increases risk of lymph node metastasis in human breast cancer. Biochem Biophys Res Commun. 2008; 370:38–43. doi: 10.1016/j.bbrc.2008.03.026.
11. Akino K, Toyota M, Suzuki H, Imai T, Maruyama R, Kusano M, Nishikawa N, Watanabe Y, Sasaki Y, Abe T, Yamamoto E, Tarasawa I, Sonoda T, et al. Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci. 2007; 98:88–95. doi: 10.1111/j.1349-7006.2006.00351.x.
12. Masuda Y, Futamura M, Kamino H, Nakamura Y, Kitamura N, Ohnishi S, Miyamoto Y, Ichikawa H, Ohta T, Ohki M, Kiyono T, Egami H, Baba H, et al. The potential role of DFNA5, a hearing impairment gene, in p53-mediated cellular response to DNA damage. J Hum Genet. 2006; 51:652–64. doi: 10.1007/s10038-006-0004-6.
13. Lage H, Helmbach H, Grottke C, Dietel M, Schadendorf D. DFNA5 (ICERE-1) contributes to acquired etoposide resistance in melanoma cells. FEBS Lett. 2001; 494:54–9.
14. Thompson DA, Weigel RJ. Characterization of a gene that is inversely correlated with estrogen receptor expression (ICERE-1) in breast carcinomas. Eur J Biochem. 1998; 252:169–77.
15. Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007; 16 Spec No 1:R50–9. doi: 10.1093/hmg/ddm018.
16. Rivenbark AG, Livasy CA, Boyd CE, Keppler D, Coleman WB. Methylation-dependent silencing of CST6 in primary human breast tumors and metastatic lesions. Exp Mol Pathol. 2007; 83:188–97. doi: 10.1016/j.yexmp.2007.03.008.
17. Huang TH, Perry MR, Laux DE. Methylation profiling of CpG islands in human breast cancer cells. Hum Mol Genet. 1999; 8:459–70.
18. Domann FE, Rice JC, Hendrix MJ, Futscher BW. Epigenetic silencing of maspin gene expression in human breast cancers. Int J Cancer. 2000; 85:805–10.
19. Bae YK, Brown A, Garrett E, Bornman D, Fackler MJ, Sukumar S, Herman JG, Gabrielson E. Hypermethylation in histologically distinct classes of breast cancer. Clin Cancer Res. 2004; 10: 5998–6005. doi: 10.1158/1078-0432.CCR-04-0667.
20. Maruvada P, Wang W, Wagner PD, Srivastava S. Biomarkers in molecular medicine: cancer detection and diagnosis. BioTechniques. 2005; 9–15.
21. Park JW, Kerbel RS, Kelloff GJ, Barrett JC, Chabner BA, Parkinson DR, Peck J, Ruddon RW, Sigman CC, Slamon DJ. Rationale for biomarkers and surrogate end points in mechanism-driven oncology drug development. Clin Cancer Res. 2004; 10:3885–96. doi: 10.1158/1078-0432.CCR-03-0785.
22. Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953; 6:963–8.
23. Heaphy CM, Bisoffi M, Fordyce CA, Haaland CM, Hines WC, Joste NE, Griffith JK. Telomere DNA content and allelic imbalance demonstrate field cancerization in histologically normal tissue adjacent to breast tumors. Int J Cancer. 2006; 119:108–16. doi: 10.1002/ijc.21815.
24. Yan PS, Venkataramu C, Ibrahim A, Liu JC, Shen RZ, Diaz NM, Centeno B, Weber F, Leu Y, Shapiro CL, Eng C, Yeatman TJ, Huang TH. Mapping geographic zones of cancer risk with epigenetic biomarkers in normal breast tissue. Clin Cancer Res. 2006; 12:6626–36. doi: 10.1158/1078-0432.CCR-06-0467.
25. Braakhuis BJM, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003; 63:1727–30.
26. Försti A, Louhelainen J, Söderberg M, Wijkström H, Hemminki K. Loss of heterozygosity in tumour-adjacent normal tissue of breast and bladder cancer. Eur J Cancer. 2001; 37:1372–80.
27. Larson PS, de Morenas AL, Cupples LA, Huang K, Rosenberg CL. Genetically abnormal clones in histologically normal breast tissue. Am J Pathol. 1998; 152:1591–8.
28. Spitzwieser M, Holzweber E, Pfeiler G, Hacker S, Cichna-Markl M. Applicability of HIN-1, MGMT and RASSF1A promoter methylation as biomarkers for detecting field cancerization in breast cancer. Breast Cancer Res. 2015; 17:125. doi: 10.1186/s13058-015-0637-5.
29. van der Auwera I, Bovie C, Svensson C, Trinh XB, Limame R, van Dam P, van Laere SJ, van Marck EA, Dirix LY, Vermeulen PB. Quantitative methylation profiling in tumor and matched morphologically normal tissues from breast cancer patients. BMC Cancer. 2010; 10:97. doi: 10.1186/1471-2407-10-97.
30. Zou D, Yoon H, Perez D, Weeks RJ, Guilford P, Humar B. Epigenetic silencing in non-neoplastic epithelia identifies E-cadherin (CDH1) as a target for chemoprevention of lobular neoplasia. J Pathol. 2009; 218:265–72. doi: 10.1002/path.2541.
31. Berman H, Zhang J, Crawford YG, Gauthier ML, Fordyce CA, McDermott KM, Sigaroudinia M, Kozakiewicz K, Tlsty TD. Genetic and epigenetic changes in mammary epithelial cells identify a subpopulation of cells involved in early carcinogenesis. Cold Spring Harb Symp Quant Biol. 2005; 70:317–27. doi: 10.1101/sqb.2005.70.051.
32. Fleischer T, Frigessi A, Johnson KC, Edvardsen H, Touleimat N, Klajic J, Riis ML, Haakensen VD, Wärnberg F, Naume B, Hell A, Helland A, Børresen-Dale A, et al. Genome-wide DNA methylation profiles in progression to in situ and invasie carcinoma of the breast with impact on gene transcription and prognosis. Genome Biol. 2014; 15:435. doi: 10.1186/PREACCEPT-2333349012841587.
33. Dworkin AM, Huang TH, Toland AE. Epigenetic alterations in the breast: Implications for breast cancer detection, prognosis and treatment. Semin Cancer Biol. 2009; 19:165–71. doi: 10.1016/j.semcancer.2009.02.007.
34. Johnson KC, Koestler DC, Fleischer T, Chen P, Jenson EG, Marotti JD, Onega T, Kristensen VN, Christensen BC. DNA methylation in ductal carcinoma in situ related with future development of invasive breast cancer. Clin Epigenetics. 2015; 7:75. doi: 10.1186/s13148-015-0094-0.
35. Umbricht CB, Evron E, Gabrielson E, Ferguson A, Marks J, Sukumar S. Hypermethylation of 14-3-3 sigma (stratifin) is an early event in breast cancer. Oncogene. 2001; 20:3348-53. doi: 10.1038/sj.onc.1204438.
36. Balch C, Montgomery JS, Paik H, Kim S, Huang TH, Nephew KP. New anti-cancer strategies: epigenetic therapies and biomarkers. Front Biosci. 2005; 10:1897–931.
37. Dammann R, Yang G, Pfeifer GP. Hypermethylation of the cpG island of Ras association domain family 1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3 locus, occurs in a large percentage of human breast cancers. Cancer Res. 2001; 61:3105–9.
38. Burbee DG, Forgacs E, Zöchbauer-Müller S, Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S, Sekido Y, Latif F, Milchgrub S, et al. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst. 2001; 93:691–9.
39. Lewis CM, Cler LR, Bu D, Zöchbauer-Müller S, Milchgrub S, Naftalis EZ, Leitch AM, Minna JD, Euhus DM. Promoter hypermethylation in benign breast epithelium in relation to predicted breast cancer risk. Clin Cancer Res. 2005; 11:166–72.
40. Wittenberger T, Sleigh S, Reisel D, Zikan M, Wahl B, Alunni-Fabbroni M, Jones A, Evans I, Koch J, Paprotka T, Lempiäinen H, Rujan T, Rack B, et al. DNA methylation markers for early detection of women’s cancer: promise and challenges. Epigenomics. 2014; 6:311–27. doi: 10.2217/epi.14.20.
41. Shan M, Yin H, Li J, Li X, Wang D, Su Y, Niu M, Zhong Z, Wang J, Zhang X, Kang W, Da Pang. Detection of aberrant methylation of a six-gene panel in serum DNA for diagnosis of breast cancer. Oncotarget. 2016; 7:18485–94. doi: 10.18632/oncotarget.7608.
42. Hoque MO, Feng Q, Toure P, Dem A, Critchlow CW, Hawes SE, Wood T, Jeronimo C, Rosenbaum E, Stern J, Yu M, Trink B, Kiviat NB, et al. Detection of aberrant methylation of four genes in plasma DNA for the detection of breast cancer. J Clin Oncol. 2006; 24:4262–9. doi: 10.1200/JCO.2005.01.3516.
43. Hammond MEH, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M, Hicks DG, Lester S, Love R, et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010; 28:2784–95. doi: 10.1200/JCO.2009.25.6529.
44. Senkus E, Kyriakides S, Ohno S, Penault-Llorca F, Poortmans P, Rutgers E, Zackrisson S, Cardoso F. Primary breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015; 26:v8–30. doi: 10.1093/annonc/mdv298.