
INTRODUCTION
The stressful biological conditions that exist within the tumor microenvironment exert strong adaptive pressure on cancer cells which in turn exploit endogenous pathways to reprogram their transcriptome, proteome, and metabolism to survive and thrive under these conditions [1-6]. Therefore, proteins that facilitate these adaptation processes are attractive drug targets as they are expected to be active only in tumor tissues, which are exposed to stress, but not in non-stressed normal tissues [2, 7, 8]. Oxidative stress is commonly associated with cancer and cancer cells have been shown to promote expression of ROS scavenging pathways in order to survive, proliferate, and resist radio- and chemotherapy [9-11]. While these basic biological principles have been extensively demonstrated and reviewed elsewhere [12], especially in the context of the transcription factor nuclear factor (erythroid-derived 2)-like 2 (NRF2 or NFE2L2) [13-15], it is still not clear which groups of AOS genes are overexpressed in multiple cancers compared with normal tissues. Similarly, it is as yet not defined which groups of AOS genes predict for bad prognosis and in different cancer entities.
Here, we systematically evaluated the mRNA expression patterns of all genes (n=285) annotated by GO(Gene Ontology) as being involved in ‘oxidative stress’ (including AOS genes) in publicly available microarray data sets and identified a sub-group of genes that is highly overexpressed in multiple cancers compared to normal tissues. Subsequently, by using multiple unsupervised analyses, we found that the glutathione and thioredoxin pathways are significantly enriched among these genes. Interestingly, high expression of a significant number of these genes is negatively correlated with survival in breast and lung carcinoma, suggesting that they might play a protective role in cancer cells as opposed to merely reflecting a transcriptional response to oxidative stress. We discuss these genes, the regulators of their expression, their specific role in cancer, and possible therapeutic strategies that can hit these targets.
Identification of oxidative stress response genes highly expressed in multiple cancers: enrichment in glutathione and thioredoxin pathways-related genes
We wondered whether specific oxidative stress response genes are highly overexpressed in cancer as compared to normal tissues. Because we are primarily interested in how cancer cells adapt to their microenvironment found within solid tumors we focused our analysis on carcinomas as they constitute the most frequent type of solid tumors. Using hierarchical clustering, we observed that the 285 genes cluster into 6 clusters (hereafter referred to as “groups”) (Figure 1; Table S1). Groups 2-5 were found to be cancer type specific (Figure 1; Table S1). While these may be interesting in the context of the corresponding cancers entity, they may also reflect genes highly expressed in the tissue of origin, and therefore will require further in-depth analysis. More interestingly, we identified a group of genes that is highly overexpressed in multiple cancers compared to normal tissues (group 6), as well as a group that is highly overexpressed in normal tissues compared to cancers (group 1).

Figure 1: Gene expression patterns of 285 oxidative stress genes in 353 normal tissues and various carcinomas (total n=994, 10 different entities). Gene expression data were retrieved from the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/gds) of published microarray studies (all Affymetrix HG-U133plus2.0). Normal tissue n=353 (GSE3526) [133]. Carcinomas: bladder n=102 (GSE31684, GSE7476), breast n=107 (GSE36774), colorectal n=177 (GSE17536), gastric cancer (ga) n=43 (GSE22377), liver (hepatocellular carcinoma) n=91 (GSE9843), kidney (ki) n=52 (GSE11151), melanoma (melan) n=101 (GSE10282, GSE15605), lung (non-small-cell lung cancer, NSCLC) n=196 (GSE37745), pancreas (pan) n=52 (GSE17891, GSE32676), ovary (ov) n=73 (GSE14001, GSE18520). All microarray data were normalized simultaneously by RMA [134] using custom brainarray (v15.0) ENTREZG CDF-files as previously described [132, 135, 136]. Hierarchical clustering of genes (1-Pearson correlation) and k-means clustering (2 signatures, 10,000 iterations) of microarray samples were performed with GENE-E software (http://www.broadinstitute.org/cancer/software/GENE-E/index.html). Gene expression data were log2 transformed for depiction in a heat-map.
GO analysis using bioprofiling.de [16] (Table S2) confirmed that in both groups ‘response to oxidative stress’ and ‘response to hydrogen peroxide’ were the top two categories (p<10-15) confirming, as expected, that both lists (groups 1 and 6) are significantly enriched in genes involved in oxidative stress response. While the first two GO categories were similar between group 1 and group 6 the third was different. ‘Aging’ was the third identified GO category in the list of genes that are highly expressed in normal tissues (group 1) (p<10-7) while ‘cellular response to hydrogen peroxide’ (p<10-12) was the third category found in the list of genes that are highly expressed in cancer (group 6) (Table S2). It is interesting to note that the expression of AOS genes that are linked to aging is a feature of normal tissue in light of the discussion on the similarities and differences between expression of stress genes in cancer and aging [10, 17].
To identify possible common biological features of the genes represented in each of the two lists we next queried common protein folds of the encoded proteins. Using Interpro (bioprofiling.de; [16]) we found that the list of genes that are highly expressed in cancer (group 6) is significantly enriched (p<10-6; Table S3) in proteins that contain ‘Alkyl hydroperoxide reductase subunit C/ Thiol specific antioxidant’ domains, ‘Thioredoxin fold’, and ‘Thioredoxin like fold’, whereas the genes that are highly expressed in normal tissues (group 1) did not result in specifically enriched protein fold(s). Moreover, using pathway and network analysis (bioprofiling.de R_Spider; [18]), we found the ‘Glutathione metabolism’ pathway among the genes highly expressed in cancers (11 genes; p=0.01) with a specific sub-group of 9 genes (p<0.005) whose products are known to interact with one another, such as glutamate cysteine ligase catalytic subunit (GCLC) and glutamate cysteine ligase modifier subunit (GCLM) (Figure 2A)[19]. Collectively, these analyses suggest that elevated glutathione synthesis and thioredoxin pathway activity are common features of cancer cells.
Identification of AOS response genes highly expressed in cancers which predict negative patient outcome
In order to identify possible drug targets within the list of genes that are up-regulated in cancer (group 6) we used the bioprofiling.de GENE_SRV tool that screens a list of genes against publicly available expression and patient survival data [20]. Specifically, this tool identifies cancer entities in which a particular gene signature is significantly enriched for predictors of patient outcome. We found significant predictive power of some genes in group 6 (highly expressed in cancers) in breast and lung cancers (p=0.035) and in chronic lymphocytic leukemia (CLL) (p=0.037). Since this study exclusively addresses AOS genes in solid tumors, we focus our discussion on the first two cancer entities. Kaplan-Meier plots for all genes that exhibited significant predictive power are summarized in Table 1 and Figure S1-3 (typical plots are shown in Figure 2B).
Table 1: List of AOS response genes highly expressed in cancers which correlate with outcome in breast or lung cancer. The cancer AOS response signature was analyzed using bioprofiling.de GENE_SRV to identify cancers in which these genes have significant predictive power. Only genes that were found to correlate with survival are shown. Gene name, ENTREZ ID, microarray probeset ID and p value are provided. Kaplan-Meier plots for all the indicated genes are displayed in Figure S1-S3.
Breast cancer |
ENTREZ ID |
||
Gene |
(probe ID) |
P-value |
|
Bad prognosis |
BTG3 |
10950 (360504) |
0.00357 |
CASP3 |
836 (540397) |
0.0000453 |
|
CDC2 |
983 (5360092) |
0.0000105 |
|
ECT2 |
1894 (5420064) |
0.00012 |
|
EGLN1 |
54583 (6130168) |
0.00586 |
|
FOXM1 |
2305 (5390044) |
2.51E-08 |
|
G6PD* |
2539 (5700072) |
0.00748 |
|
GAPDH |
2597 (1940184) |
0.00321 |
|
HMOX1* |
3162 (6180100) |
0.000294 |
|
LONP1 |
9361 (870538) |
0.0031 |
|
NUDT1 |
4521 (6180369) |
0.0016 |
|
PRDX4* |
10549 (940131) |
0.00276 |
|
PSMB5 |
5693 (3610041) |
0.00337 |
|
SELS |
55829 (7100450) |
0.00844 |
|
SERPINE1 |
5054 (6840139) |
0.00167 |
|
SRXN1* |
140809 (3190176) |
0.00336 |
|
TXNRD1* |
7296 (6220603) |
0.00000169 |
|
Good prognosis |
PON2 |
5445 (7040022) |
0.00457 |
SIRT1 |
23411 (6940021) |
0.00918 |
|
Lung cancer |
NCBI ID |
||
NCBI ID |
(probe ID) |
P-value |
|
Bad prognosis |
COL1A1 |
1277 (926) |
0.000675 |
GAPDH |
2597 (1738) |
0.00185 |
|
GCLC* |
2729 (14771) |
0.00354 |
|
GSS* |
2937 (267) |
0.00934 |
|
NQO1* |
1728 (20812) |
0.0045 |
|
RNF7 |
9616 (12099) |
0.00439 |
|
STK24 |
8428 (10957) |
0.00195 |
|
TXN* |
7295 (10753) |
0.00789 |
|
TXNRD1* |
7296 (8394) |
0.00284 |
|
Good prognosis |
NFKB1 |
4790 (3750) |
0.000849 |
* NRF2 targets |
|||
In lung cancer, 9 genes correlated with poor prognosis, including GCLC, NAD(P)H dehydrogenase (quinone) 1 (NQO1) and thioredoxin (TXN), and 1 with good outcome (Figure 2B and Figure S1). In breast cancer, 17 genes were associated with poor prognosis, such as glucose-6-phosphate dehydrogenase (G6PD), heme oxygenase (decycling) 1 (HMOX1) and thioredoxin reductase 1 (TXNRD1), and only 2 with good outcome (Table 1 and Figure S2-S3). The finding that the majority of the genes are predictors for negative patient outcome supports the model that the AOS response genes, which are up-regulated in cancer, may facilitate cancer cell adaptation to the tumor environment and/or resistance to therapy. We therefore argue that the genes identified by our analyses as being highly overexpressed in carcinomas and correlating negatively with prognosis may constitute attractive drug targets as well, which will be further discussed below.
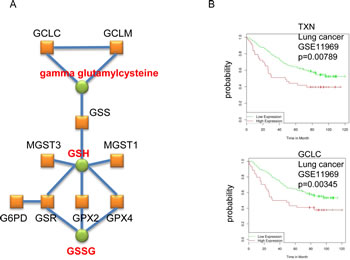
Figure 2: Enrichment of genes coding for enzymes involved in glutathione synthesis in the cancer AOS genes signature. A. The depicted gene network was identified by R_SPIDER as statistically enriched in the list of genes that are highly expressed in cancers (group 6) (Table S1). Genes are represented by red boxes, known interactions between the corresponding proteins are displayed as blue lines and metabolites by green circles. B. Typical Kaplan-Meier plots are shown.
Relevance of the glutathione and thioredoxin pathways as essential components of multiple cancers and potential drug targets
Thioredoxin pathway
The thioredoxin system is highly conserved throughout evolution and we observed that multiple members of this system are highly overexpressed in multiple cancers (Figure 1; group 6) and confer dismal prognosis in lung and breast cancers (Table 1 and Figure S1-S3). TXN is a small protein that reduces oxidized proteins and supports peroxiredoxin (PRDX)-mediated H2O2 clearance (Figure 3) [21]. It also positively regulates the activity of PTP1B, the phosphatase of the tyrosine kinase PDGF-beta, leading to increased PDGF-beta signaling [20] and it negatively regulates the tumor suppressor PTEN [22, 23]. These functions point to an oncogenic role of TXN.
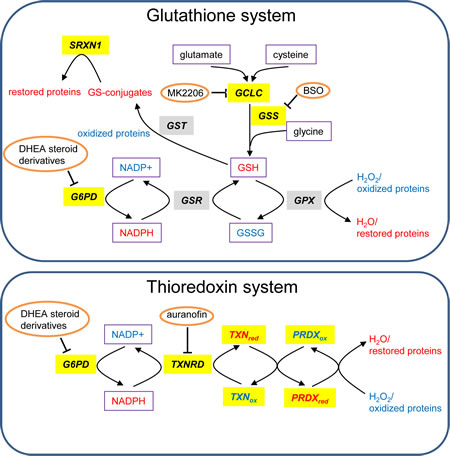
Figure 3: Glutathione and TXN systems. Genes that are highly expressed in tumors versus normal tissues are highlighted in gray and those associated with bad prognosis in lung or breast cancer are highlighted in yellow. The redox state of proteins and metabolites is depicted in color (red=reduced and blue=oxidized). Metabolites are boxed and inhibitors are circled. This scheme is adapted from [137].
In support to this notion, the expression of a number of TXN-related genes has been reported to predict negative patient outcome in multiple cancers [24]. Among the TXN-related genes we identified to be up-regulated in cancers (group 6), TXN expression was associated with reduced survival in various cancers, such as gastric, colorectal, non-small cell lung cancers and squamous cell carcinoma [25-27], whereas TXNRD1 expression was correlated with poor survival in breast cancer and squamous cell carcinoma [28, 29]. Furthermore, PRDX1 level was found to predict poor patient survival in non-small cell lung, ovarian, and breast cancers [30-32], and PRDX3 and PRDX4 expression were correlated with poor prognosis in hepatocellular carcinoma and squamous cell carcinoma respectively [33, 34].
Among TXN pathway inhibitors, the TXN inhibitor PX-12 was shown to be well-tolerated in phase I trials [35]. However, in a phase II trial it exhibited limited therapeutic benefits possibly related to its pharmacokinetics [36], which prompted the development of better TNX inhibitors. An alternative strategy to inhibit TXN is to block TXN reductases [37], such as TXNRD1, that reduce and recycle TXN (Figure 3). TXNRD1 is an interesting drug target as its gene was found in our analysis to be up-regulated in cancers compared with normal tissues (Figure 1A; group 6). Moreover its high expression correlates with worse prognosis in both lung and breast cancer (Table 1 and Figure S1-S2). Indeed, its inhibitor Auranofin [38] can induce apoptosis and inhibit cancer cell growth in vitro, and is currently tested in clinical trials for CLL (phase II). Moreover, Auranofin was suggested to be used for treatment of glioblastoma [39], breast [40], lung [41-43], and other cancers [44]. Interestingly, Auranofin is an example of drug repurposing as it is a well-tolerated FDA-approved drug being already used for treatment of rheumatoid arthritis [38].
Glutathione pathway
Glutathione is the most abundant antioxidant in the cell and is involved in resistance of cancer cells to oxidative stress arising from detachment, hypoxia, radio- and chemotherapy [45-52]. GCLC and glutathione synthetase (GSS), whose genes were identified by our analysis to be highly overexpressed in cancers (Figure 2) and to confer bad prognosis in patients (Figure S1), are both essential enzymes catalyzing the synthesis of glutathione from glutamate, cysteine and glycine (Figure 3).
Previous reports have highlighted the clinical relevance for some of the glutathione-related genes we identified to be up-regulated in melanoma only (group 5) and in all cancers (group 6). The importance for glutathione S-transferase pi 1 (GSTP1) expression as a factor of bad prognosis and of poor response to chemotherapy has been reported in head and neck, gastric, colon, breast and ovarian cancers [53-60]. In addition, high GCLC and GCLM levels were associated with poor progression-free survival in diffuse large B-cell lymphoma [61], and glutathione peroxidase (GPX) activity was found to be specifically high in prostate and lung cancers compared to corresponding normal tissues [62, 63].
The glutathione pathway can be inhibited using specific drugs such as buthionine sulfoximine (BSO). The latter is a well-known inhibitor of GCLC [64] and has been shown to have only little adverse effects in humans [65, 66]. However, its efficacy as an anticancer drug is limited possibly due to bypass effects by other detoxification pathways such as the TXN pathway. In line with this notion, it was recently demonstrated that only when both the glutathione and the TXN pathways were inhibited simultaneously, using BSO and Auranofin, respectively, there was significant inhibition of head and neck squamous cell carcinoma growth in vitro and in vivo [67]. The synergistic effects were efficiently blocked by N-acetyl cysteine (NAC), that replenishes glutathione, but not by catalase suggesting that the simultaneous inhibition of TXN and the glutathione pathways rather than redcution of total anti-oxidant cellular capacity is responsible for the growth inhibitory effect [67]. Similarly, it was shown that simultaneous inhibition of TXN and glutathione systems resulted in synergistic killing of lung cancer cells [41]. This was demonstrated using Auranofin and the AKT inhibitor MK2206, whose efficacy depends on the activity of KEAP1. KEAP1 is a known inhibitor of the transcription factor NRF2 that promotes the expression GCLC and other key enzymes in the glutathione synthesis pathway [68-71]. These data once more underscore that there is a synergistic effect caused by simultaneous block of the TXN system and the glutathione pathway. Our finding that genes enriched for both pathways are highly overexpressed in multiple cancers further supports this strategy of inhibiting both pathways simultaneously to achieve effective targeted anti-cancer therapy.
Transcription factors regulating the cancer AOS response genes and their clinical relevance
NRF2
Our first analysis is based on gene expression data that reflects the sum activities of regulators of gene expression including those of transcription factors. We observed that in the genes list that predict poor outcome, 9 are known NRF2 targets (Table 1 and Figure S1-S3). These include genes involved in glutathione and TXN pathways, G6PD that is involved in NADPH generation (Figure 2) and NQO1 and HMOX1 that encode detoxification enzymes [68, 69, 72-79]. Because NRF2 promotes the expression of oxidative stress detoxifying proteins, it is not surprising that NRF2 depletion results in increased tumor formation in mice challenged with carcinogens [80-83]. However, cancer cells also exploit NRF2 to reduce oxidative stress and resist chemotherapy [84-87]. In line with these two seemingly opposing NRF2 functions, recent data provides evidence that NRF2 knockout mice develop more K-RAS induced tumors on the one hand, but these are less aggressive on the other hand [88]. These observations support the concept that cancer cells exploit NRF2 to adapt to oxidative stress and to resist chemotherapy. This concept gained support by identification of somatic mutations in NRF2 itself and in its inhibitor, KEAP1, that lead to increased NRF2 activity in tumors (reviewed [13, 84, 87, 89, 90]). It is therefore an attractive strategy to block NRF2 in order to reduce the expression of its downstream target genes that are involved in both the glutathione and TXN pathways. Interestingly, the natural compound Brustatol was recently found to inhibit NRF2 in cells and to promote tumor sensitization to chemotherapy in vivo [91], suggesting that NRF2 is druggable and that using an NRF2 antagonist may be a feasible therapeutic strategy.
FOXM1
Another transcription factor we found to be deregulated in multiple cancers is FOXM1, an oncogenic protein known to control proliferation, DNA damage repair, angiogenesis, and AOS response [92, 93]. Indeed, our analysis showed that FOXM1 is highly expressed in multiple cancers (Figure 1) and associated with bad prognosis in breast cancer (Table 1 and Figure S2). These findings further reinforce previous studies reporting highly abnormal expression of FOXM1 in vast number of cancers and its correlation with poor prognosis [92, 94-97].
FOXM1 is known to regulate the expression of important AOS genes including catalase, superoxide dismutase 2 (SOD2) and PRDX3 [97, 98] which we found to be highly overexpressed in multiple cancers (group 6) (Table S1), at the exception of catalase, exclusively overexpressed in hepatocellular carcinoma (group 3) (Table S1). Like NRF2 [99], FOXM1 is induced by active RAS [97] and required for mutant RAS-mediated invasion, anchorage independent growth [100], and development of lung abnormalities in vivo [101].
FOXM1 can be inhibited by classic proteasome inhibitors [96, 102, 103], by piperlongumine that acts as a proteasome inhibitor [104] and promotes autophagic cell death [11], by a peptide derived from ARF [105] and by the CDK4/6 inhibitor PD0332991 [106]. Interestingly, PD0332991 is currently tested in clinical trials (phase II) in breast cancer patients emphasizing the importance of FOXM1 in breast cancer (for review see [95]). Because proteasome inhibitors are already used in the clinic to treat multiple myeloma [107, 108], it is possible that these inhibitors might prove being beneficial in breast cancers patients, whose tumors highly express FOXM1. Consistently, several ROS inducers effectively killed breast cancer cells when combined with proteasome inhibitors or siRNA-mediated knockdown of FOXM1 [103].
NF-kappaB
Our analysis revealed that among the cancer oxidative stress response genes identified (group 6), a number of them are NF-kappaB targets (Table 1). NF-kappaB is essential for proliferation, cell adhesion, inflammatory response and AOS response [109, 110], and its activity is deregulated in cancers [111]. Interestingly, a number of oxidative stress response genes are transcriptionally controlled by NF-kappaB including SOD1, SOD2, GPX1, GSTP1 and the NRF2 targets GCLC, GCLM, NQO1 and HO-1 [112, 113]. This transcriptional regulation forms the basis for the protective role of NF-kappaB under oxidative stress [112]. This is especially relevant in the tumor context as we found that a number of these NF-kappaB targets are highly upregulated in multiple cancers (group 6, Table 1), supporting the notion of an elevated NF-kappaB activity in cancers as a strategy to manage oxidative stress conditions.
Several targeting approaches are being developed to inhibit NF-kappaB activity in cancers. The current strategy is to block NF-kappaB to sensitize tumors to chemotherapy and radiotherapy, since previous reports showed that inhibiting NF-kappaB leads to radiosensitization in radioresistant cancer cells [114, 115]. This is in agreement with the capacity of NF-kappaB to support an antioxidant program of which tumor cells may take advantage to resist oxidative stress-inducing therapies [116]. Thus, few natural compounds, such as curcumin, resveratrol and genistein, have been shown to inhibit NF-kappaB and to enhance the response to chemotherapeutic agents (for review[117]). A specific inhibitor of NF-kappaB nuclear translocation, namely dehydroxy-methylepoxy-quinomicin (DHMEQ), was shown to increase antitumor activities of taxane in a mouse model of thyroid cancer [118]. In addition, NF-kappaB activity can be blocked by direct inhibition of its upstream activator IKK, and the IKK inhibitor Bay 11-7082 leads to enhanced efficacy of cisplatin or paclitaxel in an ovarian tumor model [119, 120].
The cancer oxidative stress response metabolic program: NADPH is a key factor
Our analysis showed that the TXN and glutathione pathways are up-regulated in multiple cancers at the transcriptional level and that high expression of a significant number of these genes is correlated with poor survival. Because both of these pathways rely on NADPH (Figure 3) it raises the possibility that cancer cells will be highly sensitive to NADPH depletion. Indeed, it was demonstrated that the survival of cancer cells requires activation of the AMPK pathway to maintain NADPH levels under metabolic stress, which is usually encountered within solid tumors [7, 121, 122]. Similarly, it was demonstrated that survival of cells under detachment conditions, a hallmark of transformation, is dependent on the pentose phosphate pathway that generates NADPH [10, 123]. Moreover, NRF2 was shown to promote cancer cell proliferation by increasing NADPH generation through transcriptional up-regulation of a number of enzyme-encoding genes including G6PD [74] (Figure 3). Another study showed that TAp73, a transcription factor that is a member of the p53 family [124-128], facilitates the growth of transformed and of cancer cells in vitro and in vivo by up regulating the expression of G6PD and therefore NADPH levels [129].
In our analysis, G6PD was found to be up-regulated in cancer (Figure 1) and bad prognosis in breast cancer (Table 1 and Figure S3). As G6PD fuels the TXN and glutathione pathways with NADPH (Figure 3), we speculate that G6PD might represent a highly attractive novel drug target. It is therefore encouraging that compounds that inhibit G6PD in vitro were synthesized recently [130]. However, more work is needed in order to find lead G6PD inhibitors as candidate anti-cancer drugs.
Conclusions and future perspectives
The concept that the AOS response is utilized by cancer cells to promote their proliferation, adaptation, and resistance is now widely accepted by the scientific community and, therefore, numerous attempts to target AOS response genes as a therapeutic approach have been reported [10, 116, 131]. However, targeting endogenous proteins raises the concern of adverse off-target effects. Thus it is required to determine which proteins play a critical role in cancer as compared with normal tissues, as these are expected to offer a sufficient therapeutic window for intervention. Owing to the increasing availability of patient-derived gene expression, mutation, epigenetic, and survival data, it is now possible to use bioinformatics tools to screen for such targets in large cohorts for individual cancer entities as well as across histological entities [132].
Here, we used publicly available patient-derived gene expression and survival data, and identified genes that belong to two major detoxification pathways. Specifically, we show that genes belonging to the glutathione and TXN pathways are highly overexpressed in multiple cancers versus normal tissues and demonstrate that their high expression correlates with worse patient survival, pointing to a possible role of these genes as drug targets. Moreover, transcription factors such as NRF2, FOXM1, and NF-kappaB as well as key metabolic enzymes such as G6PD that altogether drive the activity of these pathways, were identified in our analysis providing further support to the argument that these are important drug targets. Because the TXN and glutathione pathways are hyperactive in multiple cancers, we hypothesize that simultaneous inhibition of both pathways via targeting common regulators such as NRF2 or common metabolic requirements such as NADPH, may be highly efficient and should be prioritized in drug development.
ACKNOWLEDGMENTS
This work has been supported by the Medical Research Council, United Kingdom and the German Research Foundation (DFG GR3728/2-1 to TG).
REFERENCES
1. Cairns RA, Harris IS and Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011; 11(2):85-95.
2. Leprivier G, Remke M, Rotblat B, Dubuc A, Mateo AR, Kool M, Agnihotri S, El-Naggar A, Yu B, Somasekharan SP, Faubert B, Bridon G, Tognon CE, Mathers J, Thomas R, Li A, et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell. 2013; 153(5):1064-1079.
3. Ward PS and Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012; 21(3):297-308.
4. Grunewald TG, Herbst SM, Heinze J and Burdach S. Understanding tumor heterogeneity as functional compartments--superorganisms revisited. Journal of translational medicine. 2011; 9:79.
5. Venere M, Hamerlik P, Wu Q, Rasmussen RD, Song LA, Vasanji A, Tenley N, Flavahan WA, Hjelmeland AB, Bartek J and Rich JN. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 2013.
6. Matassa DS, Amoroso MR, Agliarulo I, Maddalena F, Sisinni L, Paladino S, Romano S, Romano MF, Sagar V, Loreni F, Landriscina M and Esposito F. Translational control in the stress adaptive response of cancer cells: a novel role for the heat shock protein TRAP1. Cell death & disease. 2013; 4:e851.
7. Jeon SM, Chandel NS and Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012; 485(7400):661-665.
8. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, Tsuchihara K, Bungard D, Berger SL, Jones RG, Mak TW and Zaugg K. Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type I tumor model. Cell Death Differ. 2013; 20(4):659-668.
9. Trachootham D, Alexandre J and Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nature reviews Drug discovery. 2009; 8(7):579-591.
10. Schweikert EM, Devarajan A, Witte I, Wilgenbus P, Amort J, Forstermann U, Shabazian A, Grijalva V, Shih DM, Farias-Eisner R, Teiber JF, Reddy ST and Horke S. PON3 is upregulated in cancer tissues and protects against mitochondrial superoxide-mediated cell death. Cell death and differentiation. 2012; 19(9):1549-1560.
11. Wang Y, Wang JW, Xiao X, Shan Y, Xue B, Jiang G, He Q, Chen J, Xu HG, Zhao RX, Werle KD, Cui R, Liang J, Li YL and Xu ZX. Piperlongumine induces autophagy by targeting p38 signaling. Cell Death Dis. 2013; 4:e824.
12. Gorrini C, Harris IS and Mak TW. Modulation of oxidative stress as an anticancer strategy. Nature reviews Drug discovery. 2013; 12(12):931-947
13. Shelton P and Jaiswal AK. The transcription factor NF-E2-related factor 2 (Nrf2): a protooncogene? Faseb J. 2013; 27(2):414-423.
14. Joo MS, Lee CG, Koo JH and Kim SG. miR-125b transcriptionally increased by Nrf2 inhibits AhR repressor, which protects kidney from cisplatin-induced injury. Cell death & disease. 2013; 4:e899.
15. Shah NM, Rushworth SA, Murray MY, Bowles KM and MacEwan DJ. Understanding the role of NRF2-regulated miRNAs in human malignancies. Oncotarget. 2013; 4(8):1130-1142.
16. Antonov AV, Schmidt T, Wang Y and Mewes HW. ProfCom: a web tool for profiling the complex functionality of gene groups identified from high-throughput data. Nucleic Acids Res. 2008; 36(Web Server issue):W347-351.
17. Serrano M and Blasco MA. Cancer and ageing: convergent and divergent mechanisms. Nature reviews Molecular cell biology. 2007; 8(9):715-722.
18. Antonov AV, Schmidt EE, Dietmann S, Krestyaninova M and Hermjakob H. R spider: a network-based analysis of gene lists by combining signaling and metabolic pathways from Reactome and KEGG databases. Nucleic Acids Res. 2010; 38(Web Server issue):W78-83.
19. Foller M, Harris IS, Elia A, John R, Lang F, Kavanagh TJ and Mak TW. Functional significance of glutamate-cysteine ligase modifier for erythrocyte survival in vitro and in vivo. Cell death and differentiation. 2013; 20(10):1350-1358.
20. Antonov AV. BioProfiling.de: analytical web portal for high-throughput cell biology. Nucleic Acids Res. 2011; 39(Web Server issue):W323-327.
21. Dalleau S, Baradat M, Gueraud F and Huc L. Cell death and diseases related to oxidative stress:4-hydroxynonenal (HNE) in the balance. Cell death and differentiation. 2013; 20(12):1615-1630.
22. Song Z, Saghafi N, Gokhale V, Brabant M and Meuillet EJ. Regulation of the activity of the tumor suppressor PTEN by thioredoxin in Drosophila melanogaster. Experimental cell research. 2007; 313(6):1161-1171.
23. Bassi C and Stambolic V. PTEN, here, there, everywhere. Cell death and differentiation. 2013; 20(12):1595-1596.
24. Li C, Thompson MA, Tamayo AT, Zuo Z, Lee J, Vega F, Ford RJ and Pham LV. Over-expression of Thioredoxin-1 mediates growth, survival, and chemoresistance and is a druggable target in diffuse large B-cell lymphoma. Oncotarget. 2012; 3(3):314-326.
25. Lim JY, Yoon SO, Hong SW, Kim JW, Choi SH and Cho JY. Thioredoxin and thioredoxin-interacting protein as prognostic markers for gastric cancer recurrence. World journal of gastroenterology : WJG. 2012; 18(39):5581-5588.
26. Noike T, Miwa S, Soeda J, Kobayashi A and Miyagawa S. Increased expression of thioredoxin-1, vascular endothelial growth factor, and redox factor-1 is associated with poor prognosis in patients with liver metastasis from colorectal cancer. Hum Pathol. 2008; 39(2):201-208.
27. Kakolyris S, Giatromanolaki A, Koukourakis M, Powis G, Souglakos J, Sivridis E, Georgoulias V, Gatter KC and Harris AL. Thioredoxin expression is associated with lymph node status and prognosis in early operable non-small cell lung cancer. Clin Cancer Res. 2001; 7(10):3087-3091.
28. Cadenas C, Franckenstein D, Schmidt M, Gehrmann M, Hermes M, Geppert B, Schormann W, Maccoux LJ, Schug M, Schumann A, Wilhelm C, Freis E, Ickstadt K, Rahnenfuhrer J, Baumbach JI, Sickmann A, et al. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast cancer research : BCR. 2010; 12(3):R44.
29. Zhu X, Huang C and Peng B. Overexpression of thioredoxin system proteins predicts poor prognosis in patients with squamous cell carcinoma of the tongue. Oral oncology. 2011; 47(7):609-614.
30. Kim JH, Bogner PN, Ramnath N, Park Y, Yu J and Park YM. Elevated peroxiredoxin 1, but not NF-E2-related factor 2, is an independent prognostic factor for disease recurrence and reduced survival in stage I non-small cell lung cancer. Clin Cancer Res. 2007; 13(13):3875-3882.
31. Chung KH, Lee DH, Kim Y, Kim TH, Huh JH, Chung SG, Lee S, Lee C, Ko JJ and An HJ. Proteomic identification of overexpressed PRDX 1 and its clinical implications in ovarian carcinoma. Journal of proteome research. 2010; 9(1):451-457.
32. Woolston CM, Storr SJ, Ellis IO, Morgan DA and Martin SG. Expression of thioredoxin system and related peroxiredoxin proteins is associated with clinical outcome in radiotherapy treated early stage breast cancer. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2011; 100(2):308-313.
33. Qiao B, Wang J, Xie J, Niu Y, Ye S, Wan Q and Ye Q. Detection and identification of peroxiredoxin 3 as a biomarker in hepatocellular carcinoma by a proteomic approach. International journal of molecular medicine. 2012; 29(5):832-840.
34. Chang KP, Yu JS, Chien KY, Lee CW, Liang Y, Liao CT, Yen TC, Lee LY, Huang LL, Liu SC, Chang YS and Chi LM. Identification of PRDX4 and P4HA2 as metastasis-associated proteins in oral cavity squamous cell carcinoma by comparative tissue proteomics of microdissected specimens using iTRAQ technology. Journal of proteome research. 2011; 10(11):4935-4947.
35. Ramanathan RK, Stephenson JJ, Weiss GJ, Pestano LA, Lowe A, Hiscox A, Leos RA, Martin JC, Kirkpatrick L and Richards DA. A phase I trial of PX-12, a small-molecule inhibitor of thioredoxin-1, administered as a 72-hour infusion every 21 days in patients with advanced cancers refractory to standard therapy. Investigational new drugs. 2012; 30(4):1591-1596.
36. Baker AF, Adab KN, Raghunand N, Chow HH, Stratton SP, Squire SW, Boice M, Pestano LA, Kirkpatrick DL and Dragovich T. A phase IB trial of 24-hour intravenous PX-12, a thioredoxin-1 inhibitor, in patients with advanced gastrointestinal cancers. Investigational new drugs. 2013; 31(3):631-641.
37. Peng X, Zhang MQ, Conserva F, Hosny G, Selivanova G, Bykov VJ, Arner ES and Wiman KG. APR-246/PRIMA-1(MET) inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell death & disease. 2013; 4:e881.
38. Gromer S, Arscott LD, Williams CH, Jr., Schirmer RH and Becker K. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J Biol Chem. 1998; 273(32):20096-20101.
39. Kast RE, Boockvar JA, Bruning A, Cappello F, Chang WW, Cvek B, Dou QP, Duenas-Gonzalez A, Efferth T, Focosi D, Ghaffari SH, Karpel-Massler G, Ketola K, Khoshnevisan A, Keizman D, Magne N, et al. A conceptually new treatment approach for relapsed glioblastoma: coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget. 2013; 4(4):502-530.
40. Canas A, Lopez-Sanchez LM, Valverde-Estepa A, Hernandez V, Fuentes E, Munoz-Castaneda JR, Lopez-Pedrera C, De La Haba-Rodriguez JR, Aranda E and Rodriguez-Ariza A. Maintenance of S-nitrosothiol homeostasis plays an important role in growth suppression of estrogen receptor-positive breast tumors. Breast cancer research : BCR. 2012; 14(6):R153.
41. Dai B, Yoo SY, Bartholomeusz G, Graham RA, Majidi M, Yan S, Meng J, Ji L, Coombes K, Minna JD, Fang B and Roth JA. KEAP1-dependent synthetic lethality induced by AKT and TXNRD1 inhibitors in lung cancer. Cancer Res. 2013; 73(17):5532-5543.
42. Chandler JD, Nichols DP, Nick JA, Hondal RJ and Day BJ. Selective metabolism of hypothiocyanous acid by mammalian thioredoxin reductase promotes lung innate immunity and antioxidant defense. J Biol Chem. 2013; 288(25):18421-18428.
43. Fath MA, Ahmad IM, Smith CJ, Spence J and Spitz DR. Enhancement of carboplatin-mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. Clin Cancer Res. 2011; 17(19):6206-6217.
44. Fricker SP. Strategies for the biological evaluation of gold anticancer agents. Anti-cancer agents in medicinal chemistry. 2011; 11(10):940-952.
45. Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, Marinari UM and Domenicotti C. Role of glutathione in cancer progression and chemoresistance. Oxidative medicine and cellular longevity. 2013; 2013:972913.
46. Calvert P, Yao KS, Hamilton TC and O’Dwyer PJ. Clinical studies of reversal of drug resistance based on glutathione. Chem Biol Interact. 1998; 111-112:213-224.
47. Estrela JM, Ortega A and Obrador E. Glutathione in cancer biology and therapy. Critical reviews in clinical laboratory sciences. 2006; 43(2):143-181.
48. Sceneay J, Liu MC, Chen A, Wong CS, Bowtell DD and Moller A. The antioxidant N-acetylcysteine prevents HIF-1 stabilization under hypoxia in vitro but does not affect tumorigenesis in multiple breast cancer models in vivo. PLoS One. 2013; 8(6):e66388.
49. Jardim BV, Moschetta MG, Leonel C, Gelaleti GB, Regiani VR, Ferreira LC, Lopes JR and de Campos Zuccari DA. Glutathione and glutathione peroxidase expression in breast cancer: An immunohistochemical and molecular study. Oncology reports. 2013; 30(3):1119-28. doi: 10.3892/or.2013.2540.
50. Tew KD. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994; 54(16):4313-4320.
51. Boivin A, Hanot M, Malesys C, Maalouf M, Rousson R, Rodriguez-Lafrasse C and Ardail D. Transient alteration of cellular redox buffering before irradiation triggers apoptosis in head and neck carcinoma stem and non-stem cells. PLoS One. 2011; 6(1):e14558.
52. Benassi B, Marani M, Loda M and Blandino G. USP2a alters chemotherapeutic response by modulating redox. Cell death & disease. 2013; 4:e812.
53. Shiga H, Heath EI, Rasmussen AA, Trock B, Johnston PG, Forastiere AA, Langmacher M, Baylor A, Lee M and Cullen KJ. Prognostic value of p53, glutathione S-transferase pi, and thymidylate synthase for neoadjuvant cisplatin-based chemotherapy in head and neck cancer. Clin Cancer Res. 1999; 5(12):4097-4104.
54. Schumaker L, Nikitakis N, Goloubeva O, Tan M, Taylor R and Cullen KJ. Elevated expression of glutathione S-transferase pi and p53 confers poor prognosis in head and neck cancer patients treated with chemoradiotherapy but not radiotherapy alone. Clin Cancer Res. 2008; 14(18):5877-5883.
55. Boku N, Chin K, Hosokawa K, Ohtsu A, Tajiri H, Yoshida S, Yamao T, Kondo H, Shirao K, Shimada Y, Saito D, Hasebe T, Mukai K, Seki S, Saito H and Johnston PG. Biological markers as a predictor for response and prognosis of unresectable gastric cancer patients treated with 5-fluorouracil and cis-platinum. Clin Cancer Res. 1998; 4(6):1469-1474.
56. Tan KL, Jankova L, Chan C, Fung CL, Clarke C, Lin BP, Robertson G, Molloy M, Chapuis PH, Bokey L, Dent OF and Clarke SJ. Clinicopathological correlates and prognostic significance of glutathione S-transferase Pi expression in 468 patients after potentially curative resection of node-positive colonic cancer. Histopathology. 2011; 59(6):1057-1070.
57. Jardim BV, Moschetta MG, Gelaleti GB, Leonel C, Regiani VR, de Santi Neto D, Bordin-Junior NA, Perea SA and Zuccari DA. Glutathione transferase pi (GSTpi) expression in breast cancer: an immunohistochemical and molecular study. Acta histochemica. 2012; 114(5):510-517.
58. Huang J, Tan PH, Thiyagarajan J and Bay BH. Prognostic significance of glutathione S-transferase-pi in invasive breast cancer. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2003; 16(6):558-565.
59. Kolwijck E, Zusterzeel PL, Roelofs HM, Hendriks JC, Peters WH and Massuger LF. GSTP1-1 in ovarian cyst fluid and disease outcome of patients with ovarian cancer. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2009; 18(8):2176-2181.
60. Laborde E. Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Cell death and differentiation. 2010; 17(9):1373-1380.
61. Peroja P, Pasanen AK, Haapasaari KM, Jantunen E, Soini Y, Turpeenniemi-Hujanen T, Bloigu R, Lilja L, Kuittinen O and Karihtala P. Oxidative stress and redox state-regulating enzymes have prognostic relevance in diffuse large B-cell lymphoma. Experimental hematology & oncology. 2012; 1(1):2.
62. Jerome-Morais A, Wright ME, Liu R, Yang W, Jackson MI, Combs GF, Jr. and Diamond AM. Inverse association between glutathione peroxidase activity and both selenium-binding protein 1 levels and Gleason score in human prostate tissue. The Prostate. 2012; 72(9):1006-1012.
63. Ho JC, Chan-Yeung M, Ho SP, Mak JC, Ip MS, Ooi GC, Wong MP, Tsang KW and Lam WK. Disturbance of systemic antioxidant profile in nonsmall cell lung carcinoma. The European respiratory journal. 2007; 29(2):273-278.
64. Griffith OW. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J Biol Chem. 1982; 257(22):13704-13712.
65. Bailey HH, Mulcahy RT, Tutsch KD, Arzoomanian RZ, Alberti D, Tombes MB, Wilding G, Pomplun M and Spriggs DR. Phase I clinical trial of intravenous L-buthionine sulfoximine and melphalan: an attempt at modulation of glutathione. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1994; 12(1):194-205.
66. Bahlis NJ, McCafferty-Grad J, Jordan-McMurry I, Neil J, Reis I, Kharfan-Dabaja M, Eckman J, Goodman M, Fernandez HF, Boise LH and Lee KP. Feasibility and correlates of arsenic trioxide combined with ascorbic acid-mediated depletion of intracellular glutathione for the treatment of relapsed/refractory multiple myeloma. Clin Cancer Res. 2002; 8(12):3658-3668.
67. Sobhakumari A, Love-Homan L, Fletcher EV, Martin SM, Parsons AD, Spitz DR, Knudson CM and Simons AL. Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS One. 2012; 7(10):e48175.
68. Moinova HR and Mulcahy RT. Up-regulation of the human gamma-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochem Biophys Res Commun. 1999; 261(3):661-668.
69. Wild AC, Moinova HR and Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem. 1999; 274(47):33627-33636.
70. Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S and Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. The Journal of biological chemistry. 2000; 275(21):16023-16029.
71. Chan JY and Kwong M. Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim Biophys Acta. 2000; 1517(1):19-26.
72. Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M and Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997; 236(2):313-322.
73. Kensler TW, Wakabayashi N and Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annual review of pharmacology and toxicology. 2007; 47:89-116.
74. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M and Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012; 22(1):66-79.
75. Lim JH, Kim KM, Kim SW, Hwang O and Choi HJ. Bromocriptine activates NQO1 via Nrf2-PI3K/Akt signaling: novel cytoprotective mechanism against oxidative damage. Pharmacological research : the official journal of the Italian Pharmacological Society. 2008; 57(5):325-331.
76. Alam J, Stewart D, Touchard C, Boinapally S, Choi AM and Cook JL. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. The Journal of biological chemistry. 1999; 274(37):26071-26078.
77. Kim YC, Masutani H, Yamaguchi Y, Itoh K, Yamamoto M and Yodoi J. Hemin-induced activation of the thioredoxin gene by Nrf2. A differential regulation of the antioxidant responsive element by a switch of its binding factors. J Biol Chem. 2001; 276(21):18399-18406.
78. Kim YC, Yamaguchi Y, Kondo N, Masutani H and Yodoi J. Thioredoxin-dependent redox regulation of the antioxidant responsive element (ARE) in electrophile response. Oncogene. 2003; 22(12):1860-1865.
79. Heasman SA, Zaitseva L, Bowles KM, Rushworth SA and Macewan DJ. Protection of acute myeloid leukaemia cells from apoptosis induced by front-line chemotherapeutics is mediated by haem oxygenase-1. Oncotarget. 2011; 2(9):658-668.
80. Ramos-Gomez M, Dolan PM, Itoh K, Yamamoto M and Kensler TW. Interactive effects of nrf2 genotype and oltipraz on benzo[a]pyrene-DNA adducts and tumor yield in mice. Carcinogenesis. 2003; 24(3):461-467.
81. Fahey JW, Haristoy X, Dolan PM, Kensler TW, Scholtus I, Stephenson KK, Talalay P and Lozniewski A. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99(11):7610-7615.
82. Jeong WS, Jun M and Kong AN. Nrf2: a potential molecular target for cancer chemoprevention by natural compounds. Antioxidants & redox signaling. 2006; 8(1-2):99-106.
83. Baird L and Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Archives of toxicology. 2011; 85(4):241-272.
84. Hayes JD and McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends in biochemical sciences. 2009; 34(4):176-188.
85. Hayes JD, McMahon M, Chowdhry S and Dinkova-Kostova AT. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal. 2010; 13(11):1713-1748.
86. Lau A, Villeneuve NF, Sun Z, Wong PK and Zhang DD. Dual roles of Nrf2 in cancer. Pharmacological research : the official journal of the Italian Pharmacological Society. 2008; 58(5-6):262-270.
87. Rotblat B, Melino G and Knight RA. NRF2 and p53: Januses in cancer? Oncotarget. 2012; 3(11):1272-1283.
88. Satoh H, Moriguchi T, Takai J, Ebina M and Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013; 73(13):4158-4168.
89. Taguchi K, Motohashi H and Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes to cells : devoted to molecular & cellular mechanisms. 2011; 16(2):123-140.
90. Kansanen E, Kuosmanen SM, Leinonen H and Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox biology. 2013; 1(1):45-49.
91. Ren D, Villeneuve NF, Jiang T, Wu T, Lau A, Toppin HA and Zhang DD. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108(4):1433-1438.
92. Koo CY, Muir KW and Lam EW. FOXM1: From cancer initiation to progression and treatment. Biochim Biophys Acta. 2012; 1819(1):28-37.
93. De Craene B, Denecker G, Vermassen P, Taminau J, Mauch C, Derore A, Jonkers J, Fuchs E and Berx G. Epidermal Snail expression drives skin cancer initiation and progression through enhanced cytoprotection, epidermal stem/progenitor cell expansion and enhanced metastatic potential. Cell death and differentiation. 2013; doi: 10.1038/cdd.2013.148.
94. Wang M and Gartel AL. The suppression of FOXM1 and its targets in breast cancer xenograft tumors by siRNA. Oncotarget. 2011; 2(12):1218-1226.
95. Halasi M and Gartel AL. Targeting FOXM1 in cancer. Biochemical pharmacology. 2013; 85(5):644-652.
96. Gartel AL. The oncogenic transcription factor FOXM1 and anticancer therapy. Cell Cycle. 2012; 11(18):3341-3342.
97. Park HJ, Carr JR, Wang Z, Nogueira V, Hay N, Tyner AL, Lau LF, Costa RH and Raychaudhuri P. FoxM1, a critical regulator of oxidative stress during oncogenesis. Embo J. 2009; 28(19):2908-2918.
98. Kamarajugadda S, Cai Q, Chen H, Nayak S, Zhu J, He M, Jin Y, Zhang Y, Ai L, Martin SS, Tan M and Lu J. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis. 2013; 4:e504.
99. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011; 475(7354):106-109.
100. Behren A, Muhlen S, Acuna Sanhueza GA, Schwager C, Plinkert PK, Huber PE, Abdollahi A and Simon C. Phenotype-assisted transcriptome analysis identifies FOXM1 downstream from Ras-MKK3-p38 to regulate in vitro cellular invasion. Oncogene. 2010; 29(10):1519-1530.
101. Wang IC, Snyder J, Zhang Y, Lander J, Nakafuku Y, Lin J, Chen G, Kalin TV, Whitsett JA and Kalinichenko VV. Foxm1 mediates cross talk between Kras/mitogen-activated protein kinase and canonical Wnt pathways during development of respiratory epithelium. Mol Cell Biol. 2012; 32(19):3838-3850.
102. Bhat UG, Halasi M and Gartel AL. FoxM1 is a general target for proteasome inhibitors. PLoS One. 2009; 4(8):e6593.
103. Halasi M, Pandit B, Wang M, Nogueira V, Hay N and Gartel AL. Combination of oxidative stress and FOXM1 inhibitors induces apoptosis in cancer cells and inhibits xenograft tumor growth. The American journal of pathology. 2013; 183(1):257-265.
104. Halasi M, Wang M, Chavan TS, Gaponenko V, Hay N and Gartel AL. ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem J. 2013; 454(2):201-208.
105. Kalinichenko VV, Major ML, Wang X, Petrovic V, Kuechle J, Yoder HM, Dennewitz MB, Shin B, Datta A, Raychaudhuri P and Costa RH. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004; 18(7):830-850.
106. Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, Zhai H, Vidal M, Gygi SP, Braun P and Sicinski P. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011; 20(5):620-634.
107. Kortuem KM and Stewart AK. Carfilzomib. Blood. 2013; 121(6):893-897.
108. Reinstein E and Ciechanover A. Narrative review: protein degradation and human diseases: the ubiquitin connection. Annals of internal medicine. 2006; 145(9):676-684.
109. Perkins ND. The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer. 2012; 12(2):121-132.
110. Chang CP, Su YC, Hu CW and Lei HY. TLR2-dependent selective autophagy regulates NF-kappaB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell death and differentiation. 2013; 20(3):515-523.
111. DiDonato JA, Mercurio F and Karin M. NF-kappaB and the link between inflammation and cancer. Immunological reviews. 2012; 246(1):379-400.
112. Morgan MJ and Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011; 21(1):103-115.
113. Lu SC. Regulation of glutathione synthesis. Molecular aspects of medicine. 2009; 30(1-2):42-59.
114. Lin Y, Bai L, Chen W and Xu S. The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert opinion on therapeutic targets. 2010; 14(1):45-55.
115. Ahmed KM and Li JJ. ATM-NF-kappaB connection as a target for tumor radiosensitization. Current cancer drug targets. 2007; 7(4):335-342.
116. D’Anneo A, Carlisi D, Lauricella M, Puleio R, Martinez R, Di Bella S, Di Marco P, Emanuele S, Di Fiore R, Guercio A, Vento R and Tesoriere G. Parthenolide generates reactive oxygen species and autophagy in MDA-MB231 cells. A soluble parthenolide analogue inhibits tumour growth and metastasis in a xenograft model of breast cancer. Cell death & disease. 2013; 4:e891.
117. Shen HM and Tergaonkar V. NFkappaB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis : an international journal on programmed cell death. 2009; 14(4):348-363.
118. Meng Z, Mitsutake N, Nakashima M, Starenki D, Matsuse M, Takakura S, Namba H, Saenko V, Umezawa K, Ohtsuru A and Yamashita S. Dehydroxymethylepoxyquinomicin, a novel nuclear Factor-kappaB inhibitor, enhances antitumor activity of taxanes in anaplastic thyroid cancer cells. Endocrinology. 2008; 149(11):5357-5365.
119. Mabuchi S, Ohmichi M, Nishio Y, Hayasaka T, Kimura A, Ohta T, Saito M, Kawagoe J, Takahashi K, Yada-Hashimoto N, Sakata M, Motoyama T, Kurachi H, Tasaka K and Murata Y. Inhibition of NFkappaB increases the efficacy of cisplatin in in vitro and in vivo ovarian cancer models. J Biol Chem. 2004; 279(22):23477-23485.
120. Mabuchi S, Ohmichi M, Nishio Y, Hayasaka T, Kimura A, Ohta T, Kawagoe J, Takahashi K, Yada-Hashimoto N, Seino-Noda H, Sakata M, Motoyama T, Kurachi H, Testa JR, Tasaka K and Murata Y. Inhibition of inhibitor of nuclear factor-kappaB phosphorylation increases the efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Clin Cancer Res. 2004; 10(22):7645-7654.
121. Liu PP, Liao J, Tang ZJ, Wu WJ, Yang J, Zeng ZL, Hu Y, Wang P, Ju HQ, Xu RH and Huang P. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell death and differentiation. 2013; doi: 10.1038/cdd.2013.131. Epub 2013 Oct 4.
122. Tedeschi PM, Markert EK, Gounder M, Lin H, Dvorzhinski D, Dolfi SC, Chan LL, Qiu J, Dipaola RS, Hirshfield KM, Boros LG, Bertino JR, Oltvai ZN and Vazquez A. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell death & disease. 2013; 4:e877.
123. Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P and Brugge JS. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009; 461(7260):109-113.
124. Velletri T, Romeo F, Tucci P, Peschiaroli A, Annicchiarico-Petruzzelli M, Niklison-Chirou MV, Amelio I, Knight RA, Mak TW, Melino G and Agostini M. GLS2 is transcriptionally regulated by p73 and contributes to neuronal differentiation. Cell Cycle. 2013; 12(22).
125. Dotsch V, Bernassola F, Coutandin D, Candi E and Melino G. p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol. 2010; 2(9):a004887.
126. Rufini A, Agostini M, Grespi F, Tomasini R, Sayan BS, Niklison-Chirou MV, Conforti F, Velletri T, Mastino A, Mak TW, Melino G and Knight RA. p73 in Cancer. Genes & cancer. 2011; 2(4):491-502.
127. McCubrey JA and Demidenko ZN. Recent discoveries in the cycling, growing and aging of the p53 field. Aging. 2012; 4(12):887-893.
128. Amelio I, Grespi F, Annicchiarico-Petruzzelli M and Melino G. p63 the guardian of human reproduction. Cell Cycle. 2012; 11(24):4545-4551.
129. Du W, Jiang P, Mancuso A, Stonestrom A, Brewer MD, Minn AJ, Mak TW, Wu M and Yang X. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat Cell Biol. 2013; 15(8):991-1000.
130. Hamilton NM, Dawson M, Fairweather EE, Hamilton NS, Hitchin JR, James DI, Jones SD, Jordan AM, Lyons AJ, Small HF, Thomson GJ, Waddell ID and Ogilvie DJ. Novel steroid inhibitors of glucose 6-phosphate dehydrogenase. J Med Chem. 2012; 55(9):4431-4445.
131. Traba J, Del Arco A, Duchen MR, Szabadkai G and Satrustegui J. SCaMC-1 promotes cancer cell survival by desensitizing mitochondrial permeability transition via ATP/ADP-mediated matrix Ca(2+) buffering. Cell death and differentiation. 2012; 19(4):650-660.
132. Willier S, Butt E and Grunewald TG. Lysophosphatidic acid (LPA) signalling in cell migration and cancer invasion: a focussed review and analysis of LPA receptor gene expression on the basis of more than 1700 cancer microarrays. Biology of the cell / under the auspices of the European Cell Biology Organization. 2013; 105(8):317-333.
133. Roth RB, Hevezi P, Lee J, Willhite D, Lechner SM, Foster AC and Zlotnik A. Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics. 2006; 7(2):67-80.
134. Irizarry RA, Hobbs B, Collin F, Beazer‐Barclay YD, Antonellis KJ, Scherf U and Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4(2):249-264.
135. Grunewald TG, Willier S, Janik D, Unland R, Reiss C, da Costa OP, Buch T, Dirksen U, Richter GH, Neff F, Burdach S and Butt E. The Zyxin-related protein thyroid receptor interacting protein 6 (TRIP6) is overexpressed in Ewing’s sarcoma and promotes migration, invasion and cell growth. Biology of the cell / under the auspices of the European Cell Biology Organization. 2013; 105(11):535–547, doi: 10.1111/boc.201300041
136. Miller IV, Raposo G, Welsch U, Prazeres da Costa O, Thiel U, Lebar M, Maurer M, Bender HU, von Luettichau I, Richter GH, Burdach S and Grunewald TG. First identification of Ewing’s sarcoma-derived extracellular vesicles and exploration of their biological and potential diagnostic implications. Biology of the cell / under the auspices of the European Cell Biology Organization. 2013; 105(7):289-303.
137. Bentley AR, Emrani P and Cassano PA. Genetic variation and gene expression in antioxidant related enzymes and risk of COPD: a systematic review. Thorax. 2008; 63(11):956-961.