
Introduction
Angiogenesis refers to the physiological process in which new blood vessels grow from pre-existing vessels [1, 2]. Driven by pro-angiogenic stimuli, pericytes surrounding the normally quiescent endothelial cells (ECs) detach from the basement membrane [3]. Meanwhile, junctions between ECs dilate, enabling the extravasation of plasma proteins which serve as a provisional framework of the extracellular matrix (ECM) [3]. As a result, ECs migrate, proliferate and form tubes which eventually fuse with neighboring vessels [3-5]. Angiogenesis is vital for normal physiological processes of fetal development and wound healing [3, 6, 7]. However, it is also fundamental for solid tumor growth and metastasis [8]. Tumor angiogenesis provides tumors with oxygen (O2) and nutrients, and also aids the disposal of tumor metabolites [7, 9]. Tumor sizes are limited to 2mm in diameter without neovascularization [7]. Moreover, tumor angiogenesis often results in hyperpermeable vasculature with a twisted appearance [10]. Aberrant vasculature in turn aggravates regional hypoxia and acidosis, both of which promote tumor progression [10].
Vascular endothelial growth factor (VEGF) is the predominant mediator of tumor angiogenesis [11]. Malignant cells produce VEGF to promote angiogenesis even before the formation of a visible tumor [8]. Nevertheless, due to the exceptional growth rate of cancerous cells, solid tumors still experience hypoxia despite the already increased vessel formation [8]. Oxygen insufficiency stabilizes hypoxia inducible factor-1α (HIF-1α), a constituent of the transcription factor HIF-1 [11-15]. Activated HIF-1 translocates to the cell nucleus where it binds to promoter regions to promote VEGF expression [16]. VEGF signaling is mediated by VEGF receptor-2 (VEGFR-2) on ECs [11]. Upon binding with VEGF, VEGFR-2 undergoes tyrosine phosphorylation, triggering multiple down-stream signaling events involving activation of mitogen-activated protein kinases (MAPKs) and protein kinase B (PKB)/Akt, which promote EC survival and proliferation [11, 15]. In addition, pro-inflammatory cytokines released by cancerous cells promote EC migration by increasing vascular permeability [3, 17]. Angiogenesis is regulated by a number of stimulators and inhibitors [11]. Inhibitors of angiogenesis have been developed as an attempt to improve cancer treatments [8]. For example, combined therapy with VEGF antagonists and chemotherapy effectively reduced tumor size and invasiveness [10]. On the other hand, environmental chemicals, including toxic metals, may facilitate tumor growth by stimulating angiogenesis [18, 19].
Cadmium (Cd) is a naturally occurring element that can be found both in the atmosphere and in soil [20]. Since the 1940s, Cd has been used widely in industrial processes [20]. Epidemiological evidence suggests that Cd poses a significant health threat because it substantially increases the risk of cancer, renal failure, osteoporosis, and developmental abnormality [20-25]. For non-occupationally exposed populations, Cd exposure normally results from tobacco consumption or ingestion of contaminated substances [21]. Following absorption by either lung or the intestinal epithelium, Cd enters the systemic circulation [26]. Cd exists as a mixture of free cations and metal compounds in blood [26]. Since Cd has a high affinity to thiol groups, plasma proteins containing thiol groups including albumin, metallothionein (MT), and glutathione (GSH) are considered as major carriers of Cd [26, 27]. Both GSH and MT are potent antioxidants, binding of Cd with these proteins neutralizes Cd toxicity [28-30]. Nevertheless, increased level of Cd depletes anti-oxidative enzymes, and thus induces oxidative stress which affects the surrounding cells [26, 31]. Oxidative stress associated with sub-apoptotic dose of Cd activates pro-survival signaling, leading to enhanced cell proliferation and malignant transformation [9, 22, 23, 32-34]. As a result, Cd has been characterized as a Group 1 human carcinogen by the International Agency for Research on Cancer [21].
As Cd directly alters signaling cascades in both tumor cells and ECs, it has been linked with tumor angiogenesis [1, 9, 21, 22, 35-37]. Exposure to Cd increases the production of VEGF by cancerous cells [9, 35]. Cd also directly enhances EC survival and proliferation by up-regulating the expression of VEGFR-2 [38]. Therefore, Cd-induced tumor angiogenesis contributes, at least in part, to the association between high Cd intake and increased cancer mortality [7, 21]. However, Cd has also been described as an inhibitor of angiogenesis [38-45]. The apparent discrepancy between studies calls for an in-depth review regarding the effect of Cd on tumor angiogenesis. This review will provide a detailed analysis of the interactions between Cd and tumor vasculature, and discuss potential mechanisms underlying the dose dependent effect of Cd.
Ultra-low dose Cd attenuates angiogenesis by inhibiting eNOS activity
Blood Cd concentration serves as a biomarker for Cd exposure level [21]. Data from two Swedish studies indicated that blood Cd concentration in non-occupationally exposed population may range from just above 0 μM to 0.05 μM [21, 46, 47]. Nonetheless, the blood Cd concentration of human varies remarkably subject to age, gender, diet, residential area, and smoking status [21, 47].
Cd in ultra-low concentrations ( < 0.5 μM) attenuates angiogenesis in both the wound healing assay and the chick choriollantoic membrane (CAM) assay [40]. In addition, ultra-low concentrations of Cd reduce bradykinin (BK), a powerful angiogenic agent, and mediate both tube formation in 3D matrigel matrix and ex vivo angiogenesis in CAM models [39]. Mechanisms behind these observations have not been fully understood, but such anti-angiogenic effects of Cd might be mediated partially by the blockade of eNOS activity [39, 40]. eNOS is an enzyme in ECs that catalyzes nitric oxide (NO) production [48-50]. Canonically, activation of eNOS is achieved by binding of a calcium/CaM complex to the CaM-binding region of eNOS [50-52]. Interaction with heat shock protein 90 (Hsp90), a chaperone protein, causes membrane-associated eNOS to dissociate from caveolin-1 (cav-1) while undergoing phosphorylation [48, 50, 53]. Phosphorylation of eNOS leads to a flux of electrons through its reductase domain and thus facilitates the oxidative reaction in which L-arginine is transformed to L-citrulline and NO [48, 54-57].
Upon treatment with ultra-low dose Cd, phosphorylated eNOS in human umbilical vein endothelial cells (HUVECs) is decreased [39, 40]. The reduction in activated eNOS is accompanied by a decrease in NO production [40]. Hence, Cd might directly inhibit eNOS phosphorylation, leading to reduced eNOS activation [40]. Meanwhile, when ECs are treated with ultra-low dose of Cd, BK-induced perinuclear translocation of eNOS is abolished [39]. BK is able to initiate eNOS phosphorylation [39]. Soluble BK binds to the membrane-bound BK2 receptor and activates phospholipase C-γ (PLC-γ), which up-regulates Ca2+ levels in the cytoplasm [51]. Elevated cytoplasmic Ca2+ levels facilitate the binding between calcium/CaM complex and eNOS [51]. In addition, calcium/CaM complex activates CaM kinase II (CaMKII) which directly phosphorylates eNOS [51, 58]. While membrane association is essential for eNOS activation, restricting eNOS to the caveolae-rich plasmalemma increases the binding between eNOS and cav-1 [48, 59]. Cav-1 binding inhibits the enzymatic activity of eNOS [59]. Therefore, ultra-low dose Cd decreases eNOS signaling via the inhibition of eNOS phosphorylation and perinuclear translocation [39] (Figure 1).
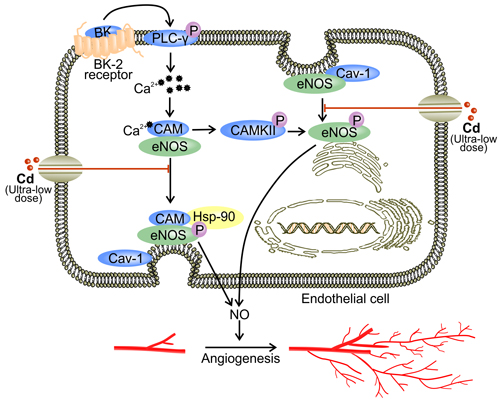
Figure 1: Ultra-low dose of cadmium inhibits angiogenesis by down-regulating eNOS activity. At a concentration lower than 0.5 μM, Cd down-regulates BK-induced eNOS activation [39]. Binding between BK and BK2 receptor initiates down-stream signaling of PLC-γ, which involves the up-regulation of intracellular Ca2+ levels and activation of CaM [51]. Activated calcium/CaM complex binds to eNOS to trigger its canonical activation involving Hsp90 [48, 50, 53]. In addition, calcium/CaM complex stimulates CaMKII which activates eNOS by direct phosphorylation [51]. Ultra-low dose Cd also impedes eNOS perinuclear translocation [39, 40]. Excessive binding of eNOS to the plasmalemma may lead to cav-1-mediated inhibition of eNOS activity [59].
Cd also appears to compete with Ca2+ for entry into cells [32, 48]. This mechanism potentially explains the decrease in intracellular Ca2+ level in Cd treated ECs [39]. Since Ca2+ is required for eNOS activation, the competition between Cd2+ and Ca2+ for passage through ion channels might be another mechanism underlying Cd-reduced NO production [48] (Figure 1). Furthermore, Cd competes with zinc (Zn) for binding sites on proteins [60]. Since myc-associated zinc-finger protein (MAZ) is a promoter of eNOS, the replacement of Zn by Cd in MAZ might attenuate eNOS activity [48, 60]. By suppressing eNOS activation, ultra-low concentrations of Cd reduce NO production by ECs [39, 40]. NO is responsible for regulating vascular tone, EC proliferation, and angiogenesis [50]. NO signaling is orchestrated via S-nitrosylation which covalently incorporates NO into a thiol group on the target protein [61]. Under normoxic conditions, S-nitrosylation stabilizes HIF-1α and initiates the transcription of VEGF [16, 61, 62]. NO also contributes to the accumulation of HIF-1α by inhibiting protein hydroxylase domain containing protein 2 (PHD 2) [63, 64]. Hence, decreased NO due to exposure to ultra-low dose Cd reduces VEGF expression. In addition, hypoxia facilitates the binding between cytochrome c oxidase and NO [61, 65, 66]. Such binding increases intracellular O2 levels by reducing mitochondrial respiration [66]. Combined with NO insufficiency, PHD is activated and promotes the proteasomal degradation of HIF-1α [65, 66]. Therefore, reduced NO level as a result of ultra-low dose Cd exposure leads to decreased VEGF production and impaired angiogenesis [61, 65].
Low-dose Cd stimulates angiogenesis by up-regulating VEGF expression
Low-dose Cd (1 μM-10 μM) promotes tumor angiogenesis in a series of experimental models [9, 35, 38-40, 67]. At this level of exposure, Cd does not induce death of vascular cells [68, 69], however, it might alter the phenotype of both non-endothelial cells and ECs [35, 70]. Low-dose Cd induces oxidative stress, which is characterized by elevated intracellular level of reactive oxygen species (ROS) [9, 29]. By binding to GSH and MT, Cd2+ impairs the ability of cells to dispose ROS [32]. In addition, free Cd2+ increases ROS formation by damaging the mitochondria and activating NADPH oxidase [32]. Furthermore, Cd may indirectly up-regulate ROS by displacing endogenous Fenton metals, such as Fe2+, from proteins [32]. By increasing ROS generation, Cd activates PKB/Akt, NF-κB, and MAPKs, resulting in endothelial cell activation and tumor angiogenesis [9, 38, 67, 71].
PKB/Akt signaling
PKB, also known as Akt, is a serine/threonine-specific protein kinase invovling in the transcriptional induction of protein expression [72, 73]. PKB/Akt is activated by phosphatidylinositol-4,5-bisphosphate-3-kinase (PI3K) while it controls many downstream components such as mammalian target of rapamycin (mTOR) [72, 73]. PKB/Akt is a primary target molecule in Cd-induced tumor angiogenesis [9]. In non-ECs, Cd (5 μM) triggers activation of PKB/Akt and its down-stream effectors mTOR and p70S6kinase (p70S6K1) [9], which mediates VEGF production [74]. Activated p70S6K1 phosphorylates eukaryotic translation initiation factor 4B (eIF4B), facilitating the recruitment of eIF4B to the pre-initiation complex of protein synthesis [75]. Subsequently, the unwinding of HIF-1 mRNA is promoted along with the induction of HIF-1 translation [16, 72, 75]. Elevated HIF-1 level leads to increased VEGF transcription [9, 16] (Figure 2). In addition, p70S6K1 phosphorylation is associated with actin filament remodeling which promotes cell migration [76]. Accordingly, Cd-conditioned ECs demonstrate enhanced tube formation in a CAM assay [9]. Both enhanced production of VEGF and augmented EC migration induced by PKB/Akt signaling might contribute to low-dose Cd-induced tumor angiogenesis.
Table 1: Dose dependent effects of Cd on tumor angiogenesis.
Cd conc. |
Mechanism of action |
Effect on angiogenesis |
Ref |
<0.5 μM |
eNOS inhibition |
Inhibitory |
39, 40 |
4 μM |
NF-κB activation |
Stimulatory |
67 |
5 μM |
Akt and ERK activation |
Stimulatory |
9 |
5 & 10 μM |
Activation of ERK, JNK, and p38 MAPK |
Stimulatory |
38 |
>10 μM |
p38 MAPK activation |
Inhibitory |
43 |
50 μM |
Blockage of VEGF signaling and increased Ang-2 expression |
Inhibitory |
41, 42 |
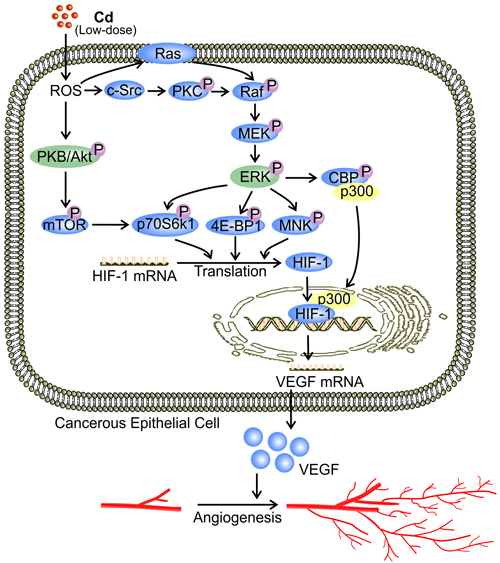
Figure 2: Low-dose Cd induces VEGF expression via activation of Akt and ERK signaling in cancerous cells. At a concentration between 1 μM and 10 μM, Cd activates PKB/Akt and ERK in cancerous cells by increasing ROS levels [9]. Activation of ERK via Raf/MEK is either Ras dependent or through c-Src-activated PKC [95]. PKB/Akt activation phosphorylates down-stream substrates including mTOR and p70S6K1 [9]. Activated ERK phosphorylates 4E-BP1, p70S6K1, and MNK, while triggering dissociation of the CEP/p300 complex, resulting in the binding of p300 with HIF-1 [16, 96]. Together, these signaling events enhance the translation and activity of HIF-1 [16, 72, 96]. Binding between HIF-1 and the promoter region of VEGF up-regulates VEGF production by cancerous cells [16].
NF-κB signaling
NF-κB is a transcription factor consisting different combinations of the Rel-family-members such as RelA (p65) and RelB (p50) [67, 77, 78]. It broadly influences gene expression to regulate cell survival, proliferation, and differentiation [77, 78]. Usually, NF-κB dimers reside in the cytosol and are bound to their inhibitory proteins, inhibitor κB (IκB) [77]. Low-dose Cd (4 μM) activates NF-κB by promoting the degradation of IκBα in human renal glomerular endothelial cells [67]. Activated NF-κB dimers then translocate to the cell nucleus where they bind to the promoter regions of pro-angiogenic proteins [67, 77]. NF-κB activation facilitates the expression of VEGFR-2 by ECs [38, 71, 79] (Figure 3). Being the predominant mediator of VEGF signaling, phosphorylated VEGFR-2 activates down-stream pro-angiogenic and pro-survival signaling including the aforementioned PLC-γ and PI3K [80]. Therefore, activation of NF-κB signaling promotes tumor angiogenesis by enriching VEGFR-2 availability.
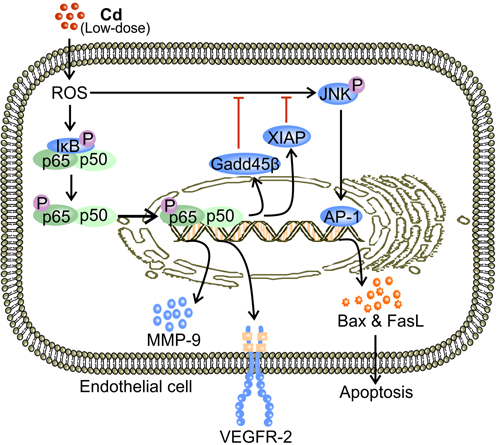
Figure 3: Low-dose Cd activates NF-κB signaling and promotes VEGFR-2 expression while inhibiting JNK-mediated apoptosis in ECs. Low-dose Cd (1-10 μM) induces formation of ROS which phosphorylates IκB [67, 77]. Phosphorylation of IκB releases NF-κB (p65/p50 dimer) which then translocates to the nucleus to promote the transcription of VEGFR-2 [71, 77]. Activation of NF-κB leads to expression of proteins that inhibit JNK, including XIAP and Gadd45β [67, 81, 82]. Therefore, Cd-activated NF-κB inhibits JNK-mediated EC apoptosis while promoting VEGFR-2 expression [67].
X-chromosome linked inhibitor of apoptosis (XIAP) and Gadd45β are products of NF-κB activation induced by low-dose Cd [67, 81, 82]. Both proteins have been independently shown to inhibit c-Jun N-terminal kinase (JNK) activation [81, 82]. Stress activated JNK is associated with nuclear translocation of activator protein-1 (AP-1) [83], which promotes the transcriptional expression of Bax and Fas ligand (FasL) [83, 84]. Bax-facilitated release of cytochrome c and FasL-engaged Fas mediate apoptosis via the intrinsic mitochondrial pathway [84-86]. In the presence of NF-κB inhibitor, low-dose Cd increased JNK phosphorylation, which resulted in a substantial decrease in viable cell count [67, 87]. On the other hand, activated NF-κB reduced the level of phosphorylated JNK and maintained the number of viable ECs [67]. Therefore, by effectively inhibiting JNK-mediated apoptosis, low-dose Cd induced NF-κB activation promotes angiogenesis by maintaining EC survival [67] (Figure 3). In addition, NF-κB activation by exposure to low-dose Cd up-regulates the expression of matrix metalloproteinase-9 (MMP-9) in ECs [88, 89] (Figure 3). MMP-9 promotes the dissociation of EC from the basement membrane, which is essential for the induction of tumor angiogenesis [3, 90]. Genetic ablation of MMP-9 prevents the initiation of tumor angiogenesis in mouse models [91]. The proteolytic activity of MMP-9 also enables it to degrade and remodel the ECM, allowing ECs to migrate [91, 92]. Moreover, ECM degradation by MMP-9 releases VEGF, the potent promoter of tumor angiogenesis [91, 93].
ERK signaling
ERK1/2 belongs to the MAPK family and has a conventional role in pro-survival signaling [94]. Similar to its effect on PKB/Akt, low-dose Cd (5 μM) increased ERK phosphorylation in human lung epithelial cells by inducing ROS formation [9]. ROS is a well-established activator of ERK mediated by the classic Ras-Raf-MEK pathway [95]. Activation of Raf may also be achieved through c-Src-activated protein kinase C (PKC) [95]. Activated ERK phosphorylates eukaryotic translation initiation factor 4E-binding protein (4E-BP1), p70S6K1, and MAP kinase interacting kinase (MNK) [9, 16, 96]. These signaling events result in increased HIF-1α mRNA translation [16]. Consequently, HIF-1α-mediated VEGF production is increased [9]. ERK is also involved in the transcriptional activation of HIF-1 [16]. ERK phosphorylates CBP/p300 and thus increases HIF-1α/p300 complex formation [16]. Enhanced HIF-1α translation and activation eventually up-regulate VEGF expression [9] (Figure 2).
Low-dose Cd also activated ERK signaling in ECs with an increase in VEGFR-2 expression [38]. Meanwhile, inhibitors of ERK signaling reduces the level of VEGFR-2 [38]. Therefore, Cd-activated ERK up-regulates VEGFR-2 in ECs [38]. The resulting enhanced VEGF signaling contributes, at least in part, to the Cd-induced increase in EC proliferation [80, 97]. Specifically, VEGF-A has been found to activate an orphan nuclear receptor transcription factor TR3 in HUVECs [80, 98]. TR3 then mediates the expression of cell cycle genes which promote EC proliferation [80, 98]. By stimulating EC proliferation, Cd-enhanced activation of ERK signaling in ECs facilitates tumor angiogenesis [1].
JNK signaling
JNK is a MAPK that mediates both pro-apoptotic and pro-survival signaling [94, 99, 100]. JNK is phosphorylated under stresses, such as elevated ROS levels [101]. Binding of c-Jun and c-fos (AP-1 complex), the downstream targets of JNK activation, to the promoter regions of DNA initiates transcriptional production of respective genes [83, 84, 102]. Upon exposure to low dose Cd, JNK phosphorylation was increased in a dose dependent manner, peaking at 10 μM, and the expression levels of both VEGF and VEGFR-2 were elevated with an increase in cell viability [38]. Inhibition of JNK activation substantially decreased VEGFR-2 expression in HUVECs [38]. JNK also mediates sustained VEGFR-2 phosphorylation, which is essential for the transduction of VEGF signaling [103] (Figure 4). Furthermore, inhibitor of JNK reduces the release of VEGF by human coronary smooth muscle cells [104]. Hence, JNK activation is essential for VEGF production by non-endothelial cells [104]. Therefore, in addition to maintaining VEGFR-2 phosphorylation, JNK activation by low-dose Cd exposure might promote tumor angiogenesis by up-regulating both VEGF and VEGFR-2 [38, 103, 104].
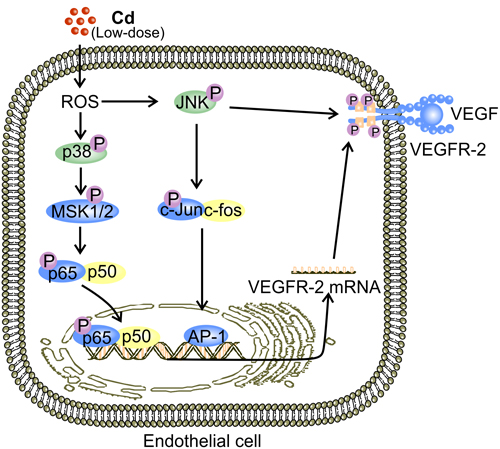
Figure 4: Putative mechanisms for low-dose Cd-activated JNK and p38 MAPK to facilitate expression and activation of VEGFR-2 in ECs. JNK-mediated elevation in VEGFR-2 expression is observed in ECs [38]. Activation of JNK by low-dose (1-10 μM) Cd-induced ROS triggers nuclear translocation of AP-1 [83, 84, 102]. Binding of AP-1 to the relevant promoters initiates transcriptional expression of a plethora of proteins including VEGFR-2 [38, 84]. ROS-induced p38 MAPK activation also accounts for elevated VEGFR-2 expression in ECs exposed to low-dose Cd possibly via MSK1/2 [94]. Eventually, NF-κB activation leads to up-regulation of VEGFR-2 [71].
p38 MAPK signaling
Similar to JNK, p38 MAPK is also a mediator of both pro-survival and pro-apoptotic signaling [105, 106]. Meanwhile, it is required for VEGF-induced EC migration [106]. Inhibition of p38 MAPK signaling also resulted in reduced VEGFR-2 expression in HUVECs [38]. Nonetheless, the pro-angiogenic activity of p38 MAPK remains to be comprehensively characterized. A potential explanation for p38 MAPK-mediated increase in VEGFR-2 expression in ECs is the ability of p38 MAPK to activate NF-kB by activating mitogen- and stress-activated kinases (MSK1/2) [38, 71, 94] (Figure 4). Importantly, it appears that members of the MAPK family, ERK, JNK, and p38 MAPK, produce a plethora of down-stream signaling events under the stimulation of low-dose Cd. The overall effect of these is enhanced VEGF signaling and tumor angiogenesis [38]. However, interactions between these signaling events remain to be fully understood.
High-dose Cd impairs angiogenesis by reducing viable ECs
Consistently through the literature, high dose Cd ( > 10 μM) attenuates angiogenesis via the induction of apoptosis [38, 41-43, 107]. As mentioned previously, both p38 MAPK and JNK mediate stress-induced apoptosis [108]. According to Jung et al., Cd (30 μM) activated all three members of the MAPK family in mouse brain microvascular endothelial cells (bEnd.3) while leading to apoptosis [43]. However, only the inhibition of p38 MAPK results in improved survival, suggesting p38 MAPK is the only active MAPK that mediates high-dose Cd-induced EC apoptosis [43]. Indeed, p38 MAPK activation increases the levels of pro-apoptotic proteins including Bax and Fas [105, 109], and is associated with cleavage of caspase-9 and the subsequent activation of caspase-3 [110-112]. p38 MAPK is also postulated to inhibit the pro-survival ERK signaling [16, 106]. Inhibited ERK activation reduces phosphorylated 4E-BP1, leading to less eIF-4E release and eventually lower rates of protein synthesis, which presumably involves decreased VEGFR-2 expression in ECs [83, 113] (Figure 5). Remarkably, with the supposed inhibition of p38 MAPK, ERK activity following Cd-exposure remains elevated [38]. The eventual apoptosis might indicate that the induced level of ERK activation is insufficient for cells to survive under stress of such intensity [38]. As described previously, Cd-stimulated JNK activation, triggering EC apoptosis [67]. Moreover, it appears that the well-known inhibition of JNK by NF-κB is conserved in Cd-treated ECs [67, 81, 82]. Inhibition of JNK activation by NF-κB might explain the failure of JNK inhibitor to preserve cell viability [43]. Future investigation of the role of NF-κB in ECs treated with high-dose Cd may help explain whether phosphorylated JNK is an essential component of Cd-induced apoptosis.
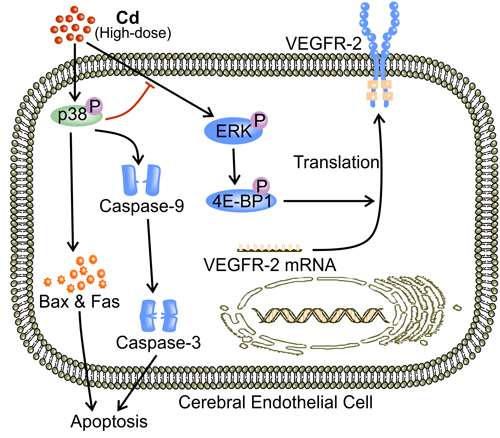
Figure 5: p38 MAPK mediates high-dose Cd induced EC apoptosis. At high concentrations ( > 10 μM), Cd triggers EC apoptosis via the activation of p38 MAPK [43]. Activation of p38 MAPK increases Bax expression and cleavage of caspase-9 [105, 110, 111]. Caspase-9 cleavage activates caspase-3 [112]. Both increased Bax level and caspase-9-activated caspase-3 are prominent mediators of apoptosis [112]. In addition, phosphorylated p38 MAPK inhibits ERK and impairs the pro-survival VEGF signaling [83, 94, 95, 106, 113].
According to Kim et al., phosphorylation of MAPKs in Cd-treated HUVECs were reduced to basal levels when the concentration of Cd exceeded 10 μM but the level of pro-caspase-3 increased with elevation in Cd concentration [38]. Hence, caspase-3 is the major contributor to Cd-related damage in ECs [38, 43]. Although activation of either JNK or p38 seems to be required for apoptosis mediated by caspase-3 [43, 114, 115], the lack of obvious change in levels of these MAPKs suggests that Cd-induced EC apoptosis might involve other activators of caspase-3 [38, 116]. Therefore, regardless of the up-stream signaling cascade involved, high-dose Cd inhibits tumor angiogenesis by inducing caspase-3-mediated apoptosis [38, 43].
With similar Cd concentrations and similar exposure time, Kim et al. observed MAPK activation patterns that were inconsistent with the results of Jung et al. [38, 43]. The only apparent difference between these studies is the variation in EC types [38, 43]. It is well established that ECs with distinct origins exhibit different gene expression patterns, enzymatic activity, and signal transduction [117-119]. In particular, differences in VEGF-induced MAPK activation have long been recognized to depend on the origin of ECs [120]. Treating primary cultures of HUVEC, human aortic EC, and human microvascular EC with the same doses of VEGF for the same time period resulted in differential ERK activation [120]. The cerebral vascular EC used by Jung and colleagues possesses distinct protein expression pattern from peripheral vascular ECs, potentially resulting in disparities in protein activation [48, 121]. Therefore, endothelial heterogeneity may explain the differences in MAPK phosphorylation pattern across these studies.
In addition, Cd in high concentrations disrupts signaling pathways that are important to vascular maintenance and growth [3, 41, 42, 122]. A high concentration of Cd significantly decreases both VEGF and VEGFR-2 expression, and thus impairs VEGF signaling [42]. In addition to promoting tumor angiogenesis, VEGF signaling protects against Cd-induced apoptosis through a number of mechanisms [70, 80]. VEGF up-regulates Bcl-2, which promotes survival by inhibiting caspase activation [42]. VEGF also activates Akt to stimulate production of pro-survival proteins such as survivin [12, 72]. Furthermore, VEGF triggers ERK1/2 activation via MEK1/2 [70, 113, 123]. ERK activation phosphorylates IEX-1 and inhibits stress-induced apoptosis [124, 125] (Figure 6). In addition, high-dose Cd inhibits the activation of VEGFR-2 tyrosine kinase (TK) activity [126] (Figure 6). Cd might chelate to ATP and form Cd-ATP which could compete with Mg-ATP for enzyme activation sites on VEGFR-2 TKs [126]. With the assumption of Cd-ATP being a slow substrate for TK activation, the accumulation of Cd-ATP could inhibit VEGFR-2 phosphorylation [126]. Alternatively, high dose Cd competes with Mg for the putative second metal-binding site on VEGFR-2 [126]. Therefore, high dose Cd inhibits the activation of VEGFR-2, preventing the down-stream pro-survival signaling transduction [126]. Together, these mechanisms aggravate the cytotoxicity of high-dose Cd while attenuating angiogenesis [42]. Furthermore, high-dose Cd (50 μM) increased the level of angiopoetin-2 (Ang-2) while impairing vascular growth in chick embryos [41]. In the absence of VEGF, high concentrations of Ang-2 inhibit Tie-2 signaling as it displaces Ang-1, the more active ligand, from the receptor [41, 127]. Since Tie-2 signaling is required for both vascular maintenance and response to angiogenic stimuli, impairment of Tie-2 signaling by high-dose Cd might attenuate angiogenesis [3, 41, 122, 128].
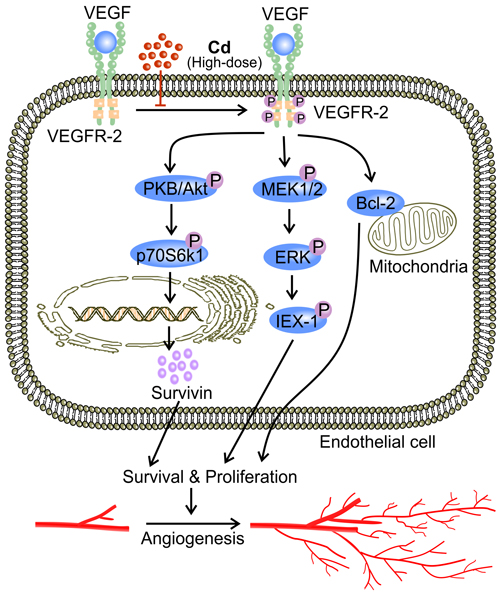
Figure 6: High-dose Cd blocks VEGF signaling and impairs EC survival. Phosphorylation of VEGFR-2 TK as a result of binding between VEGF and VEGFR-2 up-regulates inhibitors of apoptosis including Bcl and survivin [42, 72, 138]. VEGF signaling also phosphorylates ERK1/2 via MEK1/2, activating IEX-1, an inhibitor of stress-induced apoptosis [70, 113, 124, 125]. High concentrations of Cd ( > 10 μM) block VEGFR-2 activation, and impaired VEGF signaling leads to EC apoptosis and ultimately attenuated angiogenesis [42].
Mechanism underlying the dose dependency of Cd actions
The effect of Cd has been characterized as dose dependent both in vitro and in vivo [31, 129-131]. However, the mechanisms underlying such a property remain elusive. Oxidative stress, characterized by elevated level of ROS, is the primary mediator of Cd toxicity [9, 132, 133], but damage caused by low levels of oxidative stress can be neutralized by anti-oxidant enzymes [134]. After exposure to low-dose Cd, expression and activity of antioxidant enzymes including MT, catalase, glutathione S-transferase, glutathione peroxidase, and quinone oxidoreductase were substantially increased along with the cellular level of GSH [32, 134-136]. Sub-apoptotic levels of oxidative stress also trigger adaptive responses in affected cells [32]. Protection by antioxidant enzymes together with the activation of pro-survival signaling contribute to enhanced cell proliferation [137]. Excessive oxidative stress, however, overwhelms the cellular defense mechanisms and initiates apoptosis to dispose of the damaged cells [137]. Prolonged exposure to increased concentrations of Cd induces a decrease in intracellular GSH level despite the antioxidant enzyme activity, resulting in a significant reduction in cell viability [134]. Therefore, variation in the level of oxidative stress by exposure to Cd of different concentrations potentially explains the dose dependent effect of Cd.
The variations of the results might also be caused by different experimental settings. Here we propose an investigation using HUVEC as the only cell type of interest. The effects on cultured HUVECs are to be evaluated after exposure to different concentrations of Cd representing ultra-low dose, low dose, and high dose. Oxidative stress characterized by intracellular levels of ROS and GSH could be examined to validate our hypothesis that oxidative stress associated with various concentrations of Cd is responsible for the dose-dependent effects of Cd on tumor angiogenesis. Notably, the majority of studies concerning the effect of Cd on angiogenesis have been carried out only in vitro. More in vivo studies are needed to thoroughly elucidate the effects of different concentrations of Cd on angiogenesis and relevant signaling cascades.
Conclusions
As a long-recognized carcinogen, Cd affects tumor angiogenesis at all concentrations [9, 22, 38-43]. The effect of Cd, however, appears to be bi-directional, and is determined by Cd concentration [38]. Ultra-low concentrations of Cd ( < 0.5 μM) inhibit angiogenesis by blocking eNOS activation [39, 40, 49]. Subsequently, reduced level of NO is associated with PHD activation and thus the proteosomal degradation of HIF-1 during hypoxia [61, 65, 66]. The decrease in HIF-1 subsequently reduces VEGF production and angiogenesis [3, 16]. Given that hypoxia is commonly experienced by tumor cells, Cd in ultra-low concentration might in fact attenuate tumor angiogenesis [7, 80]. In contrast, low-dose Cd (1 μM-10 μM) acts on both non-endothelial cells and ECs to promote tumor angiogenesis [9, 35, 67]. Low-dose Cd-induced oxidative stress stimulates the release of VEGF by non-endothelial cells while triggering their malignant transformation [9]. It appears that phosphorylated p70S6K1, resulting from activations of PKB/Akt and ERK, is the primary mediator of this process [9]. p70S6K1 activation increases HIF-1 translation [16, 96]. ERK activation also facilitates the composition of the HIF-1/p300 complex and thus the transcriptional activation of HIF-1 [16]. Activated HIF-1 then binds to the promoter region of VEGF to up-regulate VEGF production [16, 96]. In the mean time, ECs exposed to low-dose Cd demonstrate increased expression of VEGFR-2, which is mediated by activations of NF-κB and all three MAPKs [38, 71]. Low-dose Cd activated NF-κB also up-regulates inhibitors of apoptosis to promote cell survival [67, 81, 82]. Thus, low-dose Cd facilitates tumor angiogenesis by both enhancing VEGF signaling and inhibiting EC apoptosis [9, 38, 67]. Finally, high dose Cd ( > 10 μM) inhibits angiogenesis by triggering EC apoptosis and blocking pro-angiogenic signaling pathways including VEGF and Tie-2 [3, 41, 42]. Exposure to high-dose Cd leads to EC apoptosis mediated by activation of caspase-3 [38, 43, 110, 111], but the relevant upstream signaling event differs across EC types [38, 43]. Moreover, high-dose Cd impairs the ability of non-endothelial cells to produce VEGF while inhibiting the activation of VEGFR-2 in ECs [42, 126]. With reduced VEGF availability, an increase in Ang-2 level following Cd exposure blocks Tie-2 signaling, which is essential for vascular growth [41].
Variations in levels of oxidative stress induced by different concentrations of Cd might explain the dose dependent effect of Cd on tumor angiogenesis. Cells are protected from low levels of oxidative stress owing to antioxidant enzyme activities [134, 137]. Sub-apoptotic levels of oxidative stress trigger adaptive responses, promoting cell survival and proliferation [32]. Excessive oxidative stress induced by high-dose Cd initiates apoptosis of ECs [137]. Mechanisms underlying the effects of Cd on tumor angiogenesis still need to be elucidated. Future investigations using in vivo models are needed to further validate current findings.
Abbreviations
4E-BP1, eukaryotic translation initiation factor 4E-binding protein; Ang-2, angiopoetin-2; AP-1, activator protein-1; BK, bradykinin; CAM, chick chorioallantoic membrane; CaM, camodulin; CaMKII, CaM kinase II; cav-1, caveolin-1; Cd, cadmium; EC, endothelial cell; eIF4B, eukaryotic translation initiation factor 4B; eNOS, endothelial nitric oxide synthase; ERK, extracellular signaling related kinase; FasL, Fas ligand; GSH, glutathione; HEEC, human endometrial endothelial cell; HIF, hypoxia inducible factor; Hsp90, heat shock protein 90; HUVEC, human umbilical vein endothelial cell; IκB, inhibitor κB; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MAZ, myc-associated zinc-finger protein; MMP-9, matrix metalloproteinase-9; MNK, MAP kinase interacting kinase; MSK, mitogen- and stress-activated kinases; MT, metallothionein; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor-κB; NO, nitric oxide; O2, oxygen; p70S6K, p70S6kinase; PHD, protein hydroxylase domain containing protein; PI3K, phosphatidylinositol-4,5-bisphosphate-3-kinase; PKB, protein kinase B; PKC, protein kinase C; PLC-γ, phospholipase C-γ; ROS, reactive oxygen species; rp, ribosomal protein; Ser, serine; TK, tyrosine kinase; VEGF, vascular endothelial growth factor; VEGFR-2, vascular endothelial growth factor receptor 2; XIAP, X-chromosome linked inhibitor of apoptosis; Zn, zinc.
Acknowledgments
This study was supported by the grants from Science and Technology Development Plan of Shandong Province (2013GSF11805), The National Natural Science Foundation of China (81370269, 81570255), and Shandong Taishan Scholarship (Ju Liu).
Conflicts of interest
The authors declare no conflict of interest.
References
1. Prozialeck WC, Edwards JR, Woods JM. The vascular endothelium as a target of cadmium toxicity. Life Sciences. 2006; 79:1493-1506.
2. Meissner M, Michailidou D, Stein M, Hrgovic I, Kaufmann R, Gille J. Inhibition of Rac1 GTPase downregulates vascular endothelial growth factor receptor-2 expression by suppressing Sp1-dependent DNA binding in human endothelial cells. Exp Dermatol. 2009; 18:863-869.
3. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011; 473:298-307.
4. Fan TP, Yeh J, Leung KW, Yue PY, Wong RN. Angiogenesis: from plants to blood vessels. Trends Pharmacol Sci. 2006; 27:297-309.
5. Bhat TA, Singh R. Tumor angiogenesis—a potential target in cancer chemoprevention. Food and chemical toxicology. 2008; 46:1334-1345.
6. Sage EH, Vernon RB. Regulation of angiogenesis by extracellular matrix: the growth and the glue. Journal of Hypertension Supplement. 1994; 12:S145-152.
7. Granci V, Dupertuis YM, Pichard C. Angiogenesis as a potential target of pharmaconutrients in cancer therapy. Curr Opin Clin Nutr Metab Care. 2010; 13:417-422.
8. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996; 86:353-364.
9. Jing Y, Liu LZ, Jiang Y, Zhu Y, Guo NL, Barnett J, Rojanasakul Y, Agani F, Jiang BH. Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicological Sciences. 2012; 125:10-19.
10. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011; 91:1071-1121.
11. Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008; 358:2039-2049.
12. Dvorak HS. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002; 20:4368-4380.
13. Blagosklonny MV. Antiangiogenic therapy and tumor progression. Cancer Cell. 2004; 5:13-17.
14. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971; 285:1182-1186.
15. Cook KM, Figg WD. Angiogenesis inhibitors: current strategies and future prospects. CA Cancer J Clin. 2010; 60:222-243.
16. Masoud GN, Li W. HIF-1 pathway: role, regulation and intervention for cancer therapy. Acta Pharmaceutica Sinica B. 2015; 5:378-389.
17. Zhang G, Yang L, Kim GS, Ryan K, Lu S, O’Donnell RK, Spokes K, Shapiro N, Aird WC, Kluk MJ, Yano K, Sanchez T. Critical role of sphingosine-1-phosphate receptor 2 (S1PR2) in acute vascular inflammation. Blood. 2013; 122:443-455.
18. Jarzynka MJ, Guo P, Bar-Joseph I, Hu B, Cheng SY. Estradiol and nicotine exposure enhances A549 bronchioloalveolar carcinoma xenograft growth in mice through the stimulation of angiogenesis. Int J Oncol. 2006; 28:337-344.
19. Prozialeck WC, Edwards JR, Nebert DW, Woods JM, Barchowsky A, Atchison WD. The vascular system as a target of metal toxicity. Toxicological Sciences. 2008; 102:207-218.
20. Waisberg M, Joseph P, Hale B, Beyersmann D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology. 2003; 192:95-117.
21. Jarup L, Akesson A. Current status of cadmium as an environmental health problem. Toxicology and Applied Pharmacology. 2009; 238:201-208.
22. Arroyo VS, Flores KM, Ortiz LB, Gómez-Quiroz LE, Gutiérrez-Ruiz MC. Liver and Cadmium Toxicity. J Drug Metabol Toxicol. 2012. doi: 10.4172/2157-7609.S5-001.
23. Joseph P. Mechanisms of cadmium carcinogenesis. Toxicology and Applied Pharmacology. 2009; 238:272-279.
24. Elsaid AF, Délot EC, Collins MD. Differential perturbation of the Fgf/Erk1/2 and Shh pathways in the C57BL/6N and SWV embryonic limb buds after mid-gestational cadmium chloride administration. Mol Genet Metab. 2007; 92:258-270.
25. Kapron-Bras CM, Hales BF. Heat-shock induced tolerance to the embryotoxic effects of hyperthermia and cadmium in mouse embryos in vitro. Teratology. 1991; 43:83-94.
26. Zalups RK, Ahmad S. Molecular handling of cadmium in transporting epithelia. Toxicology and Applied Pharmacology. 2003; 186:163-188.
27. Helmestam M, Stavreus-Evers A, Olovsson M. Cadmium chloride alters mRNA levels of angiogenesis related genes in primary human endometrial endothelial cells grown in vitro. Reproductive Toxicology. 2010; 30:370-376.
28. Saboli I, Breljak D, Skarica M, Herak-Kramberger CM. Role of metallothionein in cadmium traffic and toxicity in kidneys and other mammalian organs. Biometals. 2010; 23:897-926.
29. Nemmiche S. Oxidative Signaling Response to Cadmium Exposure. Toxicological Sciences. 2016; 156:4-10.
30. Cuypers A, Plusquin M, Remans T, Jozefczak M, Keunen E, Gielen H, Opdenakker K, Nair AR, Munters E, Artois TJ, Nawrot T, Vangronsveld J, Smeets K. Cadmium stress: an oxidative challenge. Biometals. 2010; 23:927-940.
31. Xiao W, Liu Y, Templeton DM. Pleiotropic effects of cadmium in mesangial cells. Toxicology and Applied Pharmacology. 2009; 238:315-326.
32. Thevenod F, Lee WK. Cadmium and cellular signaling cascades: interactions between cell death and survival pathways. Archives of Toxicology. 2013; 87:1743-1786.
33. Chen L, Xu B, Liu L, Luo Y, Zhou H, Chen W, Shen T, Han X, Kontos CD, Huang S. Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radical Biology & Medicine. 2011; 50:624-632.
34. Chen P, Duan X, Li M, Huang C, Li J, Chu R, Ying H, Song H, Jia X, Ba Q, Wang H. Systematic network assessment of the carcinogenic activities of cadmium. Toxicology and Applied Pharmacology. 2016; 310:150-158.
35. Liu F, Wang B, Li L, Dong F, Chen X, Li Y, Dong X, Wada Y, Kapron CM, Liu J. Low-Dose Cadmium Upregulates VEGF Expression in Lung Adenocarcinoma Cells. Int J Environ Res Public Health. 2015; 12:10508-10521.
36. Johnson MD, Kenney N, Stoica A, Hilakivi-Clarke L, Singh B, Chepko G, Clarke R, Sholler PF, Lirio AA, Foss C, Reiter R, Trock B, Paik S, et al. Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland. Nat Med. 2003; 9:1081-1084.
37. Stoica A, Katzenellenbogen BS, Martin MB. Activation of estrogen receptor-alpha by the heavy metal cadmium. Mol Endocrinol. 2000; 14:545-553.
38. Kim J, Lim W, Ko Y, Kwon H, Kim S, Kim O, Park G, Choi H, Kim O. The effects of cadmium on VEGF-mediated angiogenesis in HUVECs. J Appl Toxicol. 2012; 32:342-349.
39. Majumder S, Gupta R, Reddy H, Sinha S, Muley A, Kolluru GK, Chatterjee S. Cadmium attenuates bradykinin-driven nitric oxide production by interplaying with the localization pattern of endothelial nitric oxide synthase. Biochemistry and cell biology. 2009; 87:605-620.
40. Majumder S, Muley A, Kolluru GK, Saurabh S, Tamilarasan KP, Chandrasekhar S, Reddy HB, Purohit S, Chatterjee S. Cadmium reduces nitric oxide production by impairing phosphorylation of endothelial nitric oxide synthase. Biochemistry and cell biology. 2008; 86:1-10.
41. Gheorghescu A, Thompson J. Delayed vasculogenesis and impaired angiogenesis due to altered Ang-2 and VE-cadherin levels in the chick embryo model following exposure to cadmium. Pediatric Surgery International. 2016; 32:175-186.
42. Gheorghescu AK, Tywoniuk B, Duess J, Buchete NV, Thompson J. Exposure of chick embryos to cadmium changes the extra-embryonic vascular branching pattern and alters expression of VEGF-A and VEGF-R2. Toxicology and Applied Pharmacology. 2015; 289:79-88.
43. Jung YS, Jeong EM, Park EK, Kim YM, Sohn S, Lee SH, Baik EJ, Moon CH. Cadmium induces apoptotic cell death through p38 MAPK in brain microvessel endothelial cells. European Journal of Pharmacology. 2008; 578:11-18.
44. Pacini S, Punzi T, Morucci G, Gulisano M, Ruggiero M. A paradox of cadmium: a carcinogen that impairs the capability of human breast cancer cells to induce angiogenesis. Journal of Environmental Pathology, Toxicology and Oncology. 2009; 28:85-88.
45. Woods JM, Leone M, Klosowska K, Lamar PC, Shaknovsky TJ, Prozialeck WC. Direct antiangiogenic actions of cadmium on human vascular endothelial cells. Toxicology in vitro. 2008; 22:643-651.
46. Järup L, Hellström L, Alfvén T,Carlsson MD,Grubb A, Persson B, Pettersson C, Spång G, Schütz A, Elinder CG. Low level exposure to cadmium and early kidney damage: the OSCAR study. Occup Environ Med. 2000; 57:668-672.
47. Akesson A, Bjellerup P, Lundh T, Lidfeldt J, Nerbrand C, Samsioe G, Skerfving S, Vahter M. Cadmium-induced effects on bone in a population-based study of women. Environ Health Perspect. 2006; 114:830-834.
48. Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol. 2003; 284:R1-12.
49. Kolluru GK, Tamilarasan KP, Geetha Priya S, Durgha NP, Chatterjee S. Cadmium induced endothelial dysfunction: consequence of defective migratory pattern of endothelial cells in association with poor nitric oxide availability under cadmium challenge. Cell Biology International. 2006; 30:427-438.
50. Sessa WC. Regulation of endothelial derived nitric oxide in health and disease. Memórias do Instituto Oswaldo Cruz. 2005; 100:15-18.
51. Sessa WC. eNOS at a glance. J Cell Sci. 2004; 117:2427-2429.
52. Fulton D, Gratton J, Sessa WC. Post-translational control of endothelial nitric oxide synthase: why isn’t calcium/calmodulin enough? J Pharmacol Exp Ther. 2001; 299:818-824.
53. Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999; 443:285-289.
54. Zhang J, Patel J, Li YD, Block ER. Proinflammatory cytokines downregulate gene expression and activity of constitutive nitric oxide synthase in porcine pulmonary artery endothelial cells. Res Commun Mol Pathol Pharmacol. 1997; 96:71-87.
55. Harris MB, Ju H, Venema VJ, Liang H, Zou R, Michell BJ, Chen ZP, Kemp BE, Venema RC. Reciprocal phosphorylation and regulation of endothelial nitric-oxide synthase in response to bradykinin stimulation. J Biol Chem. 2001; 276:16587-16591.
56. Michell BJ, Harris MB, Chen ZP, Ju H, Venema VJ, Blackstone MA, Huang W, Venema RC, Kemp BE. Identification of regulatory sites of phosphorylation of the bovine endothelial nitric-oxide synthase at serine 617 and serine 635. J Biol Chem. 2002; 277:42344-42351.
57. Chen Z, Peng I, Sun W, Su MI, Hsu PH, Fu Y, Zhu Y, DeFea K, Pan S, Tsai MD, Shyy JY. AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circ Res. 2009; 104:496-505.
58. Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. 2001; 88:E68-75.
59. Mineo C, Shaul PW. Regulation of eNOS in caveolae. Adv Exp Med Biol. 2012; 729:51-62.
60. Chisolm JC, Handorf CR. Zinc, cadmium, metallothionein, and progesterone: do they participate in the etiology of pregnancy induced hypertension? Medical hypotheses. 1985; 17:231-242.
61. Fraisl P. Crosstalk between oxygen- and nitric oxide-dependent signaling pathways in angiogenesis. Exp Cell Res. 2013; 319:1331-1339.
62. Palmer LA, Doctor A, Chhabra P, Sheram ML, Laubach VE, Karlinsey MZ, Forbes MS, Macdonald T, Gaston B. S-nitrosothiols signal hypoxia-mimetic vascular pathology. J Clin Invest. 2007; 117:2592-2601.
63. Metzen E, Zhou J, Jelkmann W, Fandrey J, Brüne B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell. 2003; 14:3470-3481.
64. Berchner-Pfannschmidt U, Yamac H, Trinidad B, Fandrey J. Nitric oxide modulates oxygen sensing by hypoxia-inducible factor 1-dependent induction of prolyl hydroxylase 2. J Biol Chem. 2007; 282:1788-1796.
65. Mateo J, García-Lecea M, Cadenas S, Hernández C, Moncada S. Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem J. 2003; 376:537-544.
66. Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003; 302:1975-1978.
67. Zhang H, Li L, Wang Y, Dong F, Chen X, Liu F, Xu D, Yi F, Kapron CM, Liu J. NF-kappaB signaling maintains the survival of cadmium-exposed human renal glomerular endothelial cells. International Journal of Molecular Medicine. 2016; 38:417-422.
68. Dong F, Guo F, Li L, Guo L, Hou Y, Hao E, Yan S, Allen TD, Liu J. Cadmium induces vascular permeability via activation of the p38 MAPK pathway. Biochem Biophys Res Commun. 2014; 450:447-452.
69. Li L, Dong F, Xu D, Du L, Yan S, Hu H, Lobe CG, Yi F, Kapron CM, Liu J. Short-term, low-dose cadmium exposure induces hyperpermeability in human renal glomerular endothelial cells. J Appl Toxicol. 2016; 36:257-265.
70. Gupta K, Kshirsagar S, Li W, Gui L, Ramakrishnan S, Gupta P, Law PY, Hebbel RP. VEGF Prevents Apoptosis of Human Microvascular Endothelial Cells via Opposing Effects on MAPK/ERK and SAPK/JNK Signaling1. Exp Cell Res. 1999; 247:495-504.
71. Dong F, Zhou X, Li C, Yan S, Deng X, Cao Z, Li L, Tang B, Allen TD, Liu J. Dihydroartemisinin targets VEGFR2 via the NF-B pathway in endothelial cells to inhibit angiogenesis. Cancer Biol Ther. 2014; 15:1479-1488.
72. Karar J, Maity A. PI3K/AKT/mTOR Pathway in Angiogenesis. Frontiers in Molecular Neuroscience. 2011; 4:51.
73. Morales-Ruiz M, Fulton D, Sowa G, Languino LR, Fujio Y, Walsh K, Sessa WC. Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circ Res. 2000; 86:892-896.
74. Liu LZ, Hu XW, Xia C, He J, Zhou Q, Shi X, Fang J, Jiang BH. Reactive oxygen species regulate epidermal growth factor-induced vascular endothelial growth factor and hypoxia-inducible factor-1 expression through activation of AKT and P70S6K1 in human ovarian cancer cells. Free Radical Biology and Medicine. 2006; 41:1521-1533.
75. Dennis MD, Jefferson LS, Kimball SR. Role of p70S6K1-mediated phosphorylation of eIF4B and PDCD4 proteins in the regulation of protein synthesis. J Biol Chem. 2012; 287:42890-42899.
76. Qian Y, Corum L, Meng Q, Blenis J, Zheng JZ, Shi X, Flynn DC, Jiang BH. PI3K induced actin filament remodeling through Akt and p70S6K1: implication of essential role in cell migration. Am J Physiol Cell Physiol. 2004; 286:C153-163.
77. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008; 132:344-362.
78. Dutta J, Fan Y, Gupta N, Fan G, Gélinas C. Current insights into the regulation of programmed cell death by NF-kappaB. Oncogene. 2006; 25:6800-6816.
79. Cai X, Freedman SB, Witting PK. Serum amyloid A stimulates cultured endothelial cells to migrate and proliferate: inhibition by the multikinase inhibitor BIBF1120. Clinical and Experimental Pharmacology & Physiology. 2013; 40:662-670.
80. Nagy JA, Dvorak AM, Dvorak HF. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol. 2007; 2:251-275.
81. Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, Lin A. Inhibition of JNK activation through NF-kappaB target genes. Nature. 2001; 414:313-317.
82. De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature. 2001; 414:308-313.
83. Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001; 81:807-869.
84. Weston CR, Davis RJ. The JNK signal transduction pathway. Current Opinion in Cell Biology. 2007; 19:142-149.
85. Mou H, Zheng Y, Zhao P, Bao H, Fang W, Xu N. Celastrol induces apoptosis in non-small-cell lung cancer A549 cells through activation of mitochondria- and Fas/FasL-mediated pathways. Toxicology in vitro. 2011; 25:1027-1032.
86. Gum SI, Rahman M, Won JS, Cho MK. A Distinctive Pattern of Beauveria bassiana-biotransformed Ginsenoside Products Triggers Mitochondria/FasL-mediated Apoptosis in Colon Cancer Cells. Phytother Res. 2016; 30:136-143.
87. Zhang J, Guo L, Zhou X, Dong F, Li L, Cheng Z, Xu Y, Liang J, Xie Q, Liu J. Dihydroartemisinin induces endothelial cell anoikis through the activation of the JNK signaling pathway. Oncol Lett. 2016; 12:1896-1900.
88. Lian S, Xia Y, Khoi PN, Ung TT, Yoon HJ, Kim NH, Kim KK, Jung YD. Cadmium induces matrix metalloproteinase-9 expression via ROS-dependent EGFR, NF-B, and AP-1 pathways in human endothelial cells. Toxicology. 2015; 338:104-116.
89. Qin W, Lu W, Li H, Yuan X, Li B, Zhang Q, Xiu R. Melatonin inhibits IL1-induced MMP9 expression and activity in human umbilical vein endothelial cells by suppressing NF-B activation. J Endocrinol. 2012; 214:145-153.
90. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000; 2:737-744.
91. Deryugina EI, Quigley J. Pleiotropic roles of matrix metalloproteinases in tumor angiogenesis: contrasting, overlapping and compensatory functions. Biochim Biophys Acta. 2010; 1803:103-120.
92. van Hinsbergh VW, Koolwijk P. Endothelial sprouting and angiogenesis: matrix metalloproteinases in the lead. Cardiovasc Res. 2008; 78:203-212.
93. Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegué E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008; 13:206-220.
94. Cuadrado A, Nebreda A. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010; 429:403-417.
95. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007; 1773:1263-1284.
96. Bian CX, Shi Z, Meng Q, Jiang Y, Liu LZ, Jiang BH. P70S6K 1 regulation of angiogenesis through VEGF and HIF-1 expression. Biochemical and Biophysical Research Communications. 2010; 398:395-399.
97. Stoev SD, Grozeva N, Simeonov R, Borisov I, Hubenov H, Nikolov Y, Tsaneva M, Lazarova S. Experimental cadmium poisoning in sheep. Exp Toxicol Pathol. 2003; 55:309-314.
98. Zeng H, Qin L, Zhao D, Tan X, Manseau EJ, Van Hoang M, Senger DR, Brown LF, Nagy JA, Dvorak HF. Orphan nuclear receptor TR3/Nur77 regulates VEGF-A-induced angiogenesis through its transcriptional activity. J Exp Med. 2006; 203:719-729.
99. Bossy-Wetzel E, Bakiri L, Yaniv M. Induction of apoptosis by the transcription factor c-Jun. EMBO J. 1997; 16:1695-1709.
100. Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997; 275:90-94.
101. Wu CC, Bratton SB. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid Redox Signal. 2013; 19:546-558.
102. Weston CR, Davis RJ. The JNK signal transduction pathway. Current Opinion in Genetics and Development. 2002; 12: 14-21.
103. Shen K, Ji L, Lu B, Wang Z. c-Jun N-terminal kinase mediated VEGFR2 sustained phosphorylation is critical for VEGFA-induced angiogenesis in vitro and in vivo. Cell Biochem Biophys. 2012; 64:17-27.
104. Miura S, Matsuo Y, Saku K. Jun N-terminal kinase inhibitor blocks angiogenesis by blocking VEGF secretion and an MMP pathway. J Atheroscler Thromb. 2008; 15:69-74.
105. Grossi V, Peserico A, Tezil T, Simone C. p38 MAPK pathway: a key factor in colorectal cancer therapy and chemoresistance. World J Gastroenterol. 2014; 20:9744-9758.
106. Cuenda A, Rousseau S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta. 2007; 1773:1358-1375.
107. Kishimoto T, Oguri T, Yamabe S, Tada M. Effect of cadmium injury on growth and migration of cultured human vascular endothelial cells. Hum Cell. 1996; 9:43-48.
108. Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004; 23:2838-2849.
109. Wang D, Weng Q, Zhang L, He Q, Yang B. VEGF and Bcl-2 interact via MAPKs signaling pathway in the response to hypoxia in neuroblastoma. Cell Mol Neurobiol. 2009; 29:391-401.
110. Yiang GT, Yu YL, Lin KT, Chen JN, Chang WJ, Wei CW. Acetaminophen induces JNK/p38 signaling and activates the caspase-9-3-dependent cell death pathway in human mesenchymal stem cells. International Journal of Molecular Medicine. 2015; 36:485-492.
111. Xu ZR, Hu L, Cheng LF, Qian Y, Yang YM. Dihydrotestosterone protects human vascular endothelial cells from H(2)O(2)-induced apoptosis through inhibition of caspase-3, caspase-9 and p38 MAPK. European Journal of Pharmacology. 2010; 643:254-259.
112. Guerrero AD, Chen M, Wang J. Delineation of the caspase-9 signaling cascade. Apoptosis. 2008; 13:177-186.
113. Mariappan MM, Senthil D, Natarajan KS, Choudhury GG, Kasinath BS. Phospholipase Cgamma-Erk Axis in vascular endothelial growth factor-induced eukaryotic initiation factor 4E phosphorylation and protein synthesis in renal epithelial cells. J Biol Chem. 2005; 280:28402-28411.
114. Aird WC. Endothelium in health and disease. Pharmacol Rep. 2008; 60:139-143.
115. Zhuang S, Demirs JT, Kochevar IE. p38 mitogen-activated protein kinase mediates bid cleavage, mitochondrial dysfunction, and caspase-3 activation during apoptosis induced by singlet oxygen but not by hydrogen peroxide. J Biol Chem. 2000; 275:25939-25948.
116. Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999; 6:99-104.
117. Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007; 100:158-173.
118. O’Donnell J, Mille-Baker B, Laffan M. Human umbilical vein endothelial cells differ from other endothelial cells in failing to express ABO blood group antigens. J Vasc Res. 2000; 37:540-547.
119. Chi JT, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, Wang Z, Rockson SG, van de Rijn M, Botstein D, Brown PO. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci U S A. 2003; 100:10623-10628.
120. Yashima R, Abe M, Tanaka K, Ueno H, Shitara K, Takenoshita S, Sato Y. Heterogeneity of the signal transduction pathways for VEGF-induced MAPKs activation in human vascular endothelial cells. Journal of Cellular Physiology. 2001; 188:201-210.
121. Thorin E, Shatos MA, Shreeve SM, Walters CL, Bevan JA. Human vascular endothelium heterogeneity. A comparative study of cerebral and peripheral cultured vascular endothelial cells. Stroke. 1997; 28:375-381.
122. De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, Naldini L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005; 8:211-226.
123. Kroll J, Waltenberger J. The vascular endothelial growth factor receptor KDR activates multiple signal transduction pathways in porcine aortic endothelial cells. J Biol Chem. 1997; 272:32521-32527.
124. Garcia J, Ye Y, Arranz V, Letourneux C, Pezeron G, Porteu F. IEX-1: a new ERK substrate involved in both ERK survival activity and ERK activation. EMBO J. 2002; 21:5151-5163.
125. Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006; 24:21-44.
126. Parast CV, Mroczkowski B, Pinko C, Misialek S, Khambatta G, Appelt K. Characterization and kinetic mechanism of catalytic domain of human vascular endothelial growth factor receptor-2 tyrosine kinase (VEGFR2 TK), a key enzyme in angiogenesis. Biochemistry. 1998; 37:16788-16801.
127. Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997; 277:55-60.
128. Lamont RE, Vu W, Carter AD, Serluca FC, MacRae CA, Childs SJ. Hedgehog signaling via angiopoietin1 is required for developmental vascular stability. Mech Dev. 2010; 127:159-168.
129. Geoffroy-Siraudin C, Perrard M, Ghalamoun-Slaimi R, Ali S, Chaspoul F, Lanteaume A, Achard V, Gallice P, Durand P, Guichaoua MR. Ex-vivo assessment of chronic toxicity of low levels of cadmium on testicular meiotic cells. Toxicology and Applied Pharmacology. 2012; 262:238-246.
130. Lafuente A, Cano P, Esquifino A. Are cadmium effects on plasma gonadotropins, prolactin, ACTH, GH and TSH levels, dose-dependent. Biometals. 2003; 16:243-250.
131. Shao CC, Li N, Zhang ZW, Su J, Li S, Li JL, Xu SW. Cadmium supplement triggers endoplasmic reticulum stress response and cytotoxicity in primary chicken hepatocytes. Ecotoxicol Environ Saf. 2014; 106:109-114.
132. Mahto SK, Yoon TH, Shin H, Rhee SW. Multicompartmented microfluidic device for characterization of dose-dependent cadmium cytotoxicity in BALB/3T3 fibroblast cells. Biomed Microdevices. 2009; 11:401-411.
133. Pak EJ, Son GD, Yoo BS. Cadmium inhibits neurite outgrowth in differentiating human SH-SY5Y neuroblastoma cells. Int J Toxicol. 2014; 33:412-418.
134. Jin Y, Liu L, Zhang S, He R, Wu Y, Chen G, Fu Z. Cadmium exposure to murine macrophages decreases their inflammatory responses and increases their oxidative stress. Chemosphere. 2016; 144:168-175.
135. Powlin SS, Keng PC, Miller RK. Toxicity of cadmium in human trophoblast cells (JAr choriocarcinoma): role of calmodulin and the calmodulin inhibitor, zaldaride maleate. Toxicology and Applied Pharmacology. 1997; 144:225-234.
136. Breen JG, Nelson E, Miller RK. Cellular adaptation to chronic cadmium exposure: intracellular localization of metallothionein protein in human trophoblast cells (JAr). Teratology. 1995; 51:266-272.
137. Thévenod F. Cadmium and cellular signaling cascades: to be or not to be? Toxicology and Applied Pharmacology. 2009; 238:221-239.
138. Zhao P, Meng Q, Liu LZ, You YP, Liu N, Jiang BH. Regulation of survivin by PI3K/Akt/p70S6K1 pathway. Biochem Biophys Res Commun. 2010; 395:219-224.