
INTRODUCTION
Immunosuppressive cells including regulatory T cells (Tregs) are known to play beneficial roles in immune homeostasis and in preventing autoimmunity. CD4+ Tregs are characterized by the high expression of IL-2 receptor α chain (CD25) and FoxP3 transcription factor, which is critical for their development and function [1]. Tregs exert their immunosuppressive actions through various contact-dependent and independent mechanisms that are not fully understood, but require different molecules and cytokines such as Helios, immune checkpoint receptors (ICRs; such as program death 1, PD-1 and cytotoxic T-lymphocyte-associated antigen 4, CTLA-4), TGF-β, IL-10 and IL-35 among others [2, 3]. Helios was shown to be a critical regulator of stable Tregs’ suppressive activity [4] and it is a marker of activated Tregs expressing immunosuppressive molecules GARP/LAP [5]. Tregs constitutively express CTLA-4, which is important for their function [6]. Additionally, CTLA-4 blockade impairs Treg suppressive functions [7, 8]. In cancers, Tregs hamper tumor-specific immune responses by suppressing the proliferation and activation of effector T cells and hence help in tumor evasion [9]. Several studies have reported increased levels of circulating Tregs in patients with colorectal [10], gastric [11], esophageal [11], renal cell carcinoma [12], hepatocellular [13], pancreatic and breast [14, 15] cancers. Increased levels of Tregs are also reported in the tumor microenvironment (TME) of different cancers including breast [15], colon [10] and pancreatic cancers [14].
Breast cancer is one of the most common female cancers and its incidence is increasing in the developed countries each year [16]. Despite the recent advances in screening and treatment, it remains one of the leading causes of cancer-related deaths among women worldwide. One of the reasons for the unsuccessful treatment could be due to the inability of the host immune system to mount sufficient tumor-specific immune responses [17]. Breast TME consists of different types of immune cells and the composition of these immune cell infiltrates has been reported to influence the outcome of the disease in different ways [18]. For example, a high CD8+ T-lymphocyte infiltration had a favorable effect on patients’ survival [19], whereas accumulation of Tregs in the TME was associated with decreased overall survival [20, 21]. On the other hand, Mahmoud et al. did not find a dominant role of FoxP3+ cells in breast cancer prognosis in multivariate analyses [22], while Ladoire et al. found FoxP3 expression to be associated with better survival in HER2-overexpressing breast cancer patients treated with neoadjuvant chemotherapy [23]. These discrepancies can be attributed to the method used for FoxP3 detection or the interactions between different immunosuppressive molecules expressed by Tregs. Therefore, studies investigating the phenotypic characteristics of Tregs in breast cancers are warranted.
In this study we investigated the levels and phenotypes of different immune cells infiltrating primary breast cancer (PBC) tissue and compared them with adjacent non-cancerous normal tissue (NT) and peripheral blood from the same patients. The presence of different ICRs (PD-1 and CTLA-4) on intratumoral CD4+ T cells and in different FoxP3/Helios Treg subsets was also investigated. We observed preferential accumulation of CD4+FoxP3+ Tregs in the TME and their levels were expanded in peripheral blood of these patients, compared with healthy donors (HD). There was a strong positive correlation between FoxP3 and Helios expressions in the TME, and CD4+ Tregs co-expressing FoxP3 and Helios were expanded in tumor tissue compared with normal tissue and peripheral blood. Additionally, we observed increased relative frequency of PD-1/CTLA-4 co-expressing cells within FoxP3+Helios+ and FoxP3+Helios- Treg subsets, whereas the absolute percentages were significantly higher in FoxP3+Helios+ Treg subset, indicating the potent immunosuppressive potentials of the CD4+FoxP3+Helios+PD-1+CTLA-4+ Treg subset.
RESULTS
Accumulation of T cells in tumor-infiltrating leukocytes with exhaustive phenotype in breast cancer tissue
Leukocyte infiltration within the TME is considered as one of the hallmarks of cancer progression [24]. In the present study, we investigated the presence of lymphocytes infiltrating PBC tissue by immunohistochemistry (IHC) and multicolor flow cytometry. As detected by IHC, most of the patients showed higher infiltration of CD3+ cells within tumor tissue (TT) compared with NT. CD3+ cells were also observed in the stroma region in TT but majority of the staining was observed within the borders of the invasive tumors (Figure 1A). We also investigated the presence and phenotype of intratumoral lymphocytes by flow cytometry. To rule out the possibility of gating dead cells and false positive results, 7AAD viability dye was used. CD45 antibody was used to detect the presence of leukocytes and CD3, CD4 and CD8 stainings were used to detect different T cell subsets. As demonstrated in Figure 1B, there was a significant accumulation of CD45+CD3+ cells in TT (23.3 ± 6.3%), compared with adjacent non-cancerous breast tissue (1.2 ± 0.5%). Within CD3+ population, the frequency of both CD4+ (NT- 28.3 ± 10.4% vs. TT- 59.6 ± 4%) and CD8+ (NT- 16.3 ± 6.7% vs. TT- 35.3 ± 3.9%; Figure 1C) T cells increased significantly in TT compared with NT. There was also a significant accumulation of CD45+CD3- cells in the TME (TT- 2.8 ± 1.1%) compared with non-cancerous tissue (NT- 0.6 ± 0.4%); these cells could be of myeloid origin including granulocytic myeloid-derived suppressor cell(s) (G-MDSC) and neutrophils, which suppress host immune responses against cancer and hence promote cancer progression.
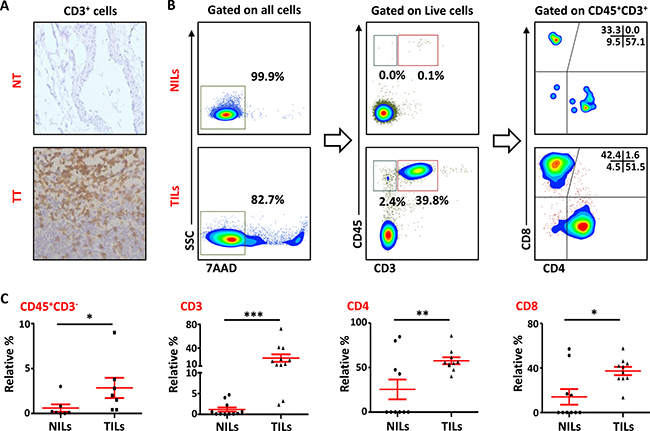
Figure 1: T-cell infiltration in normal and tumor tissues in primary breast cancer. (A). Representative images of immunohistochemical staining of tumor-infiltrating CD3+ T cells in formalin-fixed paraffin embedded breast non-tumor (NT) and tumor tissues (TT). (B). Freshly isolated immune cells infiltrating NT (NILs) and TT (TILs) from 11 PBC patients were stained with 7AAD, CD45, CD3, CD4 and CD8 antibodies for identification of T cells and their subsets. Representative flow cytometric plots of surface staining from one cancer patient are shown. 7AAD dye was used to gate live cells, followed by lymphocyte identification by CD45 and CD3 stainings. Different subsets of T cells were then characterized using CD4 and CD8 antibodies. (C). Scatter plots showing the differences in tissue-infiltrating CD45+CD3-, CD45+CD3+, CD4+ and CD8+ cells between NILs and TILs.
We also studied the phenotypical characteristics of these CD4+ T cells infiltrating TT and NT, and found that intratumoral CD4+ T cells expressed higher levels of CD25 (NT- 8.5 ± 5.9% vs. TT- 21.6 ± 6.1%), PD-1 (NT- 20.8 ± 10.5% vs. TT- 57.0 ± 6.6%) and CD39 (NT- 8.2 ± 5.9% vs. TT- 28.7 ± 5.8%) compared with NT (Figure 2A and 2C). Expression of LAP (Latency-Associated Peptide) on these cells in non-activated state was also evaluated. One of the breast cancer patients in the present cohort had exceptionally high levels of LAP in CD4+ T cells (Figure 2B), but overall there was no significant difference in LAP expression between NILs (1.7 ± 1.4%) and TILs (4.5 ± 3.5%; Figure 2A and 2C). CD8+ T cells in TILs also expressed higher levels of CD25 (NT- 0.7 ± 0.7% vs. TT- 4.6 ± 1.1%), PD-1 (NT- 27.5 ± 11.7% vs. TT- 56.7 ± 6.0%) and CD39 (NT- 0.4 ± 0.3% vs. TT- 9.0 ± 3.5%), compared with NILs (Figure 3A and 3B). Within TT, the levels of CD25 and CD39 expressing CD8+ T cells were significantly lower than CD4+ T cells, whereas there was no difference in PD-1 expression between CD4+ and CD8+ T cells (Figure 3C). The co-expression of PD-1 with CD39 in intratumoral CD4+ and CD8+ T cells was also evaluated. We noticed significantly increased levels of CD4+ and CD8+ T cells co-expressing PD-1 and CD39 in TILs compared with NILs (Figure 4A–4D).
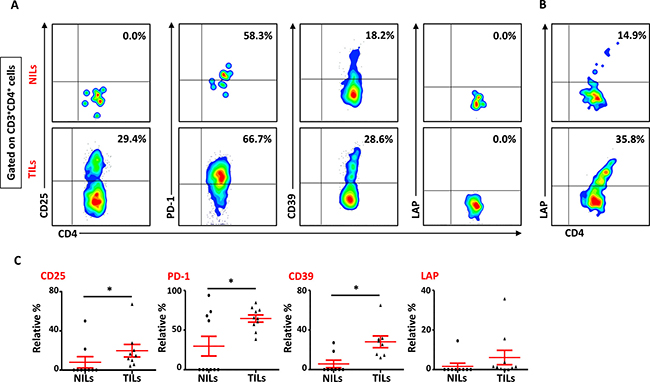
Figure 2: Phenotypic characteristics of CD4+ T cells in NILs and TILs. Freshly isolated NILs and TILs were stained for CD3, CD4, CD25, PD-1, CD39 and LAP surface markers and their relative frequencies were calculated in CD4+ T cells. (A). Representative flow cytometric plots for these markers in NILs and TILs from a cancer patient. (B). NILs and TILs from one cancer patient showed exceptionally high levels of LAP expression. (C). Scatter plots showing the differences in CD25, PD-1, CD39 and LAP between NILs and TILs.
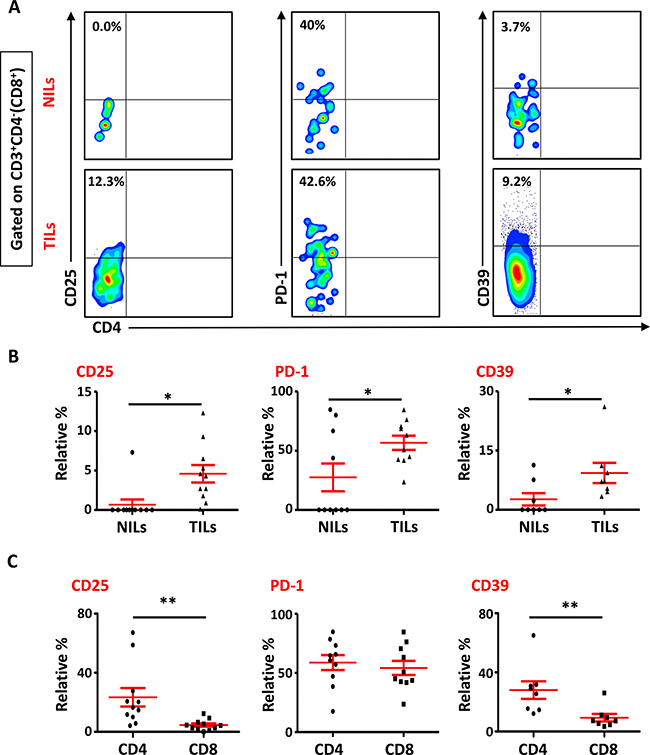
Figure 3: Phenotypic characterization of CD3+CD4- (CD8+) T cells in NILs and TILs. Freshly isolated NILs and TILs were stained for CD3, CD4, CD25, PD-1 and CD39 surface markers. Live cells were gated using 7AAD dye. Levels of CD25, PD-1 and CD39 markers were calculated in CD8+ T cells. (A). Representative flow cytometric plots for these markers in NILs and TILs from a cancer patient are shown. (B). Scatter plots comparing the differences between these surface markers in CD8+ T cells between NILs and TILs. (C). Scatter plots showing the differences in these markers between CD4+ and CD8+ populations in TILs.
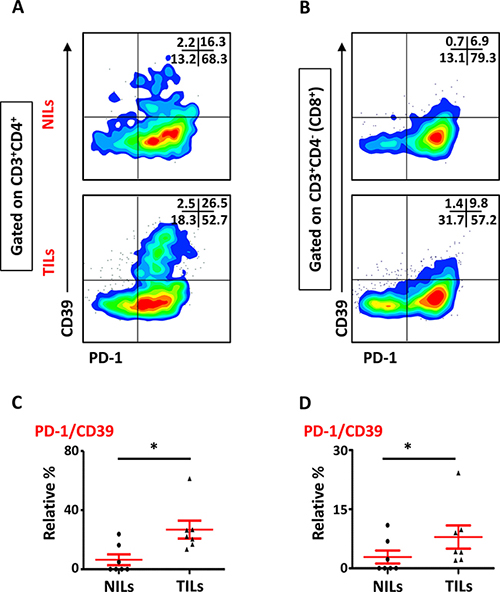
Figure 4: Expression of PD-1/CD39 in CD4+ and CD8+ T cells in NILs and TILs. Representative flow cytometric plots for co-expression of PD-1/CD39 in CD4+ (A) and CD8+ (B) T cells. Scatter plots comparing PD-1/CD39 co-expression between NILs and TILs in CD4+ (C) and CD8+ (D) T cells.
CTLA-4 and PD-1 are upregulated in CD4+ T cells in breast tumor tissue
We have recently observed that intratumoral CD4+ T cells in colorectal cancer (CRC) patients have high co-expression of ICRs including PD-1 and CTLA-4 (Syed Khaja et al. manuscript submitted). To find out if their levels are also elevated in PBC, we examined the co-expression of PD-1/CTLA-4 in CD4+ T cells in the present patient cohort. Representative flow cytometric plots of PD-1 and CTLA-4 co-expression in CD4+ T cells in PBMC of HD and PBC, NILs and TILs are shown in Figure 5A. There were no significant differences between the levels of CD4+ T cells co-expressing PD-1 and CTLA-4 in PBMC from HD (0.4 ± 0.1%) and PBC patients (0.7 ± 0.2%), but the frequency of CD4+ T cells co-expressing CTLA-4 and PD-1 increased significantly in TILs (11.5 ± 3.8%) compared with NILs (0.4 ± 0.2%) or peripheral blood (Figure 5B and 5C). Moreover, the levels of CD4+PD-1-CTLA-4+ T cells in TILs (2.6 ± 1.1%) were also significantly increased, compared with NILs (0.9 ± 0.6%) or peripheral blood (0.8 ± 0.5%). Within TT and NT, CD4+ T cells expressing PD-1 but not CTLA-4 significantly increased (TT- 49.7 ± 6.9%, NT- 57.0 ± 6.9%) compared with their levels in peripheral blood (23.9 ± 4.3%; Figure 5B). In peripheral blood of HD and PBC patients, the majority of cells did not express PD-1 or CTLA-4 and very few proportion of cells expressed only PD-1 but not CTLA-4 (Figure 5C), whereas in NILs and TILs, PD-1 expressing CD4+ T cells increased significantly. Within TT, PD-1/CTLA-4 co-expression was mainly observed in CD4+ T cells and their co-expression in CD8+ T cells was negligible (Figure 5D).
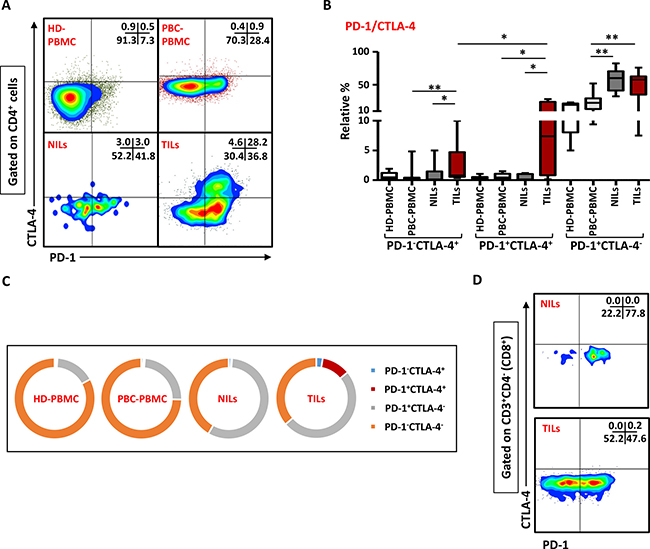
Figure 5: Expression of PD-1/CTLA-4 in CD4+ and CD8+ T cells. PBMC from HD and PBC patients, NILs and TILs were stained for CD3, CD4 and PD-1 surface markers. After fixation and permeabilization, cells were stained for intracellular CTLA-4. Live cells were gated using Fixable Viability Dye 660. Representative flow cytometric plots showing PD-1 and CTLA-4 co-expression in CD4+ T cells (A) and whisker plots (B) showing differences in their expression in HD-PBMC, PBC-PBMC, NILs and TILs. (C). Pie charts show the relative percentages of PD-1 and CTLA-4 co-expression in CD4+ T cells. (D). Representative flow cytometric plots for co-expression of PD-1/CTLA-4 in CD8+ cells in NILs and TILs are shown.
Regulatory T cells are enriched in tumor tissue and peripheral blood of PBC patients
Next, we investigated the levels of Tregs within these CD4+ T cells infiltrating breast tumors. Accumulating evidence suggests that CD4+FoxP3+ Tregs are expanded in various cancers including breast and suppress anti-tumor immunity. In line with previous studies [15, 21, 25], we observed significant accumulation of CD4+FoxP3+ Tregs in breast tumors (13.4 ± 2.3%), compared with NT (2.9 ± 1.3%; Figure 6A and 6B). We also confirmed this finding by IHC and observed significant infiltration of FoxP3+-expressing cells in TT compared with NT (Figure 6C). There was also a significant accumulation of Tregs within TME compared with matching peripheral blood samples (4.1 ± 0.9%; Figure 6A and 6B). As shown in previous studies [26], PBC patients had significantly higher levels of Tregs in peripheral blood compared with HD (2.7 ± 0.3%). We also compared FoxP3+ Treg levels between different breast cancer pathophysiological parameters such as disease stage and grade but did not find any differences in their levels, which could be due to limited sample size (data not shown).
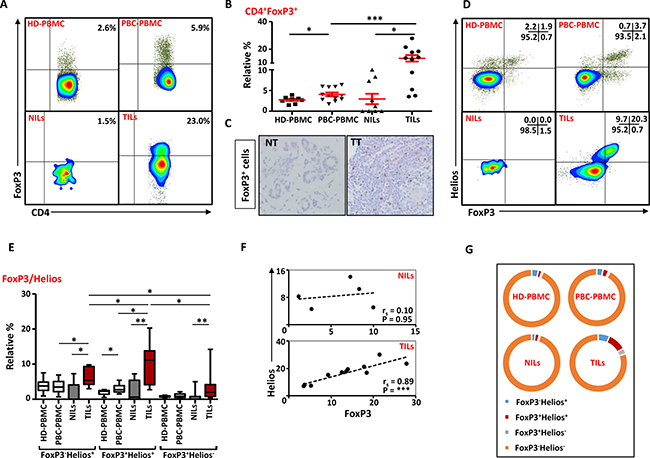
Figure 6: FoxP3 and Helios expression in CD4+ T cells. PBMC from HD and PBC patients, NILs and TILs were stained for CD3 and CD4 antibodies followed by intracellular staining for FoxP3 and Helios. Live cells were first gated using Fixable Viability Dye 660. (A). Representative flow cytometric plots of FoxP3 staining from one cancer patient are shown. (B). Scatter plot showing the differences in frequencies of FoxP3+ Tregs between different samples. (C). IHC staining of FoxP3+ expression in one NT and TT samples. (D). Flow cytometric plots of FoxP3 and Helios co-expression in CD4+ T cells from different samples and whisker plots (E) showing differences in various FoxP3 and Helios-expressing Treg subsets. (F). Non-parametric Spearman’s test showing correlations between FoxP3 and Helios expressions in NILs and TILs. (G). Pie charts show the relative percentages of different FoxP3 and Helios Treg subsets.
FoxP3+Helios+ Tregs are increased in breast tumor tissues
It has recently been reported that Helios is essential for Tregs’ stable inhibitory activity [4]. We observed that CD4+FoxP3+ Tregs in TILs expressed higher levels of Helios and the relative percentages of Tregs co-expressing FoxP3 and Helios within the TME was significantly higher than NT or peripheral blood of cancer patients (Figure 6D and 6E). Interestingly, there was a strong positive correlation between FoxP3 and Helios expressions in TILs (Spearman’s rank correlation coefficient (rs) = 0.89, p < 0.0001) but not NILs (rs = 0.10, p = 0.95; Figure 6F). Levels of tumor-infiltrating FoxP3+Helios+ Tregs were significantly higher than FoxP3-Helios+ and FoxP3+Helios- Treg subsets. Moreover, FoxP3-Helios+ and FoxP3+Helios- Treg subsets were significantly higher in TILs, compared with NILs or PBMC (Figure 6E). PBC patients also showed a significant increase in the levels of FoxP3+Helios+ Tregs in peripheral blood, compared with HD (Figure 6E). The majority of CD4+ T cells in HD-PBMC, PBC-PBMC and NILs did not express either FoxP3 or Helios, and had very few FoxP3/Helios co-expressing cells, whereas the relative proportion of FoxP3+Helios+ Tregs was significantly higher in TILs (Figure 6G).
Intratumoral FoxP3+Helios+ and FoxP3+Helios- Tregs co-express CTLA-4 and PD-1
CD4+FoxP3+ Tregs require CTLA-4 for their potent immunosuppressive activities, and CTLA-4 deficiency in Tregs impaired their suppressive functions [6]. Other studies have also defined the importance of PD-1 in Treg functions [27, 28]. In this study, we investigated any possible correlations of FoxP3 and Helios with CTLA- 4 and PD-1. There was no correlation between FoxP3 and CTLA-4 in HD PBMC (rs = 0.2, p = 0.6), and a statistically insignificant association in PBC PBMC, (rs = 0.6, p = 0.08). In TILs, there was a strong positive correlation between FoxP3 and CTLA-4 expressions (rs = 0.77, p = 0.015; Figure 7A). We also observed a strong positive correlation between Helios and CTLA-4 expressions (rs = 0.79, p = 0.01) (Figure 7A). In contrast there was an inverse correlation between CTLA-4 and Helios expressions in PBMC from HD (rs = -0.78, p = 0.017). There were no significant correlations between PD-1 or CD39 with FoxP3 or Helios in TILs, NILs and PBMC of cancer patients (data not shown).
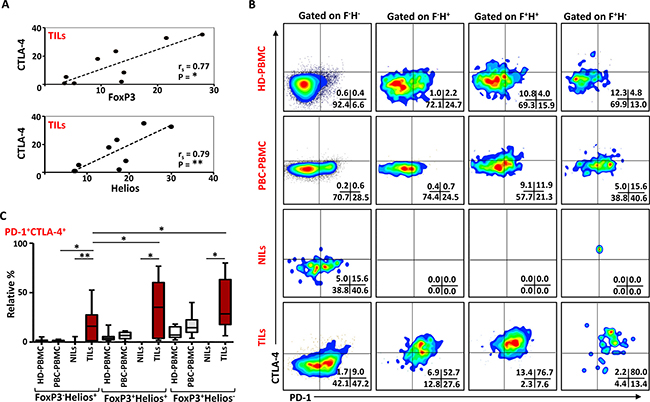
Figure 7: PD-1 and CTLA-4 expression in different FoxP3 and Helios Treg subsets. (A). Non-parametric Spearman’s test showing correlations between CTLA-4 and FoxP3, and CTLA-4 and Helios expressions in TILs. (B). Representative flow cytometric plots showing PD-1 and CTLA-4 expression in different FoxP3/Helios Treg subsets from HD-PBMC, PBC-PBMC, NILs and TILs. (C). Whisker plots comparing the levels of PD-1+CTLA4+ cells in FoxP3-Helios+, FoxP3+Helios+ and FoxP3+Helios- Treg subsets within different samples.
To further characterize these intratumoral Tregs and the different ICRs they co-express compared to NILs and peripheral blood, we examined the co-expression of PD-1 and CTLA-4 in different FoxP3/Helios Treg subsets. Representative flow cytometric plots of PD-1/CTLA-4 co-expression in different groups are shown in Figure 7B. In TILs, co-expression of PD-1 and CTLA-4 was observed in FoxP3-Helios+, FoxP3+Helios+ and FoxP3+Helios- Treg subsets, which were significantly higher compared with their levels in NILs (Figure 7B and 7C). There were no significant differences in PD-1 and CTLA-4 co-expression levels within different FoxP3/Helios Treg subsets in circulation between HD and PBC patients. Within TILs, the relative percentages of ICRs co-expression were highest in FoxP3+Helios+ and FoxP3+Helios- Treg subset, however, when investigated using absolute percentages, FoxP3+Helios+ Treg subset had the highest levels of PD-1 and CTLA-4 co-expression followed by FoxP3-Helios+ subset (data not shown).
DISCUSSION
The TME is usually enriched with infiltrating immune cells, which accumulate as a result of a possible host immune response against cancer [29]. In this study, we found a significant accumulation of CD3+ T cells within breast tumor tissues, and this increase was observed in both CD4+ and CD8+ T cell subsets. Several studies have shown that the composition of these infiltrating immune cells play essential roles in disease prognosis. A high CD8+ T-cell infiltration within the breast tumor usually indicates a tumor-specific immune response and thus can be associated with better outcome [19]. But, as an escape mechanism, tumors secrete many chemokines and cytokines that help in attracting immunosuppressive cells such as MDSC and Tregs, which suppress effector T cells and down-regulate their anti-tumor functions [29]. A significant increase in the levels of CD45+CD3- cells within tumor tissue in our study also indicated the presence of cells of myeloid origin including MDSC and neutrophils that play important roles in suppressing host immune responses against cancer, and hence promote cancer progression. We have recently reported an accumulation of myeloid cells in the TME but not in peripheral blood of this PBC patients’ cohort [30].
The expanded CD4+ T cells within the breast tumor tissue also expressed high levels of CD25, PD-1 and CD39 compared with their levels in normal breast tissue, indicating their activated but “exhaustive” state. High expression of CD25 in these cells also indicates the presence of Tregs in TILs, and elevated PD-1 and CD39 expression might suggest that these cells are exhausted and unable to mount any tumor-specific immune response. Hilchey et al. reported elevated levels of CD4+CD39+ T cells in lymph node mononuclear cells in human follicular lymphoma and blocking CD39 activity partially restored T cell hyporesponsiveness in some patients [31]. They suggested that CD39/adenosine pathway could contribute to “T-cell anergy” in tumors. In chronic lymphocytic leukemia, levels of circulating CD4+CD39+ T cells significantly increased and correlated with advanced stage of the disease [32]. In a different setting than cancer, activated T cells with high CD39 expression were prone to apoptosis in older individuals and suggested that CD4+CD39+ effector T cells do not develop into long-lived memory cells [33]. The significant increase in intratumoral CD4+ T cells co-expressing PD-1/CTLA-4 in our study further supports the unresponsiveness state of these cells. CTLA-4 and PD-1 are key negative regulators of antigen-specific T cell responses [28]. These different immune inhibitory pathways are exploited by tumor cells for escaping from host immune response, and hence blocking these pathways is an important strategy in cancer immunotherapy. Preclinical studies using monoclonal antibodies against CTLA-4 and/or PD-1 have shown promising results in some cancers, but unfortunately that was achieved in a small majority [34–36]. This unresponsiveness state might be due to the presence of MDSC, and MDSC suppression by epigenetic-modulating drugs greatly increased the treatment efficacy of dual blockade in xenograft mice models including metastatic 4T1 mouse breast tumor models [37]. Elevated levels of CD4+ T cells co-expressing CTLA-4 and PD-1 also support the dual blockade of these immunosuppressive pathways, which could be a more effective method for treating different cancers including breast [38, 39]. Data from previous in vivo studies have shown that combinatorial blockade of CTLA-4 and PD-1 is an effective method for reducing tumor-specific immunosuppression [40–42].
Accumulation of CD8+ T cells in TT would have indicated a tumor-specific immune response as a protective function against cancer, but a high PD-1 expression in these cells, compared with NT, suggested a “T-cell anergy” state and their unresponsiveness against cancer cells [43]. CD8+ T cells in TME also expressed high levels of CD39 compared with NT, indicating their terminal exhaustive state [44]. Moreover, a heavy Treg accumulation within the breast tumor tissues, as observed in our study and other earlier studies [18, 45], indicated a possible mechanism utilized by tumor cells to evade host immune responses from CD8+ T cells. Several mechanisms could contribute to the accumulation of Tregs in the TME. Tregs are usually recruited to the tumor site by chemokines and other factors, which are secreted as a result of an interplay between tumor cells and the TME, and their infiltration/expansion can lead to the suppression of effector immune cells [46]. For example, a study found that CCR4+ Tregs migrate to the tumor tissue through CCL22 secreted by ovarian cancer cells [47]. Recently, a critical role of CCL1/CCR8 axis in the accumulation of FoxP3+ Tregs in breast the TME has also been reported [15]. Treg expansion in the TME and circulation of PBC patients could also be due to either their increased proliferation or the conversion of CD4+FoxP3- cells into CD4+FoxP3+ cells in presence of TGF-β [48, 49].
The suppressive functions of FoxP3+ Tregs can be modulated by the presence of different molecules including Helios and CTLA-4. We have recently reported that Tregs co-expressing FoxP3 and Helios represent a functional subset with stronger suppressive characteristics [5], and FoxP3/Helios co-expression with GARP/LAP can be used to identify expanded Treg subsets in cancer patients [50]. In line with these data, here we report a strong positive correlation between FoxP3 and Helios and their co-expression in the majority of Tregs within the TME. Moreover, within TILs, both FoxP3 and Helios strongly correlated with CTLA-4 expression. There was also a marked increase in the relative percentage of cells co-expressing both PD-1 and CTLA-4 in FoxP3+Helios+ Treg subset, suggesting that these cells have stronger immunosuppressive potentials. Some studies have shown that Treg infiltration is associated with better prognosis in breast cancer patients [23, 51], while others have shown a reduced survival of patients with higher Treg accumulation [20, 21]. This ambiguous nature of Tregs in the prognosis of breast cancer can partly be explained by the expression of different immunosuppressive molecules and functional heterogeneity of these Tregs, especially the different cytokines they secrete [52]. In CRC, Saito et al. showed that the heterogeneity of different Treg subsets affected prognosis of the disease in different ways. CRC patients with significant accumulation of CD45RA-FoxP3lo non-Treg subset had better prognosis, compared with those with highly suppressive CD45RA-FoxP3hi Treg subset. Furthermore, these pro-inflammatory FoxP3lo non-Tregs have lower expression of CTLA-4 and TIGIT [53], indicating that immunosuppressive molecules on T cells and Tregs affect the outcome of the disease. Moreover, differences in Treg detection method can also lead to these discrepancies. We suggest that preferential accumulation of FoxP3+Helios+ Tregs co-expressing different immune inhibitory molecules might have a negative impact on breast cancer prognosis. Taken together, our results suggest that breast tumor cells might utilize immune regulatory cells such as Treg and MDSC and different suppressive pathways involving CD39, PD-1 and CTLA-4 molecules in creating an immune-subversive environment for them to survive, and a dual blockade of these immunosuppressive molecules could be a more effective approach for treating breast cancer.
MATERIALS AND METHODS
Tissue samples and processing of peripheral blood
Peripheral blood from 11 PBC patients and 9 HD was collected in tubes containing heparin. Additionally, tumor tissues (TT) and adjacent non-cancerous normal tissues (NT) were obtained from PBC patients who underwent surgery at Tawam Hospital, Al Ain, UAE and Al Noor Hospital, Abu Dhabi, UAE. All patients included in the study are treatment-naive prior to surgery. Table 1 shows the clinical and pathological characteristics of all participating subjects. The study was executed under ethical approval by Al Ain Medical District Research Ethics committee, Al Ain, United Arab Emirates (Protocol no. 13/23-CRD 244/13). All patients and HD provided written informed consent prior to sample collection. All experiments were performed in accordance with relevant guidelines and regulations.
Table 1: Characteristic features of study populations
HD |
PBC |
|
|---|---|---|
Number |
9 |
11 |
Age (median) |
27 (19–45)* |
50.5 (33–65)* |
TNM stage |
||
I |
4 (4)** |
|
II |
4 (4)** |
|
III |
3 (3)** |
|
Tumor size (cm) |
2 (0.8–4.5)* |
|
Lymph node Invasion |
5 |
|
Estrogen receptor positive |
8 |
|
Progesterone receptor positive |
7 |
|
Triple Negative |
2 |
|
Histological grade |
||
Well/moderate |
7 |
|
Poor/undifferentiated |
4 |
HD; Healthy Donor
PBC; primary breast cancer
*Data shown represent median (range).
**Samples taken from patients for investigating tissue-infiltrating immune cells by enzyme disaggregation.
Peripheral blood mononuclear cells(s) (PBMC) were isolated from fresh whole blood by density-gradient centrifugation using Histopaque-1077 (Cat. No. 10771, Sigma-Aldrich). PBMC were frozen in cryovials at a density of 5 million cells per 1 ml freezing media (50% fetal bovine serum (FBS), 40% RPMI 1640 media and 10% DMSO) to be used in batches for subsequent analysis.
Immunohistochemistry
Formalin-fixed, paraffin-embedded NT and TT sections were stained for CD3 and FoxP3 markers by immunohistochemistry. Tissue sections were de-paraffinized in xylene and rehydrated with decreasing concentrations of ethanol. Antigen retrieval was performed at 90°C for 5 min using citrate buffer (pH 6.0). After serial blocking with 3% hydrogen peroxide (in methanol) and protein block (1% bovine serum albumin, 0.05% Tween 20 in PBS), tissue sections were incubated overnight at 4°C with primary monoclonal antibodies against CD3 (M7254, Dako) and FoxP3 (14-4777-82, eBioscience). The sections were then incubated at room temperature for one hour with biotinylated immunoglobulin (E0353, Dako) and another one hour with peroxidase-conjugated streptavidin (P0397, Dako) followed by the addition of DAB+ substrate chromogen (K3468, Dako) for color development. Sections were then counterstained by hematoxylin solution.
Isolation of immune cells from breast tissues by enzyme disaggregation
Enzyme disaggregation (ED) of freshly resected breast TT and NT for immune cell isolation was performed as described previously [54]. Tissues were cut into pieces and then enzymatically digested in RPMI-1640 medium containing 1% penicillin/streptomycin and enzyme cocktail, consisting of 1mg/ml Collagenase (C0130), 100 μg/ml Hyaluronidase type V (H3506) and 30 IU/ml of Deoxyribonuclease I (D5025; all from Sigma-Aldrich) and incubated on a mixer at 37°C for 60 minutes under slow rotation. The cell suspension was then filtered through a 100 μm cell strainer (352360, BD Falcon) to remove any cellular debris and aggregates, followed by two washes with RPMI-1640 media and resuspending in complete media (RPMI-1640 media with 10% FBS and 1% penicillin/streptomycin). Cells isolated from TT (TILs, tumor-infiltrating leukocytes) and NT (NILs, non-tumor-infiltrating leukocytes) were used for flow cytometric staining.
Multicolor flow cytometry
Cells, isolated after ED, were suspended in 100μl staining solution (PBS with 2% FBS and 0.1% sodium azide) and were incubated with human Fc receptor blocking reagent (130-059-901, Miltenyi Biotec). 7AAD viability dye (00-6993-50, eBioscience) was then added to discriminate between live and dead cells. Cells were then stained with cell surface antibodies; CD45-FITC (11-0459-42, eBioscience), CD3-APC-H7 (560176, BD Biosciences), CD4-Alexa Fluor 700 (300526, BioLegend), CD25-PE/Cy7 (356108, BioLegend), LAP-PE (349604, BioLegend), PD-1-PE/Dazzle™ 594 (329940, BioLegend), and CD39-PerCP/Cy5.5 (328218, BioLegend) for 30 minutes at 4°C. After staining, cells were washed and cell pellet was resuspended in flow cytometry staining buffer (eBioscience).
For intracellular staining, Fixable Viability Dye eFluor® 660 (FVD 660; 65-0865-14, eBioscience) was added after blocking with FcR blocker (Miltenyi Biotec). Cells were then labeled with CD3-APC-H7 (BD Biosciences), CD4-Alexa Fluor 700 (BioLegend) and PD-1-PE/Dazzle™ 594 (BioLegend) for 30 minutes at 4°C. Following incubation, cells were washed twice with staining solution and fixed/permeabilized using fixation/permeabilization buffer (eBioscience) at 4ºC for 45 min. After two washes with permeabilization wash buffer, cells were blocked using mouse serum (M5905, Sigma-Aldrich) and rat serum (R9759, Sigma-Aldrich) for 10 min and stained with CTLA-4-PerCP-eFluor® 710 (46-1529-42, eBioscience), FoxP3-PE-Cy7 (25-4776-42, eBioscience) and Helios-FITC (137214, BioLegend) antibodies for another 30 minutes at 4°C. Cells were washed twice with permeabilization wash buffer (eBioscience) and resuspended in flow cytometry staining buffer. All data were acquired with a BD FACSCanto II flow cytometer using BD FACSDiva software (BD Bioscience) and analyzed on BD FACSuite software (BD Biosciences).
Statistical analyses
All statistical analyses were performed using GraphPad Prism 5.0 software (GraphPad Software, Inc.) and Microsoft Excel (Microsoft Corporation). Shapiro-wilk normality test was performed to test if data are normally distributed, followed by paired/Wilcoxon matched-pairs signed rank test or unpaired/Mann-Whitney tests were used to examine the differences within groups or between groups, respectively. Data are represented as mean ± standard error (s.e.m). Correlations between different markers were calculated using non-parametric Spearman’s correlation test. A p value < 0.05 was considered statistically significant. Flow cytometric plots show representative examples of the relative percentage of each population/subpopulation. For absolute or calculated percentages of a particular subpopulation, its relative percentage is multiplied with the relative percentage of its parent population and the resulting value was divided by 100 and presented as absolute percentage.
Abbreviations
CTLA-4: cytotoxic T-lymphocyte-associated antigen 4, CRC: colorectal cancer, ED: enzyme disaggregation, HD: healthy donors, ICR: immune checkpoint receptor, IHC: immunohistochemistry, LAP: latency-associated peptide, MDSC: myeloid-derived suppressor cells, NILs: non-tumor-infiltrating leukocytes, NT: non-tumor tissue, PBC: primary breast cancer, PBMC: peripheral blood mononuclear cells, PD-1: program death 1, TILs: tumor-infiltrating leukocytes, TME: tumor microenvironment, Tregs: regulatory T cells, TT: tumor tissue
Authors’ contributions
ASSK and ST performed experimental work, data analysis and wrote the manuscript. EE conceived the idea, designed the study, obtained funds, analyzed and interpreted data and wrote and revised the manuscript. HE, IF, NH contributed to sample collection and acquisition of patients’ clinical data. BA obtained funds and revised the manuscript. All authors were involved in the final approval of the manuscript.
ACKNOWLEDGMENTS
We are grateful to all the participating individuals for donating their samples. We also thank Ms. Ghada Bashir, Department of Medical Microbiology and Immunology, UAE University for IHC staining.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by grants from United Arab Emirates Program of Advanced Research (31M190), Terry Fox Foundation (21M094) and CMHS grant, UAE University (31M261).
REFERENCES
1. Chaudhary B, Elkord E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines (Basel). 2016; 4. doi: 10.3390/vaccines4030028.
2. Wolf D, Sopper S, Pircher A, Gastl G, Wolf AM. Treg(s) in Cancer: Friends or Foe? J Cell Physiol. 2015; 230:2598–605. doi: 10.1002/jcp.25016.
3. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol. 2012; 3: 51. doi: 10.3389/fimmu.2012.00051.
4. Kim HJ, Barnitz RA, Kreslavsky T, Brown FD, Moffett H, Lemieux ME, Kaygusuz Y, Meissner T, Holderried TA, Chan S, Kastner P, Haining WN, Cantor H. Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science. 2015; 350:334–9. doi: 10.1126/science.aad0616.
5. Elkord E, Abd Al Samid M, Chaudhary B. Helios, and not FoxP3, is the marker of activated Tregs expressing GARP/LAP. Oncotarget. 2015; 6:20026–36. doi: 10.18632/oncotarget.4771.
6. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008; 322:271–5. doi: 10.1126/science.1160062.
7. Read S, Greenwald R, Izcue A, Robinson N, Mandelbrot D, Francisco L, Sharpe AH, Powrie F. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J Immunol. 2006; 177:4376–83. doi: 10.4049/jimmunol.177.7.4376.
8. Kolar P, Knieke K, Hegel JK, Quandt D, Burmester GR, Hoff H, Brunner-Weinzierl MC. CTLA-4 (CD152) controls homeostasis and suppressive capacity of regulatory T cells in mice. Arthritis Rheum. 2009; 60:123–32. doi: 10.1002/art.24181.
9. Oleinika K, Nibbs RJ, Graham GJ, Fraser AR. Suppression, subversion and escape: the role of regulatory T cells in cancer progression. Clin Exp Immunol. 2013; 171:36–45. doi: 10.1111/j.1365-2249.2012.04657.x.
10. Ling KL, Pratap SE, Bates GJ, Singh B, Mortensen NJ, George BD, Warren BF, Piris J, Roncador G, Fox SB, Banham AH, Cerundolo V. Increased frequency of regulatory T cells in peripheral blood and tumour infiltrating lymphocytes in colorectal cancer patients. Cancer Immun. 2007; 7:7.
11. Ichihara F, Kono K, Takahashi A, Kawaida H, Sugai H, Fujii H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin Cancer Res. 2003; 9:4404–8.
12. Griffiths RW, Elkord E, Gilham DE, Ramani V, Clarke N, Stern PL, Hawkins RE. Frequency of regulatory T cells in renal cell carcinoma patients and investigation of correlation with survival. Cancer Immunol Immunother. 2007; 56:1743–53. doi: 10.1007/s00262-007-0318-z.
13. Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005; 65:2457–64. doi: 10.1158/0008-5472.CAN-04-3232.
14. Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS, Linehan DC. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002; 169:2756–61.
15. Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, Chudakov DM, Rudensky AY. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity. 2016; 45:1122–34. doi: 10.1016/j.immuni.2016.10.032.
16. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87–108. doi: 10.3322/caac.21262.
17. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007; 121:1–14. doi: 10.1111/j.1365-2567.2007.02587.x.
18. Stanton SE, Disis ML. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J Immunother Cancer. 2016; 4: 59. doi: 10.1186/s40425-016-0165-6.
19. Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AH, Ellis IO, Green AR. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol. 2011; 29:1949–55. doi: 10.1200/JCO.2010.30.5037.
20. Ohara M, Yamaguchi Y, Matsuura K, Murakami S, Arihiro K, Okada M. Possible involvement of regulatory T cells in tumor onset and progression in primary breast cancer. Cancer Immunol Immunother. 2009; 58: 441–7. doi: 10.1007/s00262-008-0570-x.
21. Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, Banham AH. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol. 2006; 24:5373–80. doi: 10.1200/JCO.2006.05.9584.
22. Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Lee AH, Ellis IO, Green AR. An evaluation of the clinical significance of FOXP3+ infiltrating cells in human breast cancer. Breast Cancer Res Treat. 2011; 127:99–108. doi: 10.1007/s10549-010-0987-8.
23. Ladoire S, Arnould L, Mignot G, Coudert B, Rebe C, Chalmin F, Vincent J, Bruchard M, Chauffert B, Martin F, Fumoleau P, Ghiringhelli F. Presence of Foxp3 expression in tumor cells predicts better survival in HER2-overexpressing breast cancer patients treated with neoadjuvant chemotherapy. Breast Cancer Res Treat. 2011; 125:65–72. doi: 10.1007/s10549-010-0831-1.
24. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009; 30:1073–81. doi: 10.1093/carcin/bgp127.
25. DeNardo DG, Coussens LM. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 2007; 9: 212. doi: 10.1186/bcr1746.
26. Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res. 2003; 9:606–12.
27. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010; 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x.
28. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012; 12:252–64. doi: 10.1038/nrc3239.
29. Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008; 27:5904–12. doi: 10.1038/onc.2008.271.
30. Toor SM, Syed Khaja AS, El Salhat H, Faour I, Kanbar J, Quadri AA, Albashir M, Elkord E. Myeloid cells in circulation and tumor microenvironment of breast cancer patients. Cancer Immunology, Immunotherapy. 2017: 1–12. doi: 10.1007/s00262-017-1977-z.
31. Hilchey SP, Kobie JJ, Cochran MR, Secor-Socha S, Wang JC, Hyrien O, Burack WR, Mosmann TR, Quataert SA, Bernstein SH. Human follicular lymphoma CD39+-infiltrating T cells contribute to adenosine-mediated T cell hyporesponsiveness. J Immunol. 2009; 183:6157–66. doi: 10.4049/jimmunol.0900475.
32. Perry C, Hazan-Halevy I, Kay S, Cipok M, Grisaru D, Deutsch V, Polliack A, Naparstek E, Herishanu Y. Increased CD39 expression on CD4(+) T lymphocytes has clinical and prognostic significance in chronic lymphocytic leukemia. Ann Hematol. 2012; 91:1271–9. doi: 10.1007/s00277-012-1425-2.
33. Fang F, Yu M, Cavanagh MM, Hutter Saunders J, Qi Q, Ye Z, Le Saux S, Sultan W, Turgano E, Dekker CL, Tian L, Weyand CM, Goronzy JJ. Expression of CD39 on Activated T Cells Impairs their Survival in Older Individuals. Cell Rep. 2016; 14:1218–31. doi: 10.1016/j.celrep.2016.01.002.
34. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363:711–23. doi: 10.1056/NEJMoa1003466.
35. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366:2443–54. doi: 10.1056/NEJMoa1200690.
36. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013; 369:122–33. doi: 10.1056/NEJMoa1302369.
37. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, Diaz LA Jr, Papadopoulos N, Kinzler KW, Vogelstein B, Zhou S. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014; 111:11774–9. doi: 10.1073/pnas.1410626111.
38. Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016; 39:98–106. doi: 10.1097/COC.0000000000000239.
39. Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, Callahan M, Wolchok JD, Halaban R, Dhodapkar MV, Dhodapkar KM. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol. 2015; 194:950–9. doi: 10.4049/jimmunol.1401686.
40. Binder DC, Schreiber H. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors--letter. Cancer Res. 2014; 74: 632. doi: 10.1158/0008-5472.CAN-13-2216.
41. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010; 107:4275–80. doi: 10.1073/pnas.0915174107.
42. Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013; 73:3591–603. doi: 10.1158/0008-5472.CAN-12-4100.
43. Wherry EJ. T cell exhaustion. Nat Immunol. 2011; 12:492–9.
44. Gupta PK, Godec J, Wolski D, Adland E, Yates K, Pauken KE, Cosgrove C, Ledderose C, Junger WG, Robson SC, Wherry EJ, Alter G, Goulder PJ, et al. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PLoS Pathog. 2015; 11: e1005177. doi: 10.1371/journal.ppat.1005177.
45. Dushyanthen S, Beavis PA, Savas P, Teo ZL, Zhou C, Mansour M, Darcy PK, Loi S. Relevance of tumor-infiltrating lymphocytes in breast cancer. BMC Med. 2015; 13: 202. doi: 10.1186/s12916-015-0431-3.
46. Whiteside TL. The role of regulatory T cells in cancer immunology. Immunotargets Ther. 2015; 4:159–71. doi: 10.2147/ITT.S55415.
47. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004; 10:942–9. doi: 10.1038/nm1093.
48. Liu VC, Wong LY, Jang T, Shah AH, Park I, Yang X, Zhang Q, Lonning S, Teicher BA, Lee C. Tumor evasion of the immune system by converting CD4+CD25- T cells into CD4+CD25+ T regulatory cells: role of tumor-derived TGF-beta. J Immunol. 2007; 178:2883–92.
49. Ondondo B, Jones E, Godkin A, Gallimore A. Home sweet home: the tumor microenvironment as a haven for regulatory T cells. Front Immunol. 2013; 4: 197. doi: 10.3389/fimmu.2013.00197.
50. Abd Al Samid M, Chaudhary B, Khaled YS, Ammori BJ, Elkord E. Combining FoxP3 and Helios with GARP/LAP markers can identify expanded Treg subsets in cancer patients. Oncotarget. 2016; 7:14083–94. doi: 10.18632/oncotarget.7334.
51. Liu C, Workman CJ, Vignali DA. Targeting regulatory T cells in tumors. FEBS J. 2016; 283:2731–48. doi: 10.1111/febs.13656.
52. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, Parizot C, Taflin C, Heike T, Valeyre D, Mathian A, Nakahata T, Yamaguchi T, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009; 30:899–911. doi: 10.1016/j.immuni.2009.03.019.
53. Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, Maeda Y, Hamaguchi M, Ohkura N, Sato E, Nagase H, Nishimura J, Yamamoto H, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. 2016; 22:679–84. doi: 10.1038/nm.4086.
54. Toor S, Syed Khaja AS, El Salhat H, Bekdache O, Kanbar J, Jaloudi M, Elkord E. Increased levels of circulating and tumor-infiltrating granulocytic myeloid cells in colorectal cancer patients. Front Immunol. 2016; 7. doi: 10.3389/fimmu.2016.00560.