
INTRODUCTION
Malignant gliomas (glioblastoma or GBM) are the most common brain tumor in adults, with approximately over 13,000 individuals in the United States dying from their cancer each year. After initial diagnosis, the median survival is only 9 months [1, 2], which improves to ~15 months following surgical resection, radiation and chemotherapy [3]. In an effort to improve clinical outcomes for people with this deadly cancer, numerous conventional and biologically-based anti-neoplastic treatments have been evaluated that specifically target the rapidly dividing cancer cells. Unfortunately, these newer therapies have resulted in only modest increases in overall survival, highlighting the urgent need to identify more effective therapies for these brain malignancies.
Over the past decades, numerous reports have described an instructive role for the tumor microenvironment in glioma formation and maintenance [4]. In this regard, glioblastomas contain a rich non-neoplastic component, including endothelial cells and immune system cells (e.g., microglia and macrophages), whose function is dictated by small molecules responsible for their recruitment and activation. One class of these proteins, chemokines, is critical for microglia and macrophage infiltration and activation through the expression of chemokine receptors on their plasma membranes. Consistent with an ecosystem paradigm in which non-neoplastic cells attracted by cancer cells in turn support neoplastic cell growth, inhibition of chemokine expression or chemokine receptor function has been shown to attenuate both low-grade and high-grade glioma growth in numerous experimental preclinical model systems in vivo. For example, CCL2 recruits regulatory T cells and myeloid-derived suppressor cells to increase GBM growth [5], as well as indirectly increases glioma invasiveness by stimulating IL-6 secretion from tumor-associated microglia (TAM) [6]. In addition, CXCL12 and its receptors CXCR4/CXCR7 function by maintaining GBM stem-like cell survival and increasing glioma cell invasion [7, 8]. Similarly, colony stimulating factor-1 (CSF-1) creates a supportive glioma microenvironment, such that silencing CSF-1-receptor signaling on macrophages is sufficient to reduce high-grade glioma growth in mice [9, 10]. Together, these experimental observations establish a fundamental role for chemokines in glioma biology.
In addition to the chemokines mentioned above, we have recently discovered that Ccl5 produced by tumor-associated microglia is a potent paracrine regulator of neurofibromatosis type 1 (NF1)-associated murine low-grade optic glioma growth [11]. In this experimental system, Ccl5 was sufficient to stimulate Nf1-deficient optic nerve glial cell proliferation in vitro, whereas Ccl5 antibody-mediated inhibition attenuated mouse optic glioma proliferation in vivo. Interestingly, CCL5 has recently emerged as a key member of an autocrine cytokine circuit critical for KRAS-driven lung cancer growth [12]. Since the NF1 protein, neurofibromin, functions as a negative regulator of RAS, specifically K-RAS in astroglial cells [13, 14], and NF1 mutation/loss characterizes the mesenchymal GBM (M-GBM) molecular subtype [15], we sought to determine whether Ccl5 expression creates a unique autocrine circuit for mesenchymal glioblastoma growth.
In this report, we describe for the first time, that increased CCL5 expression is enriched in both human and mouse M-GBM with NF1 loss. Leveraging Nf1-deficient murine M-GBM models, we further demonstrate that Ccl5 shRNA-mediated knockdown increases glioblastoma cell apoptosis in vitro, as well as extends mouse glioblastoma survival in vivo. Moreover, neurofibromin negatively regulates Ccl5 expression through RAS/AKT/mTOR pathway activation. Lastly, Ccl5 produced by the M-GBM cells acts in a cell autonomous fashion to increase M-GBM cell survival using a non-conventional CCL5 receptor, CD44, to suppress programmed cell death. Collectively, these findings broaden the concept that growth regulatory factors produced by non-neoplastic cells in the low-grade glioma microenvironment are employed to establish autocrine circuits in high-grade gliomas, thus facilitating a state of relative stromal independence.
RESULTS
Ccl5 functions in a cell-autonomous fashion to increase M-GBM survival
Based on our previous studies examining CCL5 expression and function in low-grade glioma, we first sought to determine whether CCL5 expression was higher in human GBM tumors relative to non-neoplastic brain. Using the GSE16011 [16] dataset, for which normal brain was available for comparison, CCL5 mRNA levels were higher in GBM samples relative to their non-neoplastic counterpart (Figure 1A). We next analyzed CCL5 mRNA expression in the four GBM molecular subtypes using data from the Cancer Genome Atlas (TCGA, provisional), and found that mesenchymal GBM (M-GBM) exhibited the highest levels of CCL5 mRNA expression, while proneural GBM (PN-GBM) expressed the lowest (Figure 1B).
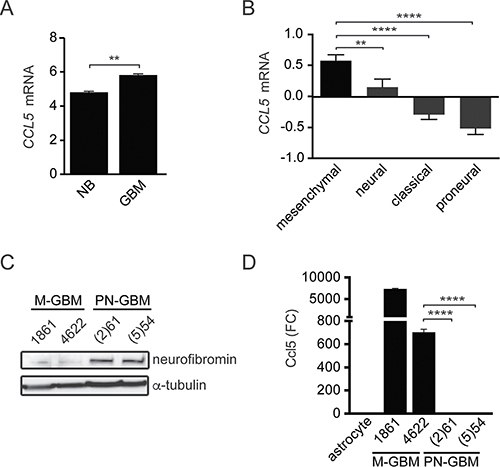
Figure 1: M-GBM express high levels of CCL5. (A) Human GBM samples (GSE16011 [16]) have elevated CCL5 mRNA levels relative to normal brain (NB). **p = 0.0023. (B) CCL5 mRNA expression (Log2 Z-score) in TCGA GBM (provisional) samples segregated by molecular subgroup reveals that mesenchymal and proneural GBM express the highest and lowest CCL5 levels, respectively. **p = 0.0095; ****p < 0.0001. n = 106, 55, 102, and 96 for the mesenchymal, neural, classical and proneural molecular GBM subgroups, respectively. (C) M-GBM cells are neurofibromin deficient, while PN-GBM cells express neurofibromin. (D) Ccl5 ELISA analysis of the culture medium reveals higher levels of Ccl5 in M-GBM cells relative to PN-GBM cells and primary astrocytes. ****p < 0.0001. FC, fold change.
This relative enrichment of CCL5 expression in the mesenchymal subtype, which frequently exhibits loss of function mutations in the NF1 tumor suppressor gene [15], prompted us to focus on murine models characterized by Nf1 loss. Using two representative mouse M-GBM cell lines (1861 and 4622), which lack Nf1 and Tp53 expression [17], and two representative mouse proneural GBM cell lines ((2)61 and (5)54), which retain neurofibromin expression (Figure 1C) [18], we found that Ccl5 levels were elevated in the culture medium of mouse M-GBM cells relative to PN-GBM cells, as well as to wild-type primary mouse astrocytes (Figure 1D).
To determine whether M-GBM cell growth was dependent on Ccl5 expression, 1861 (M-GBM) cells were grown in the presence of mouse Ccl5 (mCcl5). mCcl5 treatment increased cell growth, as revealed by ~14% increase in BrdU incorporation relative to the vehicle treatment (Figure 2A). Next, we reduced Ccl5 levels in 1861 cells by shRNA-mediated knockdown (KD). Using two independent constructs (shCcl5-1 and shCcl5-2), Ccl5 protein levels were reduced to ~20% of control levels (LacZ shRNA; Figure 2B), resulting in reduction in cell number (Figure 2C) and cell growth (BrdU incorporation; Figure 2D) as a consequence of increased apoptosis (increased cleaved caspase-3 expression and %TUNEL+ cells; Figure 2E–2G). Similar results were obtained using another M-GBM cell line (4622; Supplementary Figure 1A–1E). Importantly, treating PN-GBM cells with mCcl5 did not affect cell growth, suggesting that Ccl5 is not a regulator for PN-GBM growth (Supplementary Figure 1F).
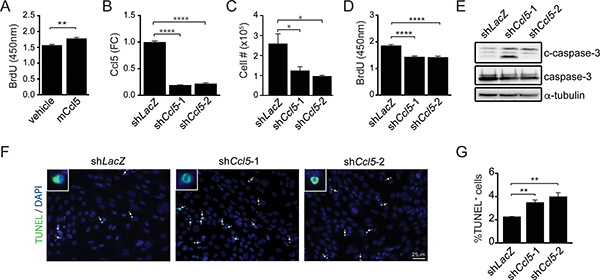
Figure 2: Ccl5 functions in a cell-autonomous fashion to increase M-GBM cell survival. (A) Increased BrdU incorporation was observed in 1861 cells following treatment with mouse Ccl5 (mCcl5). Vehicle was 0.5% BSA in PBS. **p = 0.009. (B) ELISA analysis of 1861 cell medium shows that Ccl5 KD (shCcl5-1, shCcl5-2) cells produce less Ccl5 compared to control (shLacZ) cells. ****p < 0.0001. FC, fold change. (C) Ccl5 KD cells exhibit reduced cell numbers. *p = 0.0428 (shLacZ vs shCcl5-1), and p = 0.0162 (shLacZ vs shCcl5-2). (D) Ccl5 KD cells exhibit reduced cell growth (BrdU incorporation). ****p < 0.0001. (E) Western blotting demonstrates increased cleaved caspase-3 (c-caspase-3) expression following Ccl5 KD. (F, G) Ccl5 KD increases the percentage of TUNEL+ cells (green; arrows). DAPI-stained nuclei (inset) are shown in blue. **p = 0.0039 (shLacZ vs shCcl5-1), and p = 0.0063 (shLacZ vs shCcl5-2). Scale bar, 25 μm.
Based on these in vitro results, we also sought to determine whether Ccl5 is required for M-GBM growth in vivo. Control (shLacZ) and Ccl5 KD (shCcl5-1 and shCcl5-2) 1861 cells were stereotactically implanted into the striata of 4-week-old wild-type C57BL/6 mice. Both Ccl5 KD groups demonstrated reduced Ccl5 levels (Figure 3A), reduced cell proliferation (%Ki67+ cells, Figure 3B, 3C) at time of death, as well as longer survival relative to the control (shLacZ) group (Figure 3D). In addition, tumor implants dissected at 6-weeks post-implantation demonstrated reduced Ccl5 levels, increased apoptosis (cleaved caspase-3 expression), and reduced cell proliferation (PCNA expression) in the Ccl5 KD groups (Supplementary Figure 2).
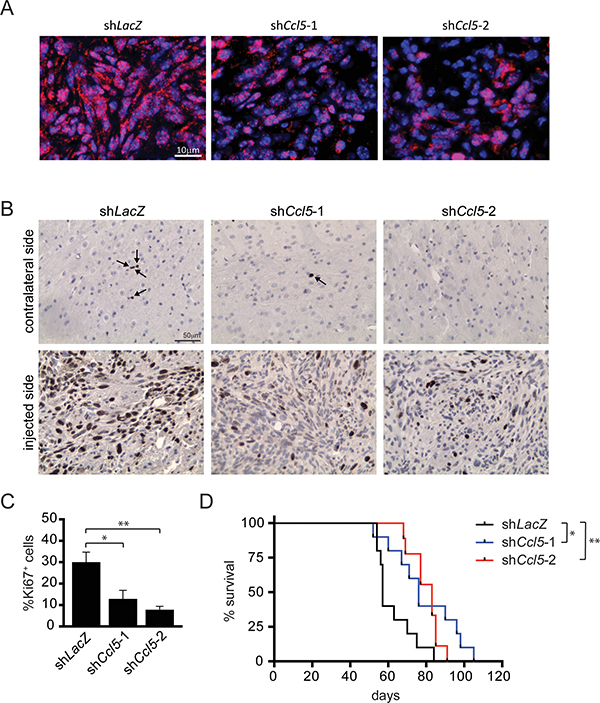
Figure 3: Ccl5 knockdown reduces M-GBM growth and prolongs mouse survival in vivo. Ccl5 KD (shCcl5-1, shCcl5-2) and control (shLacZ) 1861 cells were implanted into the striata of C57BL/6 mice. Ccl5 KD groups have fewer Ccl5+ (red; A), Ki67+ cells (B, C, arrow) relative to controls. *p = 0.0145; **p = 0.0014. Scale bars, 10 μm (A) and 50 μm (B). (D) Kaplan-Meier survival curves demonstrate that the Ccl5 KD groups have prolonged survival relative to controls. Each group contains 9–10 mice. *p = 0.0117; **p = 0.0037 (Log-rank test).
Given the role of Ccl5 as a chemokine, it may recruit monocytes to the implantation site to facilitate tumor growth; however, we found no change in the number of recruited monocytes or chemokine expression within the tumor between the control and Ccl5 KD groups (data not shown). In addition, we sought to determine whether Ccl5 produced by non-neoplastic cells contributes to M-GBM tumor growth by comparing M-GBM cell growth in wild-type versus Ccl5−/− mouse striata. Mouse survival and tumor proliferation were similar between the two groups, arguing against a major stromal source for Ccl5 in mediating the observed M-GBM survival effects (Supplementary Figure 3). Collectively, these observations demonstrate that Ccl5 positively regulates M-GBM growth in vivo and in vitro through an autocrine stimulatory mechanism.
Neurofibromin negatively regulates Ccl5 expression through suppression of AKT/mTOR pathway activation
Since increased CCL5 expression predominates in the M-GBM molecular subtype and mouse Nf1-deficient GBM cells exhibit reduced growth following Ccl5 KD, we tested the hypothesis that the NF1 protein (neurofibromin) function is responsible for regulating Ccl5 expression. Using primary brainstem astrocytes from postnatal day 1 mice (Nf1flox/flox astrocytes infected with Ad5-LacZ virus, Nf1+/+; Nf1flox/flox astrocytes infected with Ad5-Cre virus, Nf1−/−) (Supplementary Figure 4A), Ccl5 levels in the medium were increased in Nf1−/− astrocytes relative to wild-type (Nf1+/+) controls (Figure 4A). To establish causality, we re-introduced the functional domain of neurofibromin (NF1-RAS GTPase-activating protein regulatory domain [NF1-GRD; GRD-WT]; Supplementary Figure 4B). A mutant NF1-GRD construct (NF1-GRDR1276P; GRD-Mut) defective at suppressing RAS activity was used as a control for RAS-GAP activity [19]. Expression of WT, but not mutant, NF1-GRD reduced Ccl5 levels in 1861 cells (Figure 4B), demonstrating that neurofibromin negatively regulates Ccl5 expression.
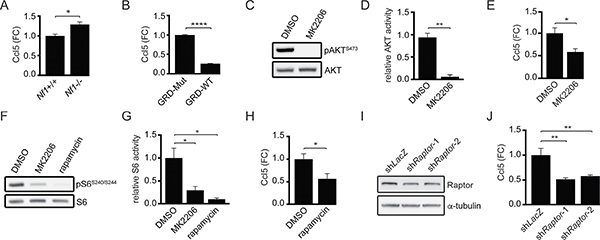
Figure 4: Ccl5 expression in M-GBM cells is regulated by neurofibromin-mediated AKT/mTOR pathway suppression. (A) ELISA analysis demonstrates greater Ccl5 levels in the medium from Nf1−/− astrocytes relative to their wild-type (Nf1+/+) counterparts. *p = 0.01. FC, fold change. (B) Wild-type NF1-GRD (GRD-WT) expression in 1861 cells reduced Ccl5 expression relative to the NF1-GRD R1276P mutant (GRD-Mut). ****p < 0.0001. (C, D) Treatment of 1861 cells with MK2206 (AKT inhibitor) reduced AKT activity (phospho-AKTS473/AKT ratio) compared to DMSO (vehicle control). **p = 0.0012. (E) ELISA reveals reduced Ccl5 expression following MK2206 treatment. *p = 0.0258. FC, fold change. (F, G) MK2206 or rapamycin (mTOR inhibitor) treatment reduced mTOR activity (phospho-S6S240/S244/S6 ratio). *p = 0.0362 (DMSO vs MK2206) and p = 0.0116 (DMSO vs rapamycin). (H) Rapamycin-treated 1861 cells had reduced Ccl5 levels in the medium compared to DMSO controls. *p = 0.0313. FC, fold change. (I) Western blotting demonstrates that shRNA-mediated Raptor KD (shRaptor-1 and shRaptor-2) reduced Raptor protein expression in 1861 cells relative to control (shLacZ) vector. (J) Raptor KD results in reduced Ccl5 levels in the medium. **p = 0.002 (shLacZ vs shRaptor-1), and p = 0.0034 (shLacZ vs shRaptor-2). FC, fold change.
Neurofibromin regulates glioma cell growth through the AKT/mTOR pathway [20, 21], such that Nf1−/− astrocytes exhibit AKT/mTOR hyperactivation relative to Nf1+/+ (wild-type) astrocytes (Supplementary Figure 4A). Consistent with this neurofibromin regulatory function, AKT/mTOR activity in 1861 cells was reduced following NF1-GRD expression (Supplementary Figure 4C). Moreover, AKT pharmacological inhibition (MK2206) of M-GBM cells similarly reduced AKT activity (Figure 4C, 4D) and Ccl5 expression (Figure 4E). In addition, AKT inhibition in 1861 cells reduced mTOR activity (Figure 4F, 4G), and mTOR inhibition using rapamycin (Figure 4F, 4G) reduced Ccl5 expression (Figure 4H). The concentrations and durations of inhibitor treatments were specifically selected so that cell growth was not affected (Supplementary Figure 4D), arguing that any potential changes in Ccl5 expression were a secondary effect of suppressed cell growth. As a complementary approach, we employed shRNA knockdown of Raptor, a molecule critical for the function of mTOR complex-1 (Figure 4I). Similar to rapamycin treatment, Raptor KD reduced Ccl5 expression (Figure 4J). However, in these experiments, cell growth was inhibited following Raptor KD (Supplementary Figure 4E), requiring that the amount of Ccl5 present in the medium be normalized to the total cell number.
Since MEK/ERK activation is also elevated following NF1 loss [22], we analyzed Ccl5 expression following PD-0325901 (PD901, MEK inhibitor) treatment. In these experiments, MEK inhibition did not change Ccl5 expression (Supplementary Figure 5). Collectively, these experiments establish RAS-GAP/AKT/mTOR signaling as the primary mechanism underlying neurofibromin regulation of Ccl5 expression.
Ccl5 controls M-GBM survival through CD44
CCL5 functions by binding to G-protein coupled receptors, including CCR (C-C chemokine receptor) 1, 3 and 5. Surprisingly, Ccr1, Ccr3 or Ccr5 mRNA levels were nearly undetectable in M-GBM cells (Supplementary Figure 6), prompting us to investigate non-conventional CCL5 receptors (GPR75, CXCR4 and CD44) as potential binding partners [23, 24]. In these experiments, only Cd44 mRNA was detected in 1861 and 4622 cells (Figure 5A), which also observed at the protein level (Figure 5B). Importantly, CD44 mRNA expression in human GBM subtypes was highest in the mesenchymal group, and was lowest in the proneural group (Figure 5C).
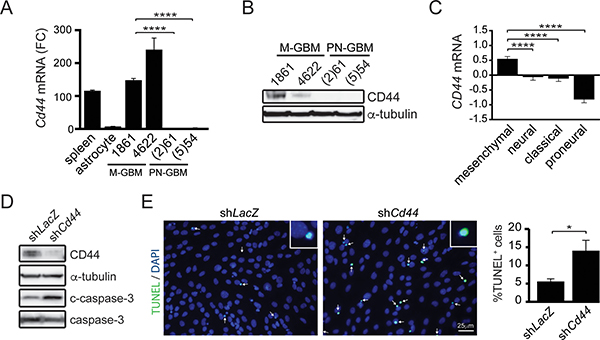
Figure 5: CD44 is required for M-GBM cell survival. (A) qPCR analysis of Cd44 mRNA in spleen (positive control), primary astrocytes, mesenchymal GBM (M-GBM) and proneural GBM (PN-GBM) cells reveals higher Cd44 mRNA levels in M-GBM cells relative to PN-GBM cells and primary astrocytes. ****p < 0.0001. FC, fold change. (B) CD44 protein is detected by Western blot in M-GBM, but not in PN-GBM, cells. (C) CD44 mRNA expression (Log2 Z-score) in TCGA GBM (provisional) samples segregated by molecular subgroup reveals that mesenchymal and proneural GBM express the highest and lowest CD44 levels, respectively. ****p < 0.0001. (D) Cd44 KD 1861 cells (shCd44) have increased cleaved caspase-3 (c-caspase-3) expression relative to control (shLacZ) cells. (E) Increased %TUNEL+ cells (inset) are observed following Cd44 KD (green; arrows). DAPI-stained nuclei are shown in blue. *p = 0.0288. Scale bar, 25 μm.
To determine whether CD44 was necessary for M-GBM cell survival, we reduced Cd44 expression in 1861 cells using shRNA. Following Cd44 KD, there was increased apoptosis (cleaved caspase-3 expression and %TUNEL+ cells) relative to controls (LacZ KD) (Figure 5D, 5E). In order to demonstrate that CD44 functions as the receptor for Ccl5 in 1861 cells, Cd44 KD cells were treated with mCcl5. Following Cd44 KD, mCcl5 could no longer increase 1861 cell growth (Figure 6A). Similarly, Ccl5 KD in Cd44 KD cells (Figure 6B) did not change cell growth (Figure 6C), establishing that the growth-promoting effects of Ccl5 require CD44 expression on M-GBM cells. In order to test whether other CD44 ligands function in a similar fashion as Ccl5, we reduced Cd44 using shRNA in Ccl5 KD cells (shCcl5 + shCd44), and compared cell proliferation to that observed in Ccl5 KD (shCcl5) and control (shLacZ) cells. It should be noted that the shLacZ and shCcl5 cells were further infected with virus carrying the shLacZ construct as an internal control for a second round of viral infection. We observed no further reduction of cell proliferation in Ccl5 KD cells following Cd44 knockdown (Figure 6D–6F), suggesting that this pro-survival pathway is specific to the interaction between Ccl5 and CD44, and is not regulated by other CD44 ligands. Taken together, these data support a model in which Nf1 loss in M-GBM cells leads to hyperactivation of the AKT/mTOR pathway, which positively regulates Ccl5 expression and increases M-GBM cell survival through CD44 signaling (Figure 6G).
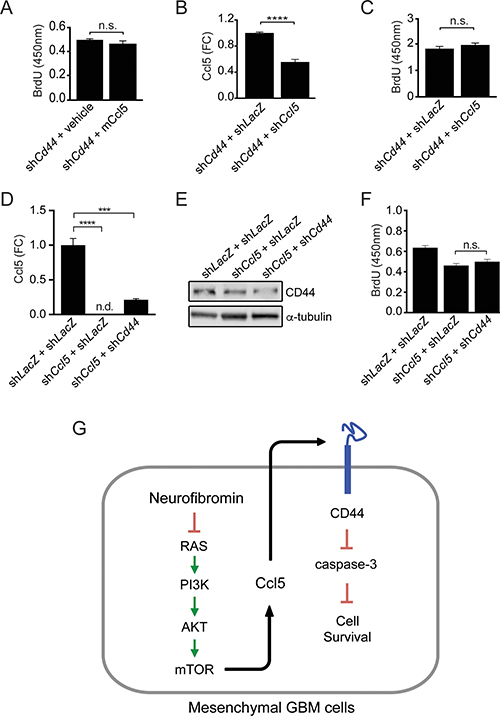
Figure 6: Ccl5 regulates M-GBM survival through CD44. (A) Cd44 KD 1861 cells treated with vehicle (0.5% BSA in PBS) or mouse Ccl5 (mCcl5) show no difference in BrdU incorporation. n.s., not significant. (B) ELISA analysis shows that shCd44 + shCcl5 cells have reduced levels of secreted Ccl5 relative to controls (shCd44 + shLacZ). ****p < 0.0001. FC, fold change. (C) Ccl5 KD in Cd44 KD cells did not change BrdU incorporation. n.s., not significant. (D) ELISA analysis shows that Ccl5 KD cells (shCcl5 + shLacZ and shCcl5 + shCd44) have reduced levels of secreted Ccl5 relative to control cells (shLacZ + shLacZ). ****p < 0.0001. ***p = 0.0002. FC, fold change. n.d., not detected. (E) Western blotting demonstrates that Cd44 KD cells (shCcl5 + shCd44) have less CD44 protein relative to other cells (shLacZ + shLacZ and shCcl5 + shLacZ). (F) Cd44 KD in Ccl5 KD cells did not change BrdU incorporation. n.s., not significant. (G) Proposed model for Ccl5 regulation of M-GBM cell survival.
DISCUSSION
Chemokines were originally described as key signaling molecules in cancer biology by virtue of their ability to recruit stromal cells (e.g., macrophages, endothelial cells) to the developing tumor [25, 26]. While this is one important function for these immune modulatory proteins, emerging evidence suggests that chemokines and cytokines may create autocrine growth regulatory circuits that facilitate tumor expansion. In this regard, CXCR2 signaling has been shown to regulate KRAS-induced pancreatic cancer growth [27], whereas IL-8 serves as an autocrine growth factor for malignant mesothelioma maintenance [28]. In the context of glioma, CXCL12/CXCL12-receptor (CXCR4/CXCR7) autoregulatory signaling has been demonstrated to be important for glioblastoma survival [8, 29, 30] and angiogenesis [31], while CX3CL1/CX3CR1 axis signaling in glioma cells controls glioma cell invasion [32].
The emergence of such autoregulatory chemokine/chemokine receptor loops relevant to glioma growth prompted us to examine a recently-identified paracrine signal (CCL5), produced by low-grade glioma-associated microglia [11], as a potential autocrine signaling molecule in high-grade glioma. While low-grade gliomas are dependent on growth-facilitatory signals from the tumor microenvironment, we hypothesize that their malignant counterparts achieve partial stromal independence by producing the very growth factors elaborated by non-neoplastic cells in low-grade gliomas. In this study, we demonstrate that M-GBM cells express high levels of CCL5 that provides an auto-stimulatory signal to increase cell survival. Similarly, in other studies, M-GBM cells express high levels of CXCL12, which forms another autoregulatory loop with its receptor CXCR4/CXCR7, to increase GBM cell proliferation [8, 29, 30]. In NF1-associated low-grade gliomas, CXCL12 also serves as a paracrine factor that drives the growth of neoplastic Nf1−/− astrocytes [33]. These findings support the idea that high-grade glioma cells achieve certain degrees of stromal independence by producing growth factors previously provided by non-neoplastic stromal cells in the low-grade glioma tumor microenvironment.
CCL5 is a potent chemokine operative in several types of cancers, including lymphoma, melanoma, prostate cancer and breast cancer [34]. However, while CCL5 function in glioma has not been fully elucidated, several reports have suggested that it might be an important chemokine in this setting. First, one of the main CCL5 receptors, CCR5, is expressed in U87 and U251 glioblastoma cell lines, such that its knockdown inhibits U87 growth in nude mice [35]. Second, CCL5 mRNA and protein expression are elevated in both tumor tissue and serum from patients with high-grade glioma [36]. Lastly, CCL5 is produced by microglia in the microenvironment of murine Nf1 optic glioma, and is increased in low-grade gliomas (pilocytic astrocytoma) relative to non-neoplastic tissue [11]. In this latter study, CCL5 treatment increased optic nerve glial proliferation in vitro, whereas antibody-mediated CCL5 inhibition reduced optic glioma growth in vivo.
CCL5 typically functions by binding to its receptors CCR1, CCR3 and/or CCR5. Surprisingly these receptors were not detectable in M-GBM cells. Instead, M-GBM cells express the non-conventional CCL5 receptor, CD44, and use this receptor to maintain tumor cell survival. CD44 is a cell surface glycoprotein that can function as a co-receptor for extracellular matrix components, growth factors and chemokines. CD44 plays several critical roles in GBM and drives GBM cell invasion, proliferation and drug resistance. CD44 expression is increased in GBM compared to low-grade gliomas and high levels of CD44 are associated with worse survival of GBM patients [37]. Inhibiting CD44 function using monoclonal antibodies or RNA silencing reduced GBM growth in vivo and prolonged mouse survival [38, 39]. The interaction between CD44 and its ligands (e.g., hyaluronic acid and osteopontin) modulates multiple aspects of glioma biology. Activation of CD44 by hyaluronic acid was shown to increase glioma cell invasion [40–43]. In addition, osteopontin-CD44 interaction induces cleavage of the intracellular domain of CD44, increases a stem cell phenotype, and increases the growth of PN-GBM cells [44]. In this study, we found that the interaction between Ccl5 and CD44 enhances cell survival in M-GBM cells. This pro-survival effect is specific to Ccl5, and does not appear to involve other CD44 ligands, since Cd44 knockdown in Ccl5 KD cells does not further reduce cell growth. Our study expands our understanding of the potential mechanisms by which CD44 regulates GBM maintenance.
CCL5 employs CD44 as an alternative receptor to facilitate the entry of HIV virus [24], where it forms a complex with CD44 and Src kinases to activate ERK signaling. In our studies, ERK activity in M-GBM cells was not reduced following Ccl5 KD (data not shown), suggesting that a distinct signaling pathway is activated following Ccl5 binding to CD44 in M-GBM cells. Previous studies have demonstrated increased cell survival following CD44 activation in several different cancers. For example, CD44 protects chronic lymphocytic leukemia cells from spontaneous and induced cell death by increasing expression of the MCL-1 pro-survival protein [45], while CD44 activation in breast cancer cells induces c-Jun mediated survival [46]. It has been suggested in GBM that CD44 also inhibits the Hippo-mediated apoptotic signaling pathway to achieve resistance to reactive oxygen species- or cytotoxic agent-induced stress [39]. Further work will be required to determine how CD44 regulates cell survival in M-GBM cells.
GBM tumors are genetically heterogeneous and can be divided into mesenchymal, classical, neural and proneural subtypes. Herein, we focused on studying the behavior of mesenchymal GBM since Ccl5 has the highest expression in this group. One common feature of the M-GBM subtype is NF1 loss/mutation, which results in impaired neurofibromin RAS suppression. Consistent with this observation, we demonstrated that neurofibromin directly regulates CCL5 expression through RAS signaling. First, we show that ectopic expression of a functional, but not mutant, NF1-GAP-related domain reduced Ccl5 expression. Second, Nf1 loss in primary astrocytes resulted in increased Ccl5 expression. Third, pharmacological inhibition of RAS downstream effector activation (AKT/mTOR) suppressed Ccl5 expression in M-GBM cells. Collectively, these results elucidate one mechanism by which Ccl5 expression is controlled by RAS.
The Ccl5-mediated autocrine circuit identified in this study may not apply to other GBM subtypes, given that they do not express high levels of either CCL5 or CD44. In fact, we have shown that treating murine PN-GBM cells with Ccl5 did not increase cell growth. While multiple signaling pathways contribute to GBM tumorigenesis, we report one in which a chemokine-driven autocrine pathway supports GBM cell survival. The regulation of chemokines by oncogenic signaling pathways suggests that other brain cancer-associated mutations might similarly modulate chemokine production. Further dissection of the chemokine/cytokine networks regulated by these tumor suppressor/oncogenic pathways might uncover unique immune modulatory programs with differential impact not only on tumor cell growth, but also on the stromal cell types in the tumor microenvironment.
Lastly, while Ccl5 silencing results in ~20% reduction in cell growth in vitro, there was a ~70% reduction in tumor cell growth following Ccl5 KD in vivo. The greater cell survival benefit in vivo raises the possibility that CCL5 may not solely function in an autocrine manner, but may also recruit monocytes (macrophages/microglia) to the tumor and enhance tumor growth in a paracrine manner. However, we found no difference in the percent of intratumoral Iba1+ monocytes in mice bearing Ccl5 KD versus control (LacZ KD) M-GBM cells (data not shown). Since it is possible that attenuated Ccl5 expression alters the functional status of tumor-associated macrophages/microglia, current studies are focused on identifying potential changes in the tumor microenvironment conferred by reduced glioblastoma tumor cell Ccl5 production in vivo.
In summary, we demonstrate, for the first time, that CCL5 functions in an autocrine growth-promoting circuit, and establish a new receptor responsible for CCL5 function in mesenchymal glioblastoma cells. We further show that key oncogenic signaling pathways, like RAS, operate to modulate chemokine expression. As such, these studies expand the role of chemokines in dictating glioma biology, and suggest that paracrine growth regulatory signals produced by non-neoplastic cells in the low-grade glioma microenvironment may be converted to autocrine tumor maintenance circuits in high-grade malignancies relevant to mesenchymal glioblastoma survival in vitro and in vivo.
MATERIALS AND METHODS
Human CCL5 and CD44 expression analysis
Expression values for CD44 and CCL5 (Glioblastoma, TCGA Provisional, mRNA Expression z-Scores) were obtained from the MSKCC computational biology cancer genomics portal (http://www.cbioportal.org), which contains annotated TCGA data [47, 48]. Tumor subtype classification was performed as previously described [15].
Mice
4-week-old male C57BL/6 mice were purchased from Taconic, and used in accordance with an approved Animal Studies Committee protocol at Washington University.
Cell culture
Mouse mesenchymal glioblastoma cells (1861 and 4622) were maintained as previously described [49], while mouse proneural glioblastoma cells ((2)61 and (5)54)) were derived from PDGFB-driven tumor-bearing mice [18] and grown in mouse neural stem cell medium (STEMCELL Technologies, 05700 and 05701), supplemented with 20 ng/ml hEGF (Sigma-Aldrich, E9644), 10 ng/ml hFGF (R&D systems, 233-FB-025) and 2 μg/ml heparin (STEMCELL Technologies, 07980). Wild-type and Nf1−/− primary astrocytes were generated and maintained as previously described [14, 50, 51]. Before performing qPCR, ELISA or Western blotting, all cells were cultured under the same condition for at least 24 hours.
Lentiviral infection
pLKO.1 plasmids with shRNAs targeting LacZ, Ccl5 (shCcl5-1 and shCcl5-2), and Cd44 were obtained from the Genome Institute at Washington University, while shRaptor-1 and shRaptor-2 plasmids were purchased (Addgene, 21339 and 21340) (Supplementary Table 1). shRNAs were prepared and cells infected as previously reported [49]. MSCV-NF1-GRD-WT and mutant control constructs [19] were transfected into 1861 cells using X-tremeGENE 9 DNA transfection reagents (Roche Life Science, XTG9-RO) according the manufacturer’s protocol.
Pharmacologic treatments
1861 cells were treated with 0.4 μM ZSTK474 (Selleckchem, S1072), 1 μM MK2206 (Selleckchem, S1078), 5 nM rapamycin (Selleckchem, S1039), 1 nM PD-0325901 (PD901; Selleckchem, S1036) or DSMO (vehicle control, Sigma-Aldrich, D8418) for 4 hours at 37°C.
BrdU incorporation
Cells were plated in 96-well plates, and treated with 250 ng/mL mCcl5 (R&D systems, 478-MR-025) or 0.5 % BSA in PBS (vehicle control) for 4 hours at 37°C. BrdU was added to the cell culture 2 hours before fixing the cells and performing the BrdU ELISA assay (Roche Life Science, 11647229001) according the manufacturer’s protocol.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)
Cells maintained in 0.5% fetal bovine serum for 24 hours were fixed with 4% PFA, and TUNEL+ cells detected using the fluorescence-based in situ cell death detection kit (Roche Life Science, 11684795910). The number of TUNEL+ cells was normalized to the total cell number (DAPI+ nuclei) using NIH ImageJ software.
Western blotting
1861 cells expressing shRNAs were maintained in 0.5% fetal bovine serum for 24 hours prior to lysis and Western blotting with appropriate primary antibodies (Supplementary Table 2) [14, 52]. Quantification of western blots were performed using ImageStudio (LI-COR). AKT and S6 activities were quantified as signal intensity of phospho-proteins versus signal intensity of total proteins.
Mouse chemokine enzyme-linked immunosorbent assay (ELISA)
Cells were serum starved for 24 hours, and the culture medium collected for mouse Ccl5 ELISA (R&D systems, MMR00). The amount of secreted Ccl5 determined by ELISA was normalized to total cell number.
Quantitative real-time PCR
qPCR was performed as described previously [11] using validated primers (Supplementary Table 3).
Intracranial injections
5 × 104 1861 cells were implanted into the striatum of 4-week-old C57BL/6 mice as previously described [49]. Mice were monitored daily, and euthanized when moribund. Kaplan-Meier survival curves were generated, and differences between control (shLacZ) and Ccl5 KD (shCcl5-1 and shCcl5-2) groups analyzed using the Log-rank test. For mCcl5 ELISA and Western blotting of the implant, tissue around the tumor implantation site was dissected 6-weeks post-implantation, followed by lysing the tissue as described previously [14, 52].
Immunohistochemistry
Implanted mouse brains were fixed at the time of death or deemed moribund by the veterinary staff, embedded in paraffin and processed as described previously [53], followed by performing immunohistochemistry using primary and secondary antibodies (Supplementary Table 2). Ccl5 signal was amplified using tyramide signal amplification as described previously [11].
Statistical analyses
Data analysis was performed using Prism GraphPad. Unpaired two-tailed Student’s t tests were used to determine the difference between two groups. All data were presented as mean values with standard error of mean (SEM). All in vitro experiments were repeated at least three times with similar results.
ACKNOWLEDGMENTS
We thank the Broad Institute RNAi Consortium (TRC), and the Hope Center Viral Vectors Core (Washington University in St. Louis) for virus preparation. We thank Courtney Corman for expert technical assistance.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work was supported by funding from the National Cancer Institute (U01-CA160882-01 to D.H. and D.H.G.). Y.P. was supported by a McDonnell Center for Cellular and Molecular Neurobiology Postdoctoral Fellowship.
REFERENCES
1. Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol. 2012; 107:359–364.
2. Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2015; 17:iv1–iv62.
3. Friedman HS, Kerby T, Calvert H. Temozolomide and treatment of malignant glioma. Clin Cancer Res. 2000; 6:2585–2597.
4. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2015; 19:20–27.
5. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, Kanojia D, Pituch KC, Qiao J, Pytel P, Han Y, Wu M, Zhang L, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 2016.
6. Zhang J, Sarkar S, Cua R, Zhou Y, Hader W, Yong VW. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis. 2012; 33:312–319.
7. Ehtesham M, Winston JA, Kabos P, Thompson RC. CXCR4 expression mediates glioma cell invasiveness. Oncogene. 2006; 25:2801–2806.
8. Gatti M, Pattarozzi A, Bajetto A, Wurth R, Daga A, Fiaschi P, Zona G, Florio T, Barbieri F. Inhibition of CXCL12/CXCR4 autocrine/paracrine loop reduces viability of human glioblastoma stem-like cells affecting self-renewal activity. Toxicology. 2013; 314:209–220.
9. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013; 19:1264–1272.
10. Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, Holland EC, Sutton JC, Joyce JA. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. 2016; 352:aad3018.
11. Solga AC, Pong WW, Kim KY, Cimino PJ, Toonen JA, Walker J, Wylie T, Magrini V, Griffith M, Griffith OL, Ly A, Ellisman MH, Mardis ER, et al. RNA Sequencing of Tumor-Associated Microglia Reveals Ccl5 as a Stromal Chemokine Critical for Neurofibromatosis-1 Glioma Growth. Neoplasia. 2015; 17:776–788.
12. Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S, Moody SE, Shen RR, Schinzel AC, Thai TC, Reibel JB, Tamayo P, Godfrey JT, et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014; 4:452–465.
13. Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992; 356:713–715.
14. Dasgupta B, Li W, Perry A, Gutmann DH. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. Cancer Res. 2005; 65:236–245.
15. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010; 17:98–110.
16. Gravendeel LA, Kouwenhoven MC, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE, Daemen A, Bleeker FE, Bralten LB, Kloosterhof NK, De Moor B, Eilers PH, van der Spek PJ, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009; 69:9065–9072.
17. Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000; 26:109–113.
18. Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling Adult Gliomas Using RCAS/t-va Technology. Transl Oncol. 2009; 2:89–95.
19. Hiatt KK, Ingram DA, Zhang Y, Bollag G, Clapp DW. Neurofibromin GTPase-activating protein-related domains restore normal growth in Nf1−/− cells. J Biol Chem. 2001; 276:7240–7245.
20. Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci USA. 2005; 102:8573–8578.
21. Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005; 65:2755–2760.
22. Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME, Eaves D, Widemann B, Kim MO, Dombi E, Sabo J, Hardiman Dudley A, Niwa-Kawakita M, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013; 123:340–347.
23. Liu B, Hassan Z, Amisten S, King AJ, Bowe JE, Huang GC, Jones PM, Persaud SJ. The novel chemokine receptor, G-protein-coupled receptor 75, is expressed by islets and is coupled to stimulation of insulin secretion and improved glucose homeostasis. Diabetologia. 2013; 56:2467–2476.
24. Roscic-Mrkic B, Fischer M, Leemann C, Manrique A, Gordon CJ, Moore JP, Proudfoot AE, Trkola A. RANTES (CCL5) uses the proteoglycan CD44 as an auxiliary receptor to mediate cellular activation signals and HIV-1 enhancement. Blood. 2003; 102:1169–1177.
25. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004; 4:540–550.
26. Slettenaar VI, Wilson JL. The chemokine network: a target in cancer biology? Adv Drug Deliv Rev. 2006; 58:962–974.
27. Purohit A, Varney M, Rachagani S, Ouellette MM, Batra SK, Singh RK. CXCR2 signaling regulates KRAS(G(2)D)-induced autocrine growth of pancreatic cancer. Oncotarget. 2016; 7:7280–7296. doi: 10.18632/oncotarget.6906.
28. Galffy G, Mohammed KA, Dowling PA, Nasreen N, Ward MJ, Antony VB. Interleukin 8: an autocrine growth factor for malignant mesothelioma. Cancer Res. 1999; 59:367–371.
29. Zhou Y, Larsen PH, Hao C, Yong VW. CXCR4 is a major chemokine receptor on glioma cells and mediates their survival. J Biol Chem. 2002; 277:49481–49487.
30. Hattermann K, Held-Feindt J, Lucius R, Muerkoster SS, Penfold ME, Schall TJ, Mentlein R. The chemokine receptor CXCR7 is highly expressed in human glioma cells and mediates antiapoptotic effects. Cancer Res. 2010; 70:3299–3308.
31. Ping YF, Yao XH, Jiang JY, Zhao LT, Yu SC, Jiang T, Lin MC, Chen JH, Wang B, Zhang R, Cui YH, Qian C, Wang J, et al. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J Pathol. 2011; 224:344–354.
32. Sciume G, Soriani A, Piccoli M, Frati L, Santoni A, Bernardini G. CX3CR1/CX3CL1 axis negatively controls glioma cell invasion and is modulated by transforming growth factor-beta1. Neuro Oncol. 2010; 12:701–710.
33. Warrington NM, Woerner BM, Daginakatte GC, Dasgupta B, Perry A, Gutmann DH, Rubin JB. Spatiotemporal differences in CXCL12 expression and cyclic AMP underlie the unique pattern of optic glioma growth in neurofibromatosis type 1. Cancer Res. 2007; 67:8588–8595.
34. Aldinucci D, Colombatti A. The inflammatory chemokine CCL5 and cancer progression. Mediators Inflamm. 2014; 2014:292376.
35. Zhao L, Wang Y, Xue Y, Lv W, Zhang Y, He S. Critical roles of chemokine receptor CCR5 in regulating glioblastoma proliferation and invasion. Acta Biochim Biophys Sin (Shanghai). 2015; 47:890–898.
36. Moogooei M, Shamaei M, Khorramdelazad H, Fattahpour S, Seyedmehdi SM, Moogooei M, Hassanshahi G, Kalantari Khandani B. The Intricate Expression of CC Chemokines in Glial Tumors: Evidence for Involvement of CCL2 and CCL5 but Not CCL11. Acta Med Iran. 2015; 53:770–777.
37. Jijiwa M, Demir H, Gupta S, Leung C, Joshi K, Orozco N, Huang T, Yildiz VO, Shibahara I, de Jesus JA, Yong WH, Mischel PS, Fernandez S, et al. CD44v6 regulates growth of brain tumor stem cells partially through the AKT-mediated pathway. PLoS One. 2011; 6:e24217.
38. Breyer R, Hussein S, Radu DL, Putz KM, Gunia S, Hecker H, Samii M, Walter GF, Stan AC. Disruption of intracerebral progression of C6 rat glioblastoma by in vivo treatment with anti-CD44 monoclonal antibody. J Neurosurg. 2000; 92:140–149.
39. Xu Y, Stamenkovic I, Yu Q. CD44 attenuates activation of the hippo signaling pathway and is a prime therapeutic target for glioblastoma. Cancer Res. 2010; 70:2455–2464.
40. Merzak A, Koocheckpour S, Pilkington GJ. CD44 mediates human glioma cell adhesion and invasion in vitro. Cancer Res. 1994; 54:3988–3992.
41. Monaghan M, Mulligan KA, Gillespie H, Trimble A, Winter P, Johnston PG, McCormick D. Epidermal growth factor up-regulates CD44-dependent astrocytoma invasion in vitro. J Pathol. 2000; 192:519–525.
42. Radotra B, McCormick D. Glioma invasion in vitro is mediated by CD44-hyaluronan interactions. J Pathol. 1997; 181:434–438.
43. Radotra B, McCormick D. CD44 is involved in migration but not spreading of astrocytoma cells in vitro. Anticancer Res. 1997; 17:945–949.
44. Pietras A, Katz AM, Ekstrom EJ, Wee B, Halliday JJ, Pitter KL, Werbeck JL, Amankulor NM, Huse JT, Holland EC. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell. 2014; 14:357–369.
45. Herishanu Y, Gibellini F, Njuguna N, Hazan-Halevy I, Farooqui M, Bern S, Keyvanfar K, Lee E, Wilson W, Wiestner A. Activation of CD44, a receptor for extracellular matrix components, protects chronic lymphocytic leukemia cells from spontaneous and drug induced apoptosis through MCL-1. Leuk Lymphoma. 2011; 52:1758–1769.
46. Chen L, Bourguignon LY. Hyaluronan-CD44 interaction promotes c-Jun signaling and miRNA21 expression leading to Bcl-2 expression and chemoresistance in breast cancer cells. Mol Cancer. 2014; 13:52.
47. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404.
48. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:l1.
49. Chen YH, Cimino PJ, Luo J, Dahiya S, Gutmann DH. ABCG1 maintains high-grade glioma survival in vitro and in vivo. Oncotarget. 2016; 7:23416–24. doi: 10.18632/oncotarget.8030.
50. Sandsmark DK, Zhang H, Hegedus B, Pelletier CL, Weber JD, Gutmann DH. Nucleophosmin mediates mammalian target of rapamycin-dependent actin cytoskeleton dynamics and proliferation in neurofibromin-deficient astrocytes. Cancer Res. 2007; 67:4790–4799.
51. Smithson LJ, Gutmann DH. Proteomic analysis reveals GIT1 as a novel mTOR complex component critical for mediating astrocyte survival. Genes Dev. 2016; 30:1383–1388.
52. Apicelli AJ, Uhlmann EJ, Baldwin RL, Ding H, Nagy A, Guha A, Gutmann DH. Role of the Rap1 GTPase in astrocyte growth regulation. Glia. 2003; 42:225–234.
53. Dasgupta B, Gutmann DH. Neurofibromin regulates neural stem cell proliferation, survival, and astroglial differentiation in vitro and in vivo. J Neurosci. 2005; 25:5584–5594.