
INTRODUCTION
Breast cancer remains the most common female malignancy, despite progress in diagnostic techniques and multimodality therapy [1, 2]. Oncogene activation and tumor suppressor gene (TSG) inactivation are major molecular events responsible for transforming normal mammary epithelia into tumorous epithelia [3–5]. Increasing evidences suggest that suppression of candidate TSG expression in many cancers, including breast cancer, is mainly the result of promoter CpG methylation [6–8]. It is therefore crucial to understand the epigenetic inactivation of novel TSGs in breast cancers.
Interferon (IFN) regulatory factor 8 (IRF8, also named IFN consensus sequence binding protein, ICSBP) is an IRF family transcription factor located at chromosomal region 16q24.1 [9]. IRF8 has been validated as a downstream target of the IFN-γ/signal transducer and activator of transcription 1 (STAT1) signaling pathway [10]. We previously showed that IRF8 expression was inversely correlated with hypermethylation of its promoter region in various cancer cell types, including breast cancer cells [11]. Moreover, the promoter of IRF8 has been shown to be methylated in gastric, colon, and lung carcinomas, and in myelogenous leukemia and multiple myeloma [12–16]. Furthermore, disruption of IRF8 function reduced tumor cell sensitivity to apoptosis and increased their metastatic potential, indicating its role as a TSG in multiple cancers [10, 17]. Although some evidence suggests that IRF8 enhanced cell proliferation, motility and invasion via TGF-β signaling [18, 19], its function and underlying mechanism in breast cancer remain unclear.
Wnt/β-catenin signaling pathways are involved in cell proliferation, stem cell characteristics, migration, and metastasis, and are activated in numerous cancers, including breast cancers [3, 20, 21]. Several studies have shown a negative correlation between IRF8 and β-catenin signaling in myeloid cells and leukemia [22, 23], indicating that IRF8 may at least partly suppress β-catenin signaling in solid tumors.
This study showed that the IRF8 promoter was frequently hypermethylated in primary breast cancers, and IRF8 served as a main downstream factor in the IFN-γ/STAT1 signaling pathway, with a possible role in enhancing the anti-tumor effect of IFN-γ. Furthermore, IRF8 acted as a candidate TSG in breast cancer, at least partly by suppressing the β-catenin signaling pathway. These results indicate that IRF8 serves as a candidate TSG that is frequently hypermethylated in breast cancers.
RESULTS
IRF8 is an independent prognostic factor for breast cancer
To investigate the expression of IRF8 in molecular subtypes of breast cancer, we analyzed expression data for IRF8 in The Cancer Genome Atlas 2012 (TCGA) breast cancer database using the online cBioPortal (http://www.cbioportal.org/) [24, 25], classified according to estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER-2) statuses, mutation and expression of 50 genes (PAM50) (Figure 1A). There was no significant difference in IRF8 expression between triple-negative breast cancers (TNBCs) (n = 379) and non-TNBCs (n = 42) in the 2015 TCGA breast cancer database (p > 0.05) (Figure 1B). We also analyzed the expression of IRF8 using online Oncomine software (https://www.oncomine.org/), and showed that IRF8 was downregulated in invasive ductal breast cancers (n = 389) compared with normal breast tissues (n = 61) (p = 0.017) (Figure 1C). IRF8 has been shown to be a downstream target gene of IFNγ–STAT1 signaling [10], and we identified a negative correlation between STAT1 and IRF8 expression using bc-GenExMiner v4.0 online software (n = 5474, r = 0.45, p < 0.0001) (Figure 1D) [26, 27]. Importantly, high expression of IRF8 was associated with relatively high distant-metastasis-free survival (DMFS) (p = 0.03001) and overall survival (OS) (p = 0.03223) in breast cancers, especially in ER-negative tumors (DMFS, p = 0.00394, OS, p = 0.02847) and grade 3 tumors (DMFS, p = 0.01267, OS, p = 0.00975) (Figure 1E). These results suggest that IRF8 may be an independent prognostic factor in breast cancer, especially in patients with ER-negative tumors.
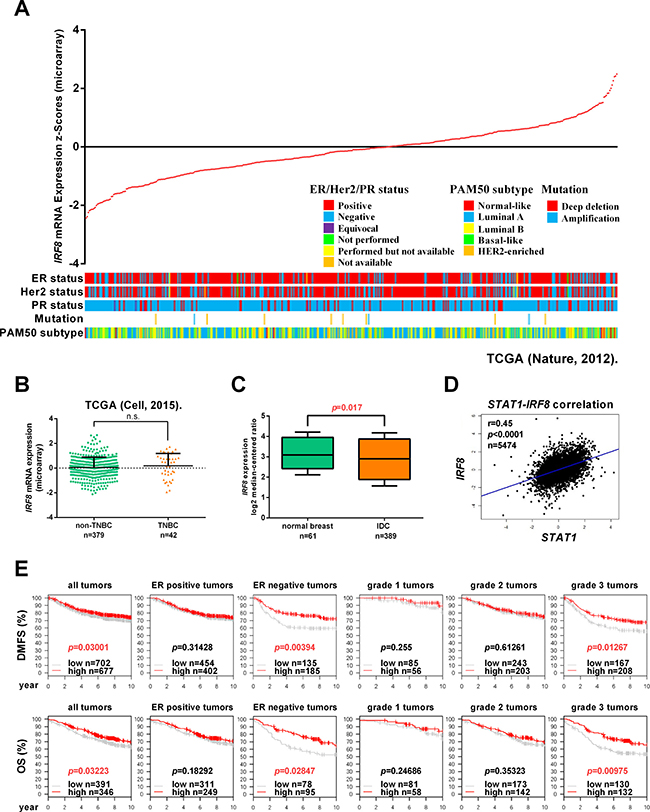
Figure 1: IRF8 is an independent prognostic factor for breast cancer. (A) Information on IRF8 expression was extracted from The Cancer Genome Atlas (TCGA) breast cancer database using cBioPortal online software. Tumors were arranged in ascending order of IRF8 expression in relation to ER status, PR status, HER2 status, mutation, and PAM50 classification. (B) IRF8 expression was compared between triple-negative breast cancers (TNBCs) (n = 379) and non-TNBCs (n = 42) in the TCGA breast cancer database (p > 0.05). (C) IRF8 expression was compared between normal breast tissues (n = 61) and invasive ductal carcinomas (IDC) (n = 389) in the TCGA breast cancer database using Oncomine online software (p = 0.017). (D) The correlation between STAT1 and IRF8 expression was analyzed using Breast Cancer Gene-Expression Miner v4.0 (bc-GenExMiner v4.0) (r = 0.45, p < 0.0001, n = 5474). (E) Distant-metastasis-free survival (DMFS) was compared between high- and low-IRF8-expressing breast tumors (p = 0.03001) for ER-positive tumors (p = 0.31428), ER-negative tumors (p = 0.00394), grade 1 tumors (p = 0.255), grade 2 tumors (p = 0.61261), and grade 3 tumors (p = 0.01267). Overall survival (OS) was compared between high- and low-IRF8-expressing breast tumors (p = 0.03223) for ER-positive tumors (p = 0.18292), ER-negative tumors (p = 0.02847), grade 1 tumors (p = 0.24686), grade 2 tumors (p = 0.35323), and grade 3 tumors (p = 0.00975).
Promoter methylation contributes to IRF8 downregulation in breast cancer cells
Expression of IRF8 has been shown to be silenced or downregulated due to promoter hypermethylation in multiple cancers, including breast cancers [11]. We evaluated IRF8 expression in a panel of breast cancer cell lines and three normal breast tissue samples by RT-PCR. IRF8 expression was silenced in BT549, MDA-MB-231, and T47D cells, but not in SK-BR-3 cells or normal breast tissues (Figure 2A). In addition, IRF8 expression was downregulated in primary breast cancers according to real-time PCR (n = 12, p = 0.0241), compared with adjacent non-cancerous tissues (n = 7) (Figure 2B).
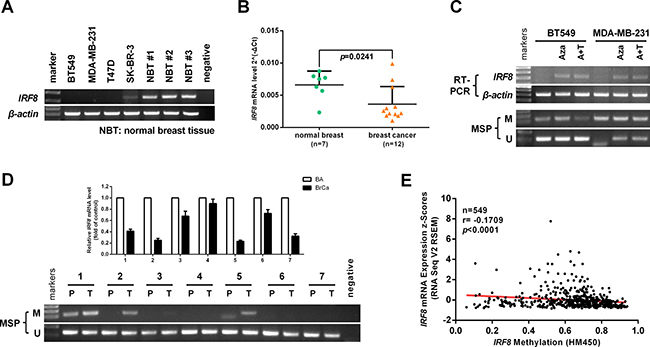
Figure 2: Promoter methylation contributes to IRF8 downregulation in breast cancer cells. (A) IRF8 expression in a panel of human breast cancer cell lines and three normal breast tissue samples was detected by RT-PCR. β-actin was used as a control. (B) Expression of IRF8 in normal breast tissues (n = 7) and breast cancer tissues (n = 12) detected by real-time PCR, with β-actin as a control. Mean ± SD, p = 0.0241. (C) The expression and methylation status of IRF8 were measured in BT549 and MDA-MB-231 cells treated with 5-aza-2′-deoxycytidine (Aza) with or without trichostatin A (TSA) by RT-PCR and methylation-specific PCR (MSP). M: methylated, U: unmethylated. (D) Expression and methylation status of IRF8 in 7 paired breast cancers and matched non-cancerous breast tissues were detected by real-time PCR and MSP, respectively. (E) The correlation between IRF8 methylation and expression in the TCGA breast cancer database was analyzed using cBioPortal online software (n = 549, r = –0.1709, p < 0.0001).
Furthermore, IRF8 expression was reactivated by treatment with 5-aza-2′-deoxycytidine (Aza) with or without the histone deacetylase inhibitor TSA in BT549 and MDA-MB-231 cells, accompanied by decreased methylation of IRF8 (Figure 2C). We also evaluated the expression and promoter methylation of IRF8 in 7 paired breast cancer tissues and matched non-cancerous adjacent tissues. 42.9% (3/7) tumors had IRF8 promoter methylation accompanied by decreased expression of IRF8 expression (Figure 2D). Similarly, there was a negative correlation between IRF8 promoter methylation and its expression in TCGA breast cancer database (n = 549, r = –0.1709, p < 0.0001) (Figure 2E).
These results indicated that IRF8 was downregulated mainly as a result of promoter methylation in breast cancers.
IRF8 is frequently methylated in primary breast cancer
It has been demonstrated that IRF8 is frequently methylated in breast cancers [11]. We therefore determined the IRF8 promoter methylation status by MSP and analyzed the correlation between methylation status and clinicopathological features in primary breast cancer samples (n = 114) and surgical-margin tissues (n = 12). Among these patients, the IRF8 promoter was hypermethylated in 49.12% (56/114) of breast cancer tissues (Figure 3A and Table 1) and 16.67% (2/12) of surgical-margin tissues (Figure 3B and Table 1). However, no correlations were found between IRF8 promoter methylation and age, tumor size, grade, lymph node metastasis, distant metastasis, ER, PR, or HER-2 (Table 2). Overall, these results demonstrated that the IRF8 promoter was frequently hypermethylated in breast cancers, but this correlation needs further confirmation in a larger sample group.
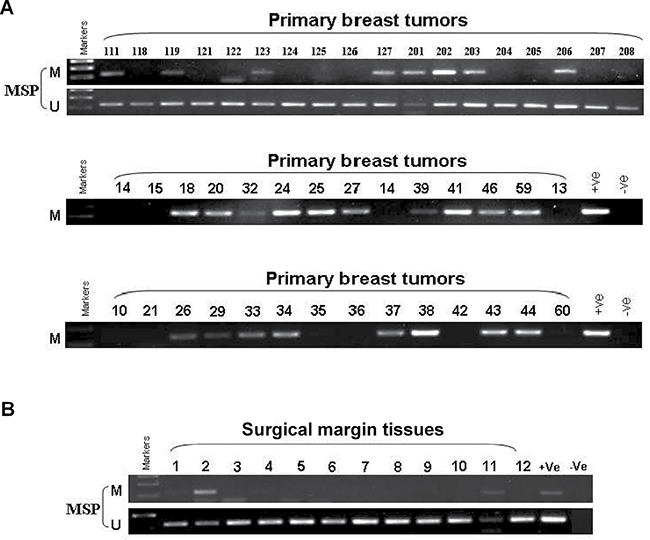
Figure 3: Promoter methylation status of IRF8 in primary breast cancers. Representative analysis of IRF8 promoter hypermethylation in (A) primary breast cancer tissues (n = 114) and (B) surgical-margin tissues (n = 12) in breast cancer patients. M, methylated; U, unmethylated.
Table 1: The promoter methylation status of IRF8 in primary breast cancer-associated tissues
Samples |
IRF8 methylation status |
Frequency of methylation |
|
|---|---|---|---|
Methylated |
Unmethylated |
||
BrCa (n = 114) |
56 |
58 |
49.12% (56/114) |
BA (n = 12) |
2 |
10 |
16.67% (2/12) |
BrCa, Breast Cancer.
BA, Breast Cancer Adjacent Tissue.
Table 2: The correlation between IRF8 promoter methylation and clinicopathological feature in breast cancers
Clinicopathological |
Numbers |
IRF8 methylation status |
P value |
||
|---|---|---|---|---|---|
Methylated |
Unmethylated |
||||
Age |
≤ 40 |
15 |
6 |
9 |
0.720 |
> 40 |
88 |
44 |
44 |
||
unknown |
11 |
6 |
5 |
||
Tumor grade |
I |
10 |
4 |
6 |
0.629 |
II |
73 |
39 |
34 |
||
III |
6 |
2 |
4 |
||
unknown |
25 |
11 |
14 |
||
Tumor size |
≤ 2.0 cm |
35 |
12 |
23 |
0.079 |
> 2.0 cm ≤ 5.0 cm |
61 |
36 |
25 |
||
> 5.0 cm |
7 |
2 |
5 |
||
unknown |
11 |
6 |
5 |
||
Lymph node metastasis |
Positive |
48 |
23 |
25 |
0.796 |
Negative |
54 |
26 |
28 |
||
unknown |
12 |
7 |
5 |
||
ER status |
Positive |
55 |
31 |
24 |
0.460 |
Negative |
35 |
17 |
18 |
||
unknown |
24 |
10 |
14 |
||
PR status |
Positive |
41 |
22 |
19 |
0.647 |
Negative |
49 |
24 |
25 |
||
unknown |
24 |
10 |
14 |
||
HER2 status |
Positive |
12 |
6 |
6 |
0.777 |
++ |
41 |
21 |
20 |
||
Negative |
36 |
19 |
17 |
||
unknown |
25 |
10 |
15 |
||
ER, Estrogen Receptor; PR, Progesterone Receptor; HER2, Human Epidermal Growth Factor 2.
Using the χ2 tests and Fisher’s exact teast.
IRF8 suppresses cell proliferation in vitro and in vivo
Inactivation of TSGs, including by promoter methylation, is often associated with breast-tumor progression [6]. Several studies have indicated that IRF8 acts as a functional tumor suppressor by inhibiting cell proliferation [11, 16, 28]. Furthermore, the negative correlation between IRF8 promoter methylation and its transcriptional level also suggests that IRF8 might act as a tumor suppressor suppressing promoter methylation in breast cancer. To test this hypothesis, we performed colony-formation assays using MDA-MB-231, T47D, and BT549 cells, CCK-8 assays using MDA-MB-231 and T47D cells, and EdU incorporation using MDA-MB-231 cells. IRF8 expression was completely silenced by promoter methylation in all these cells [11]. As expected, colony formation (Figure 4A), cell viability (Figure 4B), and EdU incorporation (Figure 4C) were significantly suppressed in IRF8-expressed cells compared with control cells.
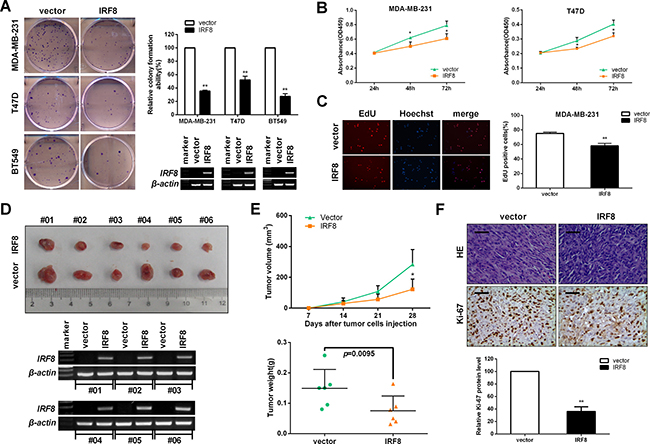
Figure 4: IRF8 suppresses cell proliferation in vitro and in vivo. Cells were transfected with vector and IRF8 plasmid, and selected with neomycin after 48 h. IRF8 expression was determined by RT-PCR. Cell proliferation was measured by (A) colony-formation assay, (B) CCK-8 assay at 24, 48, and 72 h, and (C) EdU incorporation assay. Data were based on three independent assays, and representative images are shown. Mean ± SD, *p < 0.05, **p < 0.01. (D) Tumors derived from vector- and IRF8-expressing MDA-MB-231 cells in mice, each group has 6 mice, and expression of IRF8 validated by RT-PCR. (E) Tumor volume and tumor weight in vector- and IRF8-expressing xenografts in MDA-MB-231 cells. Mean ± SD, *p < 0.05. (F) Hematoxylin and eosin and Ki-67 staining of IRF8-expressing tumors compared with vector-containing tumors. Data were based on three independent assays, and representative images are shown. Mean ± SD, **p < 0.01.
We further tested the anti-proliferation effect of IRF8 in vivo, in tumor-bearing nude mice with MDA-MB-231 xenografts (Figure 4D). Compared with the control (pcDNA3.1) group (n = 6), tumor volume and tumor weight were suppressed in the IRF8-expression group (n = 6) (Figure 4E), accompanied by inhibition of proliferation-related Ki-67 antigen (Figure 4F). These results indicated that IRF8 suppressed cell proliferation of breast cancer in vitro and in vivo.
IRF8 induced G2/M cell cycle arrest and apoptosis
Cell cycle arrest and apoptosis contribute to the inhibition of tumor cell proliferation. Based on existing data in renal cancer [28], we hypothesized that IRF8 might suppress cell growth by modulating the cell cycle and apoptosis. We tested this hypothesis by flow cytometry analysis to determine the cell cycle and apoptosis in vector- or IRF8-transfected MDA-MB-231 and T47D cells. The G2/M phase was significantly increased accompanied by decreased S and G0/G1 phases in IRF8-transfected cells compared with vector-transfected cells. This effect may have occurred by upregulating p21 and inhibiting p-cdc25C (Figure 5A, 5C). Moreover, the percentage of apoptotic cells was significantly increased in IRF8-transfected breast cancer cells by cleaving PARP (Figure 5B, 5C), indicating the pro-apoptotic effect of IRF8.
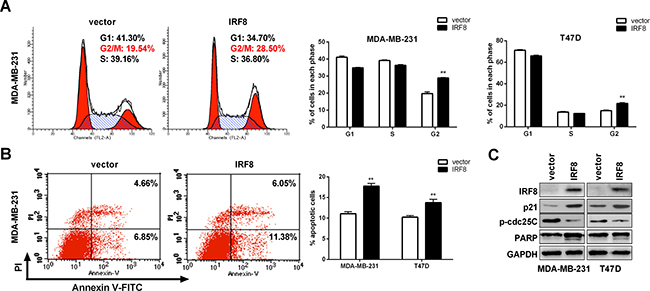
Figure 5: IRF8 induced G2/M cell cycle arrest and apoptosis in MDA-MB-231 and T47D cells. Cell cycle distribution was measured in vector- and IRF8-expressing MDA-MB-231 and T47D cells (A). Representative flow cytometry plots (left) and histograms of cell cycle alterations (right). The percentage of apoptotic cells was measured in vector- and IRF8-expressing MDA-MB-231 cells. Annexin V-positive cells indicated apoptotic cells. Representative flow cytometry plots (left) and histograms of apoptosis alterations (right). The level of p21, p-cdc25C and cleaved PARP were measured by immunoblotting in vector- and IRF8-expressing MDA-MB-231 and T47D cells. Data were based on three independent assays in MDA-MB-231 and T47D cells, respectively. Mean ± SD, **p < 0.01.
IRF8 inhibited breast cancer cell migration and invasion
The lung-metastatic potency of tumor cells was previously shown to be enhanced when IRF8 function was disrupted in BALB/c mice [29], indicating that downregulation of IRF8 expression may contribute to breast cancer cell migration and invasion. To test this hypothesis, we performed wound-healing and Transwell assays to assess the effects of IRF8 on cell migration and invasion. Wound-healing assays showed that cell migration was significantly inhibited by ectopic IRF8 expression in MDA-MB-231 cells, but not in T47D cells (Figure 6A). In addition, ectopic IRF8 expression markedly inhibited MDA-MB-231 cell invasion through a Matrigel barrier, with 10% FBS as an attractant (Figure 6C). This effect may have been mediated by modulating the morphology of MDA-MB-231 cells (Figure 6B), and by downregulating matrix metalloproteinases (MMP) 2 and 9, and vascular endothelial growth factor (VEGF) (Figure 6D). In contrast, cell migration and invasion were enhanced in cells transfected with IRF8-K79E, which disrupted the function of IRF8 (Figure 6E–6G). These results suggested that the effects of IRF8 on cell migration and invasion may depend on the molecular type of breast cancer, especially in ER-negative tumors.
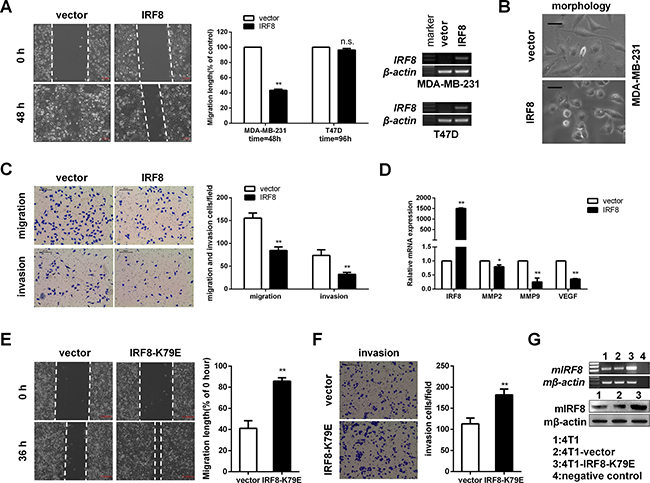
Figure 6: IRF8 suppressed cell migration and invasion in MDA-MB-231 and 4T1 cells in vitro. (A) Wound-healing assay. Confluent monolayers of vector- and IRF8-transfected MDA-MB-231 and T47D cells were scarred and migration distance was measured after 48 h and 96 h respectively. Data were based on three independent assays, and representative images are shown. mean ± SD, **p < 0.01. (B) Cell morphology of vector- and IRF8-expressing MDA-MB-231 cells. Bar = 50 μm. (C) Invaded MDA-MB-231 cells in the lower side of the chamber with Matrigel as a barrier were fixed and stained. Numbers of cells were counted by phase-contrast microscopy. Data were based on three independent assays, and representative images are shown. Mean ± SD, **p < 0.01. (D) The expression of matrix metalloproteinases (MMP) 2 and 9, and vascular endothelial growth factor (VEGF) were evaluated under IRF8 overexpression by real-time PCR, with β-actin as an internal control. Data were based on three independent assays. Mean ± SD, *p < 0.05, **p < 0.01. (E) Migration distances of vector- and IRF8-K79E-expressing 4T1 cells after 36 h. Data were based on three independent assays. Mean ± SD, **p < 0.01. (F) cell invasion by vector- and IRF8-K79E-expressing 4T1 cells after 24 h. Data were based on three independent assays. Mean ± SD, **p < 0.01. (G) IRF8 expression was measured by RT-PCR and immunoblotting in 4T1 cells.
IFR8 acts as a downstream target gene of IFN-γ
IRF8 acts as a candidate TSG in breast cancer, but its underlying mechanism remains unclear. IRF8 has been shown to be a downstream target gene of the IFN-γ/STAT1 signaling pathway, and methylation of its promoter can block this inducibility [10, 11]. IRF8 was induced by IFN-γ treatment via phosphorylation of STAT1 in SK-BR-3 cells (without IRF8 promoter methylation) (Figure 7A), while restoration of IRF8 upregulated the expression of JAK1 and JAK2 in MDA-MB-231 cells (Figure 7B), and also enhanced pSTAT1 levels and apoptosis under IFN-γ treatment (Figure 7C, 7D). However, there was no significant difference in pSTAT1 between vector- and IRF8-expressing MDA-MB-231 cells (Figure 7F). Moreover, pharmacological demethylation upregulated IRF8 and pSTAT1, and suppressed the levels of Bcl-2 and active-β-catenin (Figure 7E). These data suggest that promoter methylation of IRF8 disrupts its pro-apoptotic effect in breast cancers.
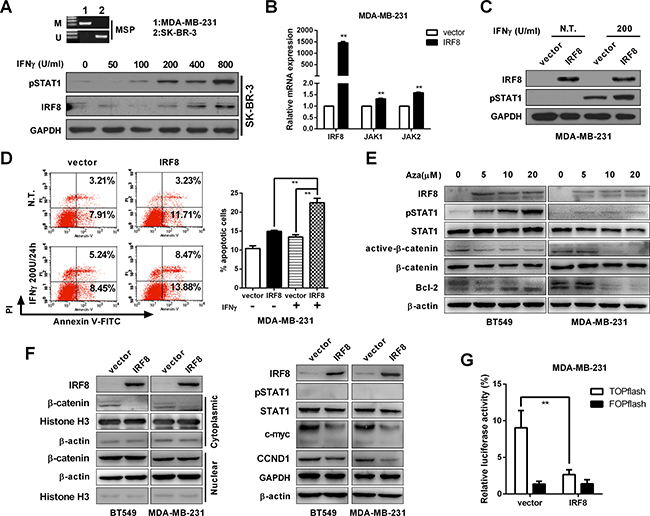
Figure 7: IRF8 expression enhanced the pro-apoptotic effect of IFN-γ and suppressed β-catenin signaling. (A) Promoter methylation of IRF8 was measured by MSP in MDA-MB-231 and SK-BR-3 cells. SK-BR-3 cells were treated with different concentrations of IFN-γ (50, 100, 200, 400, 800 U/ml) or vehicle control (DMSO) for 24 h, and the expression levels of pSTAT1 and IRF8 were detected by immunoblotting. (B) Expression levels of JAK1 and JAK2 were evaluated under IRF8 overexpression by real-time PCR in MDA-MB-231 cells. Data were based on three independent assays. mean ± SD, **p < 0.01. (C) Expression of pSTAT1 was evaluated under IRF8 overexpression with or without IFN-γ by immunoblotting in MDA-MB-231 cells. GAPDH was used as an internal loading control. (D) The percentage of apoptotic cells was measured under treatment with IRF8 and/or IFN-γ (200 U/24 h) in MDA-MB-231 cells. Annexin V-positive cells indicated apoptotic cells. Data were based on three independent assays, and representative images are shown. Mean ± SD, **p < 0.01. (E) Protein levels of IRF8, pSTAT1, STAT1, active-β-catenin, β-catenin and Bcl2 were measured in BT549 and MDA-MB-231 cells treated with Aza at concentrations of 5, 10, and 20 μM for 3 days. β-actin was used as the control. Data were based on three independent assays, and representative images are shown. (F) Effect of ectopic IRF8 expression on β-catenin signaling and its downstream target genes c-myc and CCND1 in BT549 and MDA-MB-231 cells, with GAPDH and β-actin as an internal loading control. (G) TOP/FOP luciferase reporter activity assay in vector- and IRF8-expressing cells. Data were based on three independent assays. Mean ± SD, **p < 0.01.
Effect of IRF8 on β-catenin signaling in breast cancer
IRF8 has been shown to play an important role in the IFN–γ/STAT1 signaling pathway. However, IRF8, as a transcription factor, may thus modify other signaling pathways, such as β-catenin signaling in breast cancers. The current results showed that ectopic expression of IRF8 suppressed β-catenin activation and downregulated the downstream target genes of β-catenin signaling, including CCND1 and c-myc (Figure 7F). Furthermore, the effect of IRF8 on TcF transcriptional activity was determined by luciferase reporter assay. Luciferase activities were inhibited by IRF8 in TOPflash, but not in FOPflash, with Renilla reniformis luciferase activities as an internal control (Figure 7G).
DISCUSSION
IRF8 is an IRF with a conserved DNA-binding domain at the N terminal and a IRF-association domain at the C terminal [9]. It can regulate the target gene directly, or as a collaborator through the IFN-stimulated response element, and thus plays an important role in regulating the immune response, and cell growth and differentiation [30]. Evidence has suggested that IRF8 acts as a TSG in multiple cancers, including leukemia, renal cancer, and soft tissue sarcoma, via regulating cell proliferation, apoptosis, cell cycle distribution, cell invasion, and metastasis [16, 17, 28]. In contrast, IRF8 also has been shown to promote HL-60 proliferation via the transforming growth factor (TGF)-β receptor/TAK-1/p38 pathway [18], and enhance cell motility and invasion by repressing TGF-β signaling in U2OS cells [19], as well as acting as an independent adverse factor in acute myeloid leukemia [31]. However, the role and underlying mechanism of IRF8 in breast cancer remains unclear. In this study, we demonstrated that IRF8 was downregulated in breast cancers, mainly as a result of promoter hypermethylation. We also showed that IRF8 may act as a candidate TSG by inducing cell cycle arrest and apoptosis, and inhibiting cell migration and invasion in breast cancer by inhibiting β-catenin signaling. Meanwhile, IRF8 also act as a downstream target gene of IFN-γ/STAT1 signaling and enhances the pro-apoptotic effect of IFN-γ by regulating STAT1 phosphorylation.
The expression of IRF8 was downregulated or silenced in breast cancer cell lines and primary breast cancers due to promoter hypermethylation. Furthermore, its expression may be an independent factor for DMFS and OS in ER-negative and grade 3 tumors. It has been reported that 13 genes were hypermethylated in hormone receptor (HR)+, luminal A, or p53 wild-type breast cancers, while 9 genes were hypermethylated in HR−, basal-like, or p53 mutant tumors, indicating differences in DNA methylation patterns among breast cancer subtypes [32]. However, it seems there was no obviously correlation between ER status and the expression of IRF8, and there was no significant difference in IRF8 expression between ER-positive (n = 323) and ER-negative (n = 92) tumors (data not shown), or between TNBCs and non-TNBCs. In this study, the IRF8 promoter was methylated in 49.12% (56/114) of breast cancers, which was slightly higher than in breast cancers in previous studies (36%, 5/14) [11]. Moreover, our results were in line with several other cancers, including multiple myeloma, nasopharyngeal, esophageal, lung, and renal cancers [11, 12, 15, 28]. Notably, IRF8 methylation was associated with tumor grade in renal cancer [28]. However, the current study found no correlation between IRF8 methylation and clinicopathological features, including ER status, based on 114 breast cancer cases. Further studies with more patients, especially with ER-negative tumors, and more detailed follow-up information may be necessary to clarify this relationship.
Previous studies reported that IRF8 acted as a TSG in solid and non-solid tumors [15, 28]. Our data showed that IRF8 performed as a candidate tumor suppressor by inducing G2/M phase cell cycle arrest and apoptosis in MDA-MB-231 and T47D cells, consistent with the function of IRF8 in renal cancer [28], and also by inhibiting cell migration and invasion in MDA-MB-231, but not in T47D cells. The effects of IRF8 thus seemed to be more pronounced in ER-negative breast cancer cells, supporting the prognostic effect of IRF8 expression in patients with ER-negative breast cancers.
IRF8 has previously been shown to be an inducible gene of IFN-γ/STAT1 signaling, and its promoter methylation disrupted the IFN-γ response [10, 11]. In this study, IRF8 was induced by IFN-γ in SK-BR-3 cells (without IRF8 promoter methylation) but not in MDA-MB-231 cells (with IRF8 promoter methylation) (data not shown). In addition, we showed that IRF8 enhanced the anti-tumor activity of IFN-γ in MDA-MB-231 cells, possibly via upregulated expression JAK1 and JAK2. Meanwhile, pSTAT1 was not induced by ectopic IRF8 in the absence of IFN-γ. However, accumulated data have shown that IFN-γ-induced genes, such as STAT1, promote tumor growth, metastasis, and resistance to therapy. The synergistic effect between IFN-γ and demethylating agents in cancer treatment may thus need further study [33].
IRF8, as a transcription factor, also exerts its effect by modulating the downstream target gene and/or signal pathway directly. Oncogenes (YAP1, Survivin) were shown to be repressed and TSGs (p21, PTEN, CASP1) upregulated under IRF8 overexpression in renal cancer [28], and negative feedback occurred between IRF8 and β-catenin in leukemia [23]. The current study revealed a relationship between IRF8 and β-catenin, and indicated that β-catenin signaling was suppressed by ectopic expression of IRF8, followed by decreased luciferase activities and downregulation of CCND1 and c-myc. Thus, β-catenin signaling was implicated in the anti-tumor effect of IRF8 in multiple cancers, including breast cancers.
In conclusion, expression of the IFN-γ-inducible gene IRF8 may be downregulated due to promoter methylation, impairing its anti-tumor effect by modulating β-catenin signaling in breast cancers.
MATERIALS AND METHODS
Primary breast samples
Primary breast-associated tissues were obtained from the Department of Endocrine and Breast Surgery, the First Affiliated Hospital of Chongqing Medical University, P.R. China, from January 2013 to October 2014, stored at −80°C in Chongqing Key Laboratory of Molecular Oncology and Epigenetics, the First Affiliated Hospital of Chongqing Medical University, Chongqing, China, and confirmed by pathology. None of the patients had received preoperative anti-tumor treatment, and written informed consent was obtained from each patient before surgery. This research was approved by the Institute Ethics Committee of the First Affiliated Hospital of Chongqing Medical University.
Cell culture and transfection
Four cell lines were used in this study, including BT549, MDA-MB-231, SK-BR-3 and T47D [34]. All cells were cultured in RPMI 1640 or DMEM (Gibco-BRL, Grand Island, NY, USA), supplemented with 10% fetal bovine serum (Gibco-BRL), and 100 U/ml penicillin–streptomycin (Gibco-BRL), and maintained at 37°C with 5% CO2. All transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. pcDNA3.1, pcDNA3.1-IRF8 and pcDNA3.1-IRF8-K79E plasmids were transfected into cells at a concentration of 4 μg, and selected by neomycin at 48 h after transfection.
RNA and DNA extraction
Total RNA and genomic DNA were extracted using TRIzol reagent (Invitrogen) and a QIAamp DNA Mini Kit (Qiagen, Duesseldorf, Germany), respectively, as described previously [35]. Their concentrations were measured with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and their levels were determined by gel electrophoresis.
Reverse transcription, semi-quantitative and quantitative polymerase chain reaction
Reverse transcription, semi-quantitative polymerase chain reaction (PCR) was performed as described previously, using Go-Taq polymerase (Promega, Madison, WI, USA) [11]. Quantitative PCR was performed using a SYBR® Green PCR Master Mix kit (Invitrogen) and an Applied Biosystem 7500 Real-time PCR System (Applied Biosystems, Foster City, CA, USA). β-actin served as a control. The relative expression of IRF8 was evaluated using the 2(-ΔCt) method. All assays were performed three times, independently. Primers are listed in Supplementary Table 1.
Bisulfite treatment and methylation-specific PCR
Bisulfite treatment and methylation-specific PCR (MSP) were performed as described previously [11, 36, 37]. Briefly, bisulfite-treated DNA was amplified to evaluate the methylation status of IRF8 by MSP with primers IRF8-m1 and IRF8-m2 to detect methylated DNA, and IRF8-u1 and IRF8-u2 to detect unmethylated DNA. Primers are listed in Supplementary Table 1.
Colony-formation assay
Anchorage-dependent growth was evaluated by colony-formation assay. IRF8-expressing cells and control (pcDNA3.1) cells (200, 500, or 1000) were re-plated in six-well plates with the indicated concentrations of neomycin. Surviving colonies (>50 cells) were counted after 2 weeks following fixation and staining. All experiments were performed three times, independently.
Cell-viability assay
Cell viability was evaluated using a CCK-8 kit (Beyotime Institute of Biotechnology, Jiangsu, China). Briefly, IRF8-expressing cells and control (pcDNA3.1) cells (MDA-MB-231 and T47D) were seeded in 96-well plates after transfection, and cell viability was measured with the CCK-8 kit at 24, 48, and 72 h. All experiments were performed three times, independently.
5-Ethynyl-2’-deoxyuridine assay
Cell proliferation was detected using 5-ethynyl-2’-deoxyuridine (EdU) kits (RiboBio, Guangzhou, China). Briefly, cell proliferation was measured by analyzing EdU incorporation during DNA synthesis after transfection. All assays were performed three times, independently.
Wound-healing and Transwell assays
Cell migration and invasion ability were evaluated by wound-healing and Transwell assays, respectively. Briefly, IRF8- or IRF8-K79E-expressing cells and control (pcDNA3.1) cells were plated in six-well plates, and wounded using sterile tips once confluent. Cell migration was semi-quantified by measuring the migration distance under phase-contrast microscopy (Leica DMI4000B, Milton Keynes, Buckinghamshire, UK). Transwell chambers (Corning Life Sciences, NY, USA) with a pore size of 8 μm were used to evaluate cell migration and cell invasion, with a Matrigel (BD Biosciences, San Jose, CA, USA) barrier. Cells in the lower side of the chamber were counted after fixation and staining under phase-contrast microscopy (Leica). All assays were performed three times, independently
Flow cytometry analysis
Cell cycle distribution and the percentage of apoptotic cells were analyzed by flow cytometry [38]. Briefly, cells were stained with propidium iodide (PI) to analyze cell cycle, after transfection and fixation. Cells were double stained with annexin V–fluorescein isothiocyanate/PI to detect apoptosis. All assays were evaluated using a Cell Quest kit (BD Biosciences). All assays were performed three times, independently.
Immunoblotting
Immunoblotting was performed as described previously [39]. Briefly, whole cells were lysed in RIPA lysis buffer (Beyotime Institute of Biotechnology) with a protease inhibitor cocktail (Pierce, Cramlington, UK). Lysates were separated by 10%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes (Merck Millipore, Billerica, MA, USA), and incubated overnight at 4°C with the following primary antibodies: IRF8 (Santa Cruz Biotechnology, CA, USA), p21, p-cdc25C, poly (ADP-ribose) polymerase (PARP), pSTAT1, STAT1, β-catenin, β-actin, Bcl-2, Histone H3 (Cell Signaling Technology, Danvers, MA, USA), active-β-catenin (Merck Millipore), CCND1, c-myc, GAPDH (Epitomics, Burlingame, CA, USA), followed by incubation with secondary antibody. The bands were visualized using ECL Plus Detection Reagents (RPN2132; GE Healthcare Life Science, Buckinghamshire, UK). All assays were performed three times, independently.
In vivo tumor model
The anti-tumor function of the target gene was evaluated using an in vivo model. Vector- and IRF8-expressing MDA-MB-231 cells (5×106) were injected subcutaneously into nude mice (n = 6 in each group). The tumor volumes were monitored (volume = 0.5 × length × width2 per week), and the weights of the xenografts were measured after sacrifice. All procedures for constructing the tumor model were approved by the Institute Ethics Committee of the First Affiliated Hospital of Chongqing Medical University.
Immunohistochemistry
Standard streptavidin–peroxidase immuno histochemistry was performed using an UltraSensitive TM SP Kit (Maixin-Bio, Fujian, China) according to the manufacturer’s instructions. Sections were dewaxed, rehydrated, and blocked, and then incubated with a primary antibody against Ki-67 (1:50 dilution). The sections were then treated with a secondary antibody and stained with diaminobenzidine. The staining was assessed by a trained pathologist using Image-Pro Plus (IPP, version 6.0). All assays were performed three times, independently.
Dual-luciferase reporter assay
The effect of IRF8 on TcF transcriptional activities was determined by luciferase reporter assay [40]. Briefly, the TcF-responsive luciferase construct TOPflash or FOPflash (containing a mutant TCF/LEF binding site) was cotransfected with IRF8 or control vector, with Renilla luciferase reporter pRL-TK aa an internal control (Promega). Luciferase activities were determined 48 h later using a dual-luciferase reporter assay kit (Promega). All assays were performed three times, independently.
Statistical analysis
Data were analyzed using SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA) and presented as mean ± standard deviation (SD). Two-tailed Student t-tests were used to determine p values. Correlations between methylation status and clinicopathological features were analyzed using χ2 and Fisher’s exact tests. p < 0.05 was considered significant.
ACKNOWLEDGMENTS AND FUNDING
This study was supported by National Natural Science Foundation of China (#31420103915, #81572769, #81402178). VC special fund from CUHK. We thank Prof. Kebin Liu (Department of Biochemistry and Molecular Biology, Medical College of Georgia, Augusta, Georgia) for generously providing pcDNA3.1-IRF8-K79E and pcDNA3.1 plasmids.
CONFLICTS OF INTEREST
The authors declear that they have no conflicts of interest.
REFERENCES
1. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in china, 2015. CA Cancer J Clin. 2016; 66:115–32.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017; 67:7–30.
3. Ying Y, Tao Q. Epigenetic disruption of the wnt/ss-catenin signaling pathway in human cancers. Epigenetics. 2009; 4:307–12.
4. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nature reviews. Genetics. 2002; 3:415–28.
5. Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nature reviews. Cancer. 2006; 6:107–16.
6. Xiang TX, Yuan Y, Li LL, Wang ZH, Dan LY, Chen Y, Ren GS, Tao Q. Aberrant promoter cpg methylation and its translational applications in breast cancer. Chinese journal of cancer. 2013; 32:12–20.
7. Li L, Li C, Mao H, Du Z, Chan WY, Murray P, Luo B, Chan AT, Mok TS, Chan FK, Ambinder RF, Tao Q. Epigenetic inactivation of the cpg demethylase tet1 as a DNA methylation feedback loop in human cancers. Scientific reports. 2016; 6:26591.
8. Du Z, Li L, Huang X, Jin J, Huang S, Zhang Q, Tao Q. The epigenetic modifier chd5 functions as a novel tumor suppressor for renal cell carcinoma and is predominantly inactivated by promoter cpg methylation. Oncotarget. 2016; 7:21618–30. doi: 10.18632/oncotarget.7822.
9. Nguyen H, Hiscott J, Pitha PM. The growing family of interferon regulatory factors. Cytokine & growth factor reviews. 1997; 8:293–312.
10. Yang D, Thangaraju M, Greeneltch K, Browning DD, Schoenlein PV, Tamura T, Ozato K, Ganapathy V, Abrams SI, Liu K. Repression of ifn regulatory factor 8 by DNA methylation is a molecular determinant of apoptotic resistance and metastatic phenotype in metastatic tumor cells. Cancer research. 2007; 67:3301–9.
11. Lee KY, Geng H, Ng KM, Yu J, van Hasselt A, Cao Y, Zeng YX, Wong AH, Wang X, Ying J, Srivastava G, Lung ML, Wang LD, et al. Epigenetic disruption of interferon-gamma response through silencing the tumor suppressor interferon regulatory factor 8 in nasopharyngeal, esophageal and multiple other carcinomas. Oncogene. 2008; 27:5267–76.
12. Suzuki M, Ikeda K, Shiraishi K, Eguchi A, Mori T, Yoshimoto K, Shibata H, Ito T, Baba Y, Baba H. Aberrant methylation and silencing of irf8 expression in non-small cell lung cancer. Oncology letters. 2014; 8:1025–30.
13. Yamashita M, Toyota M, Suzuki H, Nojima M, Yamamoto E, Kamimae S, Watanabe Y, Kai M, Akashi H, Maruyama R, Sasaki Y, Yamano H, Sugai T, et al. DNA methylation of interferon regulatory factors in gastric cancer and noncancerous gastric mucosae. Cancer science. 2010; 101:1708–16.
14. McGough JM, Yang D, Huang S, Georgi D, Hewitt SM, Rocken C, Tanzer M, Ebert MP, Liu K. DNA methylation represses ifn-gamma-induced and signal transducer and activator of transcription 1-mediated ifn regulatory factor 8 activation in colon carcinoma cells. Molecular cancer research. 2008; 6:1841–51.
15. Tshuikina M, Jernberg-Wiklund H, Nilsson K, Oberg F. Epigenetic silencing of the interferon regulatory factor icsbp/irf8 in human multiple myeloma. Experimental hematology. 2008; 36:1673–81.
16. Hu X, Yang D, Zimmerman M, Liu F, Yang J, Kannan S, Burchert A, Szulc Z, Bielawska A, Ozato K, Bhalla K, Liu K. Irf8 regulates acid ceramidase expression to mediate apoptosis and suppresses myelogeneous leukemia. Cancer research. 2011; 71:2882–91.
17. Yang D, Wang S, Brooks C, Dong Z, Schoenlein PV, Kumar V, Ouyang X, Xiong H, Lahat G, Hayes-Jordan A, Lazar A, Pollock R, Lev D, et al. Ifn regulatory factor 8 sensitizes soft tissue sarcoma cells to death receptor-initiated apoptosis via repression of flice-like protein expression. Cancer research. 2009; 69:1080–8.
18. Sung JY, Kim H, Kim YN, Na YS, Park BK. Interferon consensus sequence binding protein-induced cell proliferation is mediated by tgf-beta signaling and p38 mapk activation. Laboratory investigation; a journal of technical methods and pathology. 2011; 91:1304–13.
19. Sung JY, Park SY, Kim JH, Kang HG, Yoon JH, Na YS, Kim YN, Park BK. Interferon consensus sequence-binding protein (icsbp) promotes epithelial-to-mesenchymal transition (emt)-like phenomena, cell-motility, and invasion via tgf-beta signaling in u2os cells. Cell death & disease. 2014; 5:e1224.
20. Curtin JC, Lorenzi MV. Drug discovery approaches to target wnt signaling in cancer stem cells. Oncotarget. 2010; 1:563–577. https://doi.org/10.18632/oncotarget.191.
21. King TD, Suto MJ, Li Y. The wnt/beta-catenin signaling pathway: A potential therapeutic target in the treatment of triple negative breast cancer. Journal of cellular biochemistry. 2012; 113:13–8.
22. Huang W, Zhou W, Saberwal G, Konieczna I, Horvath E, Katsoulidis E, Platanias LC, Eklund EA. Interferon consensus sequence binding protein (icsbp) decreases beta-catenin activity in myeloid cells by repressing gas2 transcription. Molecular and cellular biology. 2010; 30:4575–94.
23. Scheller M, Schonheit J, Zimmermann K, Leser U, Rosenbauer F, Leutz A. Cross talk between wnt/beta-catenin and irf8 in leukemia progression and drug resistance. The Journal of experimental medicine. 2013; 210:2239–56.
24. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Science signaling. 2013; 6:l1.
25. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012; 2:401–4.
26. Jezequel P, Campone M, Gouraud W, Guerin-Charbonnel C, Leux C, Ricolleau G, Campion L. Bc-genexminer: An easy-to-use online platform for gene prognostic analyses in breast cancer. Breast cancer research and treatment. 2012; 131:765–75.
27. Jezequel P, Frenel JS, Campion L, Guerin-Charbonnel C, Gouraud W, Ricolleau G, Campone M. Bc-genexminer 3.0: New mining module computes breast cancer gene expression correlation analyses. Database. 2013; 2013:bas060.
28. Zhang Q, Zhang L, Li L, Wang Z, Ying J, Fan Y, Xu B, Wang L, Liu Q, Chen G, Tao Q, Jin J. Interferon regulatory factor 8 functions as a tumor suppressor in renal cell carcinoma and its promoter methylation is associated with patient poor prognosis. Cancer letters. 2014; 354:227–34.
29. Zimmerman MA, Rahman NT, Yang D, Lahat G, Lazar AJ, Pollock RE, Lev D, Liu K. Unphosphorylated stat1 promotes sarcoma development through repressing expression of fas and bad and conferring apoptotic resistance. Cancer research. 2012; 72:4724–32.
30. Tamura T, Ozato K. Icsbp/irf-8: Its regulatory roles in the development of myeloid cells. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 2002; 22:145–52.
31. Pogosova-Agadjanyan EL, Kopecky KJ, Ostronoff F, Appelbaum FR, Godwin J, Lee H, List AF, May JJ, Oehler VG, Petersdorf S, Pogosov GL, Radich JP, Willman CL, et al. The prognostic significance of irf8 transcripts in adult patients with acute myeloid leukemia. PloS one. 2013; 8:e70812.
32. Conway K, Edmiston SN, May R, Kuan PF, Chu H, Bryant C, Tse CK, Swift-Scanlan T, Geradts J, Troester MA, Millikan RC. DNA methylation profiling in the carolina breast cancer study defines cancer subclasses differing in clinicopathologic characteristics and survival. Breast cancer research. 2014; 16:450.
33. Shakya R, Gonda T, Quante M, Salas M, Kim S, Brooks J, Hirsch S, Davies J, Cullo A, Olive K, Wang TC, Szabolcs M, Tycko B, et al. Hypomethylating therapy in an aggressive stroma-rich model of pancreatic carcinoma. Cancer research. 2013; 73:885–96.
34. Xiang T, Fan Y, Li C, Li L, Ying Y, Mu J, Peng W, Feng Y, Oberst M, Kelly K, Ren G, Tao Q. Dact2 silencing by promoter cpg methylation disrupts its regulation of epithelial-to-mesenchymal transition and cytoskeleton reorganization in breast cancer cells. Oncotarget. 2016; 7:70924–35. doi: 10.18632/oncotarget.12341.
35. Yin X, Xiang T, Mu J, Mao H, Li L, Huang X, Li C, Feng Y, Luo X, Wei Y, Peng W, Ren G, Tao Q. Protocadherin 17 functions as a tumor suppressor suppressing wnt/beta-catenin signaling and cell metastasis and is frequently methylated in breast cancer. Oncotarget. 2016; 7:51720–32. doi: 10.18632/oncotarget.10102.
36. Tao Q, Huang H, Geiman TM, Lim CY, Fu L, Qiu GH, Robertson KD. Defective de novo methylation of viral and cellular DNA sequences in icf syndrome cells. Human molecular genetics. 2002; 11:2091–102.
37. Ying J, Li H, Seng TJ, Langford C, Srivastava G, Tsao SW, Putti T, Murray P, Chan AT, Tao Q. Functional epigenetics identifies a protocadherin pcdh10 as a candidate tumor suppressor for nasopharyngeal, esophageal and multiple other carcinomas with frequent methylation. Oncogene. 2006; 25:1070–80.
38. Xiang T, Li L, Yin X, Yuan C, Tan C, Su X, Xiong L, Putti TC, Oberst M, Kelly K, Ren G, Tao Q. The ubiquitin peptidase uchl1 induces g0/g1 cell cycle arrest and apoptosis through stabilizing p53 and is frequently silenced in breast cancer. PloS one. 2012; 7:e29783.
39. Shi Y, Luo X, Li P, Tan J, Wang X, Xiang T, Ren G. Mir-7–5p suppresses cell proliferation and induces apoptosis of breast cancer cells mainly by targeting reggamma. Cancer letters. 2015; 358:27–36.
40. Li L, Ying J, Tong X, Zhong L, Su X, Xiang T, Shu X, Rong R, Xiong L, Li H, Chan AT, Ambinder RF, Guo Y, et al. Epigenetic identification of receptor tyrosine kinase-like orphan receptor 2 as a functional tumor suppressor inhibiting beta-catenin and akt signaling but frequently methylated in common carcinomas. Cellular and molecular life sciences. 2014; 71:2179–92.