
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is currently the fourth leading cause of cancer-related mortality in developed countries despite its comparably low incidence of less than 3 %, clearly demonstrating the lack of effective therapeutic strategies. The five-year survival rate is around 7 % for all stages of the disease and drops to below 2 % and a median survival of less than a year for patients with metastatic disease, mostly due to late diagnosis at the stage of inoperability and the unusually high resistance of PDAC to conventional radiation and chemotherapy [1, 2]. Despite intensive scientific and industrial efforts, so far no significant extension of survival could be achieved by any of the numerous therapy approaches tested [3].
The genetic and morphological changes in the carcinogenesis of PDAC are well known and include the initiation and progression of premalignant lesions to invasive and metastatic pancreatic cancer [3–6]. The genetic hallmark of PDAC development is an activating mutation in the KRAS oncogene, followed by other genetic changes, commonly including inactivation of the tumor suppressors TP53, CDKN2A (P16INK4A) and SMAD4, and activation of several growth factor receptors, such as the epidermal growth factor receptor (EGFR) [3, 7].
Against this background, several complex genetically modified mouse models of PDAC that mirror the typical changes found in human patients, have been generated in recent years [3, 4, 8–10]. One such model is the Ptf1a+/Cre;Kras+/LSL-G12D;Trp53loxP/loxP (Kras;p53) mouse. Here, PDAC is induced by pancreas-specific activation of constitutively active KrasG12D in combination with conditional deletion of Trp53 [8]. To restrict these genetic modifications to the pancreas, mice with the mutated alleles are interbred with animals that express the Cre recombinase driven by the pancreas-specific promoter for Ptf1a, a subunit of pancreas transcription factor 1 (Ptf1) that is required to commit cells to a pancreatic fate during embryonic development [4, 11]. Thus, the activation of the oncogenic KrasG12D via excision of a transcriptionally inhibitory LSL (loxP-STOP-loxP) construct and deletion of the floxed tumor suppressor Trp53 occur in the pancreas only, leading to ductal lesions with complete penetrance [4, 12]. The development of endogenous mouse models away from the usual transplant models represents a significant step in the evolution of preclinical models [13]. The morphological and molecular composition of endogenous tumors far better reflects human disease, making them highly suitable to predict the clinical effectiveness of a specific treatment strategy.
The sodium iodide symporter (NIS; SLC5A5) mediates the uptake of iodide into thyroid follicular cells allowing both diagnostic and therapeutic application of radioiodide in thyroid cancer patients [14, 15]. In our previous work, we have extensively investigated the dual reporter/therapy capacity of NIS in various non-thyroidal tumors and have proven the feasibility of extrathyroidal radioiodide therapy after tumor-selective NIS gene transfer [16–26]. Transfection of cancer cells with the NIS gene allows non-invasive monitoring of functional NIS expression and in vivo biodistribution before the application of a therapeutic dose of radioiodide. One of the major hurdles of efficient and safe application of the NIS gene therapy concept in the clinical setting is optimal tumor-specific targeting in the presence of low toxicity and high transfection efficiency of gene delivery vectors, with the ultimate goal of systemic vector application.
In a previous study, we used synthetic polyplexes based on pseudodendritic oligoamines with high intrinsic tumor affinity for NIS gene therapy in a syngeneic neuroblastoma mouse model as well as a subcutaneous human hepatocellular carcinoma mouse model [16, 18]. After systemic NIS gene transfer, the tumor-selective accumulation of radioiodide was sufficient for a significant therapeutic effect. In addition to an intrinsic tumor affinity due to the so-called enhanced permeability and retention (EPR) effect based on “leaky” tumor vasculature, the tumor targeting of polyplexes can be further optimized by the attachment of tumor-specific ligands. To this end, in a subsequent study, we used LPEI-PEG-GE11 polymers composed of linear polyethylenimine (LPEI), shielded by polyethylene glycol (PEG) and coupled to the synthetic peptide GE11 as an EGFR-specific ligand for NIS gene delivery [17]. After systemic application of these polymers condensed with NIS DNA, tumor-specific radioiodide accumulation demonstrated effective and EGFR-specific tumor targeting in a high EGFR-expressing xenograft mouse model of hepatocellular carcinoma. After the injection of a therapeutic dose of 131I, tumoral iodide uptake was sufficiently high for a significant delay of tumor growth and prolongation of animal survival [17].
Based on our previous work and the well-known characteristic upregulation of EGFR in PDAC, we investigated the potential of EGFR-targeted polyplexes for systemic NIS gene therapy in an advanced endogenous mouse model of PDAC as a next step towards clinical application.
RESULTS
Iodide uptake studies in vitro
In order to optimize transfection conditions for LPEI-PEG-GE11 polymers condensed with the NIS plasmid (LPEI-PEG-GE11/NIS) in high EGFR-expressing PDAC cell explants derived from Kras;p53 mice (Figure 1A), radioiodide uptake activity was evaluated 24 h after polyplex application (data not shown). A conjugate to plasmid (c/p) ratio of 0.8 resulted in highest transfection efficiency at lowest cytotoxicity. Therefore, this c/p ratio was used in all subsequent experiments. Twenty-four hours after transfection with LPEI-PEG-GE11/NIS, cell explants from three different mice showed a 22-26-fold increase in 125I accumulation as compared to cells incubated with the empty control vector LPEI-PEG-GE11 (Figure 1B). Transfection with untargeted LPEI-PEG-Cys/NIS (targeting ligand GE11 replaced by a cysteine residue) resulted in significantly lower iodide uptake activity compared to EGFR-targeted LPEI-PEG-GE11/NIS (Figure 1B). In both cases, iodide uptake was blocked upon additional treatment with the NIS-specific inhibitor perchlorate.
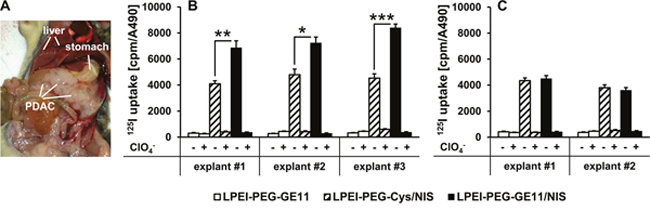
Figure 1: Iodide uptake in PDAC cell explants in vitro. Kras;p53 mice develop PDAC that occupies a large portion of the abdominal cavity below the stomach (A). (B) PDAC cell explants from three separate mice (three technical replicates per mouse) transfected in vitro with LPEI-PEG-GE11/NIS showed a significant increase in perchlorate- (ClO4--) sensitive 125I accumulation compared to transfection with LPEI-PEG-Cys/NIS (mean ± S.E.M.; *p<0.05; **p<0.01; ***p<0.001). No iodide uptake above background levels was observed in cells transfected with LPEI-PEG-GE11 alone. (C) Transfection of EGFR-ablated PDAC cell explants from two mice (three technical replicates per mouse) with LPEI-PEG-GE11/NIS and LPEI-PEG-Cys/NIS showed no significant differences between transfection with targeted or untargeted polyplexes, demonstrating EGFR-specificity of the targeting ligand GE11 (mean ± S.E.M.).
To further verify EGFR-specificity of the targeting ligand GE11, we performed additional iodide uptake studies in EGFR-ablated PDAC cell explants from Ptf1a+/Cre;Kras+/LSL-G12D;Trp53loxP/loxP;Egfrfl/fl (Kras;p53;Egfr) mice. No significant difference between transfection with targeted LPEI-PEG-GE11/NIS or untargeted LPEI-PEG-Cys/NIS polyplexes was observed (Figure 1C). Polyplex-mediated NIS gene transfer did not affect cell viability for any of the treatment conditions compared to untreated cells as measured by MTS assay (data not shown).
123I scintigraphy and 124I PET imaging of EGFR-targeted NIS gene delivery
Functional NIS expression in mice with high EGFR-expressing PDAC was imaged by whole body 123I gamma camera and 124I PET imaging. Polyplexes were administered intravenously (i.v.) either 24 or 48 h before injection of the respective radionuclide for imaging.
In vivo 123I gamma camera imaging revealed high levels of NIS-mediated radionuclide accumulation in pancreatic tumors both at 24 and 48 h after systemic injection of EGFR-targeted LPEI-PEG-GE11/NIS (Figures 2A, 2B). Tumors accumulated 10.8 ± 0.7 % of the injected dose per gram (ID/g) with an average biological half-life of 4 h at 24 h and 14.2 ± 1.4 % ID/g with an average biological half-life of 4.5 h at 48 h (Figure 2E). For 131I, a tumor-absorbed dose of 74.7 mGy/MBq/g tumor with an effective half-life of 3.2 h (24 h after polyplex administration) and 96.5 mGy/MBq/g tumor, effective half-life 4.5 h (48 h after polyplex administration), was calculated. In contrast, injection of non-coding control polyplexes LPEI-PEG-GE11/antisenseNIS (NIS sequence back to front) resulted in no significant tumoral radioiodide accumulation (Figure 2C). In addition to 123I uptake in the tumor, radioiodide accumulation was also observed in the stomach, the thyroid and the salivary glands, as they physiologically express NIS, as well as in the urinary bladder due to renal radionuclide elimination (Figures 2A-2C). To further confirm that tumoral iodide uptake was indeed NIS-mediated, LPEI-PEG-GE11/NIS-injected mice were additionally treated with the competitive NIS-inhibitor perchlorate 30 min before 123I administration, which completely blocked polyplex-mediated tumoral iodide accumulation in addition to physiological uptake in the stomach, the thyroid gland and the salivary glands (Figure 2D).
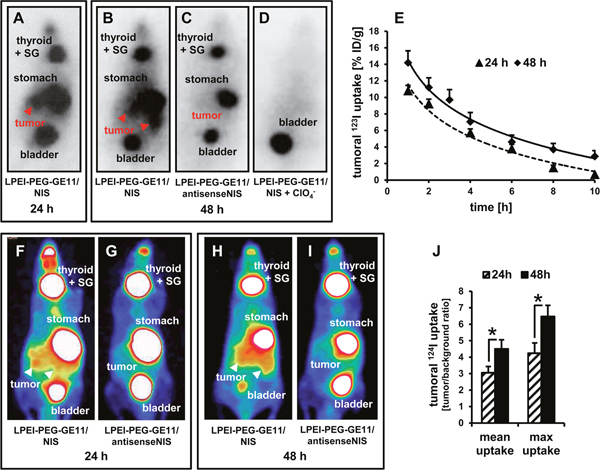
Figure 2: In vivo imaging of NIS-mediated iodide uptake. 123I scintigraphy revealed pancreatic tumoral radioiodide uptake 24 h (A) and 48 h (B) after injection of mice with LPEI-PEG-GE11/NIS that was not seen after injection with non-coding LPEI-PEG-GE11/antisenseNIS (C) Iodide uptake was perchlorate-sensitive (D) and therefore indeed NIS-mediated. (E) Radionuclide retention time in tumors was determined by serial scanning over 10 h (mean ± S.E.M.; 24 h: n=9, 48 h: n=7). 124I PET-imaging confirmed findings of scintigraphy and allowed better differentiation between tumoral and stomach radioiodide uptake (F, H) After injection of the control vector LPEI-PEG-GE11/antisenseNIS (G, I), no pancreatic iodide uptake activity above background levels could be detected. Significantly higher radioiodide accumulation 48 h after gene transfer as compared to 24 h was confirmed by PET (mean ± S.E.M.; n=5 each; *p<0.05) (J) SG: salivary glands.
To better distinguish tumoral uptake from iodide accumulation in the stomach, we additionally employed three-dimensional high-resolution 124I PET to image radioiodide biodistribution. Again, systemic injection of LPEI-PEG-GE11/NIS resulted in strong transfection of tumor tissue at both time points (Figures 2F, 2H), an effect that was not seen in mice treated with LPEI-PEG-GE11/antisenseNIS (Figures 2G, 2I). Quantification of tumoral 124I uptake again revealed significantly higher radioiodide accumulation 48 h after i.v. injection of LPEI-PEG-GE11/NIS as compared to 24 h after NIS gene transfer (Figure 2J).
Ex vivo analysis of NIS expression in PDAC
48 h after polyplex administration, mice were sacrificed and dissected. Tumors and non-target organs (liver, lung) were analyzed for NIS mRNA expression by quantitative real-time PCR (qPCR). A 20-fold increase in NIS mRNA expression was detected in PDAC lesions from mice injected with LPEI-PEG-GE11/NIS as compared to untreated tumors (Figure 3A). In contrast, no significant NIS mRNA expression above background levels was observed in non-target organs and tumors of mice treated with the control vector LPEI-PEG-GE11/antisenseNIS (Figure 3A). In tumors from LPEI-PEG-GE11/NIS-treated mice, areas of NIS-specific immunoreactivity were observed surrounding ductal lesions by immunohistochemical and immunofluorescence staining using a human NIS-specific antibody (Figure 3B). NIS staining was found to be both cell membrane-associated and cytoplasmic. In contrast, tumors from mice treated with the control vector LPEI-PEG-GE11/antisenseNIS showed no NIS-specific immunoreactivity (Figure 3B).
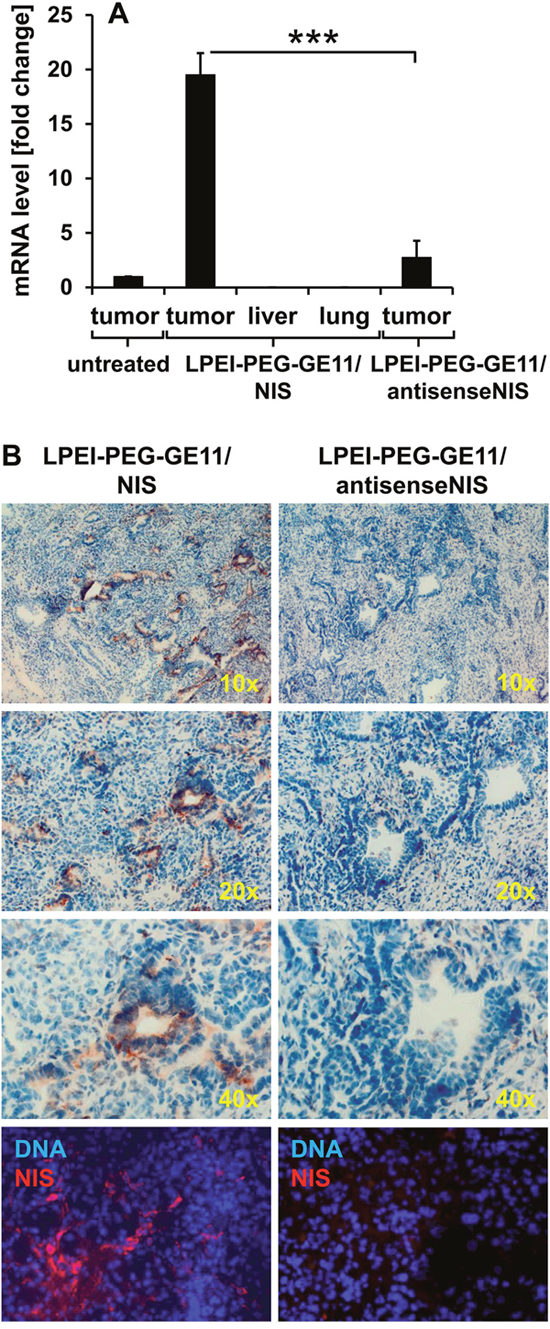
Figure 3: Analysis of NIS mRNA and protein distribution ex vivo. NIS-specific qPCR analysis revealed a 20-fold increase of NIS mRNA expression in pancreatic tumors of mice injected with LPEI-PEG-GE11/NIS as compared to tumors of untreated mice. In contrast, NIS mRNA was not increased in non-target organs and in tumors of mice injected with the control vector LPEI-PEG-GE11/antisenseNIS (mean-fold change ± S.E.M.; ***p<0.001) (A) Both immunohistochemical (B, upper three panels; magnification: 10×, 20× and 40×) and immunofluorescence staining (B, bottom panel; magnification: 200×) of sections of pancreatic tumors revealed areas of NIS-specific immunoreactivity after systemic application of LPEI-PEG-GE11/NIS. In contrast, tumors treated with the control vector LPEI-PEG-GE11/antisenseNIS showed no NIS-specific immunoreactivity.
NIS-mediated 131I therapy of PDAC
PDAC-bearing mice were treated with three cycles of LPEI-PEG-GE11/NIS followed by 131I 48 h later – the optimal time point for radionuclide injection based on the imaging studies. Controls were injected with non-coding LPEI-PEG-GE11/antisenseNIS and 131I or saline only. Tumor progression was monitored by magnetic resonance imaging (MRI). Mice in the therapy group showed a significant stabilization of tumor growth and, in two cases, even a reduction in tumor volume (Figures 4A, 4D), while aggressive tumor growth was observed in both control groups (Figures 4B-4D). This led to an enhanced survival in the therapy group that lived up to 28 days post therapy start with a median survival of 25 days, as compared to the antisenseNIS group that survived up to 13 days with a median survival of 11 days and saline controls that lived up to 21 days, median survival 21 days (Figure 4E). The effect on mouse survival was, however, not significant.
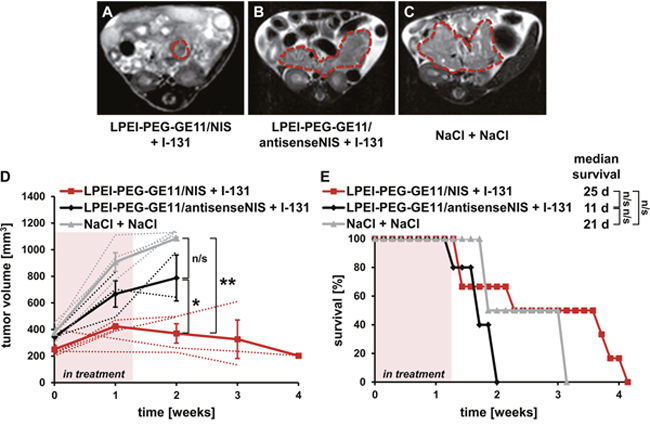
Figure 4: Therapeutic application of 131I after NIS gene transfer in vivo. Kras;p53 mice were treated with three cycles of i.v. injection of polyplexes on days 0/4/7 followed by i.p. injection of 55.5 MBq 131I 48 h later, on days 2/6/9. Tumor sizes were monitored weekly by MRI. Exemplary MRI images of endpoint tumor sizes from an LPEI-PEG-GE11/NIS + 131I- (A), an LPEI-PEG-GE11/ antisenseNIS + 131I- (B) and a NaCl + NaCl-treated Kras;p53 mouse are shown (C). Tumors are highlighted by red dotted lines. (D) Mice treated with LPEI-PEG-GE11/NIS + 131I (n=6) showed a stabilization in tumor volume compared to control groups LPEI-PEG-GE11/antisenseNIS + 131I (n=3; mean ± S.E.M.; *p<0.05) and NaCl + NaCl (n=4; **p<0.01). Mean tumor volumes (solid lines) and volumes for individual mice (dotted lines) are shown. (E) Injection of LPEI-PEG-GE11/NIS + 131I led to an increased overall and median survival in the therapy group (n=6) compared to control groups injected with LPEI-PEG-GE11/antisenseNIS + 131I (n=3; n/s) or NaCl + NaCl (n=4; n/s).
DISCUSSION
While the incidence of PDAC is gradually increasing, the prognosis of patients with pancreatic cancer has not significantly changed over the last 20 years – despite numerous advances in diagnostic imaging, surgical techniques and chemotherapeutic strategies [3, 27]. Intensified chemotherapy protocols in patients with advanced pancreatic cancer show a significant, yet still unsatisfactory survival benefit [28]. So far, no targeted agent or approach has changed this fatal course of the disease, even though preclinical trials in in vitro cell culture systems and in vivo xenograft models had shown promising results [3, 28]. These set-backs can mainly be attributed to the complexity of the disease. The homogeneous molecular equipment, simple stromal architecture and immune deficiency of xenograft models limits their transferability to the clinical setting. Endogenously grown tumors, in contrast, are genetically and morphologically heterogeneous, less vascularized and harbor a far more complex microenvironment with high immunosuppression and extensive desmoplasia [29, 30]. Genetically engineered mouse models that closely reflect the key aspects of pancreatic carcinogenesis have been shown to correlate well with data from clinical trials and provide an exciting new platform to predict human tumor responses to treatment [13].
After the proof-of-principle of our polyplex-mediated NIS gene therapy concept in different subcutaneous xenograft tumor models [16–18], the genetically engineered mouse model of PDAC used in this study provides an important step towards further development towards clinical application. Based on the known activity of EGFR in PDAC and this model, we chose EGFR-targeted LPEI-PEG-GE11 polymers as delivery vehicles for the NIS gene [17, 31, 32].
Transfection of PDAC explant cell cultures with LPEI-PEG-GE11/NIS led to significant perchlorate-sensitive and therefore NIS-mediated radioiodide accumulation. The empty vector LPEI-PEG-GE11 did not result in iodide accumulation above background levels, further confirming NIS-dependency of radioiodide accumulation. Iodide uptake was significantly reduced after transfection with non-targeted LPEI-PEG-Cys/NIS, demonstrating improved transfection efficiency using the targeting ligand GE11. EGFR-specificity of targeting was further substantiated by the observation that in EGFR-negative cultures derived from Kras;p53;Egfr mice, no significant difference between transfection with EGFR-targeted or non-targeted vectors was observed. Translating these promising in vitro results to systemic vector application in vivo, intravenous administration of LPEI-PEG-GE11/NIS resulted in a significant perchlorate-sensitive tumor-specific iodide uptake in mice harboring endogenous PDAC tumors, as demonstrated by 123I gamma camera imaging. Three-dimensional 124I PET imaging with increased sensitivity and resolution was employed for more accurate quantification of tumoral radioiodide uptake, as radionuclide signals from pancreatic lesions partially overlap with stomach signals based on physiological gastric NIS expression. Results from PET imaging confirmed gamma camera imaging results with strong radioiodide signals in pancreatic tumors. Control experiments with LPEI-PEG-GE11/antisenseNIS showed no significant tumoral radioiodide accumulation above background levels, confirming NIS-specificity of tumoral tracer uptake. These molecular imaging data were further corroborated by NIS-specific immunohistochemistry and immunofluorescence as well as qPCR analysis.
Both the abundance and the permeability of the tumor’s vasculature are crucial for sufficient transgene delivery into the tumor [33, 34]. One of the main factors thought to hamper efficient drug delivery to PDAC, is its highly desmoplastic stroma alongside its high interstitial pressure and poor vascularization [35]. Thus, the enhanced permeability and retention effect that is caused by the irregular, “leaky” tumor vasculature and is usually exploited for passive targeting of therapeutic agents to tumor sites, is limited in PDAC [34, 36]. For this reason, an additional tumor-targeting strategy is particularly important. Our imaging data convincingly demonstrate that targeting our polyplexes to EGFR allows strong transfection of pancreatic tumors with NIS. In a previous study, using the same vector construct in a subcutaneous hepatocellular carcinoma xenograft model, a tumor-absorbed dose of 47 mGy/MBq/g was calculated for 131I 24 h after polyplex administration [17], while in the current study, a dose of 74.7 mGy/MBq/g tumor 24 h post polyplex injection was achieved. We mainly attribute this significantly enhanced tumoral radioiodide uptake to the very high EGFR expression in PDAC. NIS staining was restricted to areas of high EGFR expression surrounding ductal lesions [7]. This focal pattern of transgene expression further underlines the advantage of NIS as therapy gene in this setting, as the high radionuclide bystander effect allows destruction of tumor cells beyond transfected cells.
Building on these promising results, the next logical step was to evaluate the therapeutic effectiveness of 131I in the PDAC mouse model after LPEI-PEG-GE11-mediated systemic NIS gene delivery. We were able to demonstrate stabilization, and, in two cases, even a pronounced reduction, of tumor growth after application of three cycles of LPEI-PEG-GE11/NIS followed by 131I. Mouse survival was prolonged in the therapy group, especially compared to the non-coding LPEI-PEG-GE11/antisenseNIS-treated control group, although without reaching statistical significance, despite the strong effects on tumor growth. Interestingly, while animals in the saline group had to be sacrificed due to compromised well-being owing to excessive tumor growth, the non-coding control group showed signs of ill health at much lower tumor volumes and had to be sacrificed. Similarly, effects on animal health were observed in the therapy group, though to a lower extent. We attribute this observation to toxicity of the LPEI-based conjugates, possibly combined with effects from 131I. Due to the stabilization of tumor growth in therapy animals, they fared better than the non-coding control group that was potentially affected by side effects from polyplex and radioiodide injection in addition to rapid tumor growth. To date, the use of LPEI-based polymers did not affect animal health in any of our previous studies, nor was viability of PDAC cell explants affected in the current study. LPEI has been shown to exhibit certain cytotoxic effects both in vitro and in vivo [37–41], though LPEI-based polyplexes have already been tested in a clinical trial for bladder cancer therapy and no adverse effects were reported [42]. Similarly, we have so far only encountered side effects from 131I in one previous study with the objective to radioablate mouse thyroids under intentional stimulation of thyroidal radioiodide uptake [43]. Symptoms developed with a delay of seven days after radioiodide application, while in the current study, animal health deteriorated from the beginning of treatment [43]. However, our earlier work was done in subcutaneous xenograft models, where tumor growth per se has no impact on animal health. In contrast, Kras;p53 mice with their extremely aggressive pancreatic tumor growth and subsequent rapid health deterioration, seem to react more unfavorably to the polyplexes and/or radioiodide treatment. LPEI is seen as the “gold standard” for non-viral DNA delivery, as it shows such high transfection efficiency and flexibility at relatively low toxicity, compared to other viral and non-viral gene delivery approaches. To further refine our approach and solve the toxicity issue, we are currently developing sequence-defined polymers with higher biocompatibility for targeted NIS gene delivery [26].
In conclusion, our data clearly show the high potential of EGFR-targeted nanoparticle vectors to target the NIS gene to PDAC. After systemic application of LPEI-PEG-GE11/NIS, we were able to reach sufficient iodide concentrations at the tumor site to (1) produce a strong enough signal to image pancreatic tumors in situ and (2) provoke a therapeutic effect. Based on its role as potent and well characterized reporter gene, NIS allows non-invasive imaging and detailed characterization of in vivo biodistribution of functional NIS expression as an essential prerequisite for exact planning and monitoring of clinical gene therapy trials with the aim of individualization of the NIS gene therapy concept in the clinical setting.
MATERIALS AND METHODS
Establishment of genetically modified mice
Establishment of the Kras;p53 (Ptf1a+/Cre;Kras+/LSL-G12D;Trp53loxP/loxP) and Kras;p53;Egfr (Ptf1a+/Cre;Kras+/LSL-G12D;Trp53loxP/loxP;Egfrfl/fl) strains has been described previously [4, 11, 12, 44, 45]. Mouse strains were maintained on a mixed C57BL/6;129/Sv background. Animals were kept under specific pathogen-free conditions with access to mouse chow and water ad libitum. Both male and female mice at 5-7.5 weeks of age were used for experiments. The experimental protocol was approved by the regional governmental commission for animals (Regierung von Oberbayern, Munich, Germany).
Preparation and culture of PDAC cell explants
Cell explants from primary PDAC of Kras;p53 and Kras;p53;Egfr mice were isolated as described previously [46] and cultured in DMEM high glucose medium (Invitrogen, Karlsruhe, Germany) supplemented with 10% fetal bovine serum (v/v; PAA, Colbe, Germany), 100 U/mL penicillin/100 μg/mL streptomycin (Invitrogen) and 1% non-essential amino acids (v/v; Invitrogen). Cells were maintained at 37°C and 5% CO2 in an incubator with 95% humidity. Cell culture medium was replaced every second day and explants were passaged at 85% confluency.
Plasmid and polymer synthesis and polyplex formation
The human NIS-encoding plasmid and LPEI-based conjugates were cloned and synthesized, respectively, as described previously [17]. Plasmid DNA was condensed with polymers at indicated c/p ratios (w/w) in HEPES-buffered glucose (HBG: 20 mmol/L HEPES, 5% glucose (w/v), pH 7.4) as described previously [47] and incubated at room temperature for 20 min before use. Final DNA concentrations were 2 μg/mL for in vitro studies and 200 μg/mL for in vivo studies.
Transient transfection
For in vitro transfection experiments, PDAC cell explants were grown to 60-80% confluency. Explants were incubated for 4 hours with polyplexes in the absence of serum and antibiotics followed by incubation with complete growth medium for 24 h. Either LPEI-PEG-GE11/NIS (EGFR-targeting of NIS due to the EGFR-specific ligand GE11), LPEI-PEG-Cys/NIS (no active targeting of NIS to EGFR, as the ligand GE11 is replaced by a cysteine residue), or LPEI-PEG-GE11 alone (polymer without NIS DNA) were added in c/p ratios as indicated. Transfection efficiency was evaluated by measurement of iodide uptake activity as described below. Transfections were done in triplicate for each separate explant.
In vitro 125I uptake assay
Following transfections, iodide uptake of PDAC cell explants was determined at steady-state conditions as described previously [48, 49]. Results were normalized to cell viability that was measured using the commercially available MTS-assay (Promega, Mannheim, Germany) as described previously [50].
Radioiodide uptake after systemic NIS gene transfer in vivo
For the proof-of-principle of NIS-mediated tumor-specific radioiodide accumulation in vivo, polyplexes (LPEI-PEG-GE11/NIS, c/p 0.8) were applied via the tail vein at a DNA dose of 2.5 mg/kg (50 μg DNA in 250 μL HBG). Mice received 18.5 MBq 123I (GE Healthcare, Braunschweig, Germany) intraperitoneally (i.p.) 24 h (n=9) or 48 h (n=7) after polyplex injection and radioiodide distribution was monitored by serial imaging on a gamma camera (Forte, ADAC Laboratories, Milpitas, CA, USA) equipped with a VXHR (ultra-high resolution) collimator as described previously [50]. Regions of interest were quantified and expressed as a fraction of the total amount of applied radionuclide per gram tumor tissue. The retention time within the tumor was determined by serial scanning after radioiodide injection. A subset of mice (n=2 for each time point) was pretreated i.p. with 2 mg of the competitive NIS inhibitor sodium perchlorate (NaClO4; Sigma-Aldrich, Taufkirchen, Germany) 30 min before 123I administration. Dosimetric calculations for 131I were made using the Medical Internal Radiation Dose (MIRD) technique and a RADAR dose factor (http://www.doseinfo-radar.com). In order to achieve better discrimination between uptake in the tumor and the adjacent stomach, 24 or 48 h after i.v. injection of polyplexes (LPEI-PEG-GE11/NIS, each time point n=5; LPEI-PEG-GE11/antisenseNIS, each time point n=1) mice received 10 MBq 124I (Perkin Elmer, Waltham, MA, USA) i.p. and radioiodide biodistribution was monitored by static acquisition 3 h post injection using a micro PET system (Inveon, Siemens Preclinical Solutions, Erlangen, Germany). Mean tumoral radioiodide uptake was calculated in MBq/mL by manually placing 3D regions of interest in the tumor.
Analysis of NIS mRNA expression by quantitative real-time PCR
Total RNA was isolated from PDAC or non-target tissues (liver, lung) using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s recommendations. Single-stranded oligo (dT)-primer cDNA was generated using Super Script III Reverse Transcriptase (Invitrogen). qPCR was performed with the cDNA from 1 μg RNA using SYBR Green PCR master mix (Qiagen) in a Rotor Gene 6000 (Corbett Research, Morthlake, New South Wales, Australia). The following primers were used: NIS, forward 5′-ACACCTTCTGGACCTTCGTG-3′, reverse 5′-GTCGCAGTCGGTGTAGAACA-3′ and GAPDH, forward 5′-GAGAAGGCTGGGGCTCATTT-3′, reverse 5′-CAGTGGGGACACGGAAGG-3′. Relative expression levels were calculated using the comparative ΔΔCt method and internal GAPDH for normalization.
Analysis of tissue sections
Immunohistochemical and immunofluorescence staining of NIS was performed using a mouse monoclonal antibody directed against human NIS (kindly provided by John C Morris, Mayo Clinic, Rochester, MN, USA) as described previously [20, 51].
Radioiodide therapy
Starting when mice were around 30 d of age, tumor sizes were assessed weekly by high resolution MRI on a 3T clinical scanner (Philips Ingenia 3.0T; Royal Philips Electronics, Eindhoven, The Netherlands). Once tumors reached the inclusion size of 200-450 mm3, therapy trials were started. To this end, 48 h after systemic administration of LPEI-PEG-GE11/NIS or, as control, LPEI-PEG-GE11/antisenseNIS, a therapeutic dose of 55.5 MBq 131I (GE Healthcare) was administered i.p. (LPEI-PEG-GE11/NIS + 131I n=6; LPEI-PEG-GE11/antisenseNIS + 131I n=3). A second control group received saline only (n=4). The cycle consisting of systemic NIS gene transfer followed by radioiodide was repeated for a total of three times on days 0/2, 4/6 and 7/9. Mice from all groups were sacrificed when at least one endpoint criterion was reached. Endpoint criteria included a tumor volume >1000 mm3, a body weight loss >15%, as well as a number of general physical, clinical and behavioral criteria. Body condition was monitored by independent animal care personnel blind to treatment and hypothesis.
Statistics
Results are reported as mean ± S.E.M., mean-fold change ± S.E.M. or, for survival plots, percent. Statistical significance was generally tested by two-tailed Student’s t-test except for the therapy study. For tumor volumes, one-way ANOVA was performed, followed by Tukey’s Honestly Significant Difference test. Statistical significance of Kaplan-Meier plots was analyzed by log-rank test. p-values <0.05 were considered statistically significant (*p<0.05; **p<0.01; ***p<0.001; n/s not significant).
ACKNOWLEDGMENTS
We are grateful to Sissy M Jhiang (Ohio State University, Columbus, OH, USA) for supplying the full-length human NIS complementary DNA and to John C Morris (Mayo Clinic, Rochester, MN, USA) for providing the NIS mouse monoclonal antibody. We also thank Nathalie Schwenk (LMU Munich, Munich, Germany) for her help with in vitro and ex vivo studies, Sybille Reder, Stefan Ambros, Klemens Scheidhauer and Jakob Allmann (Klinikum rechts der Isar der Technischen Universität München, Munich, Germany) for assistance with PET and MRI studies, Wolfgang Rödl (LMU Munich, Munich, Germany) for conjugate synthesis as well as Doris Mayr (LMU Munich, Munich, Germany) for preparation of paraffin-embedded slides.
FUNDING
This work was supported by grants from the Deutsche Forschungsgemeinschaft within the Collaborative Research Center SFB 824 to C.S. (project C8) and J.T.S. (project C4) and within the Priority Programme SPP1629 to C.S. and P.J.N. (SP 581/6-1, SP 581/6-2, NE 648/5-2), as well as a grant from the Wilhelm-Sander-Stiftung to C.S. (2014.129.1), a grant within the European Commission Seventh Framework Program (FP7/CAM-PaC under grant agreement n°602783) and a grant from the German Cancer Consortium (DKTK) to J.T.S., a grant from the excellence cluster Nanosystems Initiative Munich to E.W. as well as a grant from the Center for Nanoscience (CeNS) to M.O. The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest that could be perceived as prejudicing the impartiality of the research reported.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65:5-29. doi: 10.3322/caac.21254.
2. Narayanan V, Weekes CD. Molecular therapeutics in pancreas cancer. World J Gastrointest Oncol. 2016; 8:366-379. doi: 10.4251/wjgo.v8.i4.366.
3. Mazur PK, Siveke JT. Genetically engineered mouse models of pancreatic cancer: unravelling tumour biology and progressing translational oncology. Gut. 2012; 61:1488-1500. doi: 10.1136/gutjnl-2011-300756.
4. Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003; 4:437-450. doi: 10.1016/S1535-6108(03)00309-X.
5. Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM, Deng CX, Hruban RH, Adsay NV, Tuveson DA, Hingorani SR. Kras (G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell. 2007; 11:229-243. doi: 10.1016/j.ccr.2007.01.017.
6. Siveke JT, Schmid RM. Chromosomal instability in mouse metastatic pancreatic cancer--it's Kras and Tp53 after all. Cancer Cell. 2005; 7:405-407. doi: 10.1016/j.ccr.2005.04.025.
7. Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC, Pal D, Briel T, Herner A, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012; 22:304-317. doi: 10.1016/j.ccr.2012.07.024.
8. Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, Hanahan D, Redston MS, Chin L, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A. 2006; 103:5947-5952. doi: 10.1073/pnas.0601273103.
9. Schneider G, Siveke JT, Eckel F, Schmid RM. Pancreatic cancer: basic and clinical aspects. Gastroenterology. 2005; 128:1606-1625. doi: 10.1053/j.gastro.2005.04.001.
10. Siveke JT, Einwachter H, Sipos B, Lubeseder-Martellato C, Kloppel G, Schmid RM. Concomitant pancreatic activation of Kras (G12D) and Tgfa results in cystic papillary neoplasms reminiscent of human IPMN. Cancer Cell. 2007; 12:266-279. doi: 10.1016/j.ccr.2007.08.002.
11. Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002; 32:128-134. doi: 10.1038/ng959.
12. Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000; 14:994-1004. doi: 10.1101/gad.14.8.994.
13. Singh M, Lima A, Molina R, Hamilton P, Clermont AC, Devasthali V, Thompson JD, Cheng JH, Bou Reslan H, Ho CC, Cao TC, Lee CV, Nannini MA, et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol. 2010; 28:585-593. doi: 10.1038/nbt.1640.
14. Carrasco N. Iodide transport in the thyroid gland. Biochim Biophys Acta. 1993; 1154:65-82. doi: 10.1016/0304-4157(93)90017-I.
15. Jhiang SM, Cho JY, Ryu KY, DeYoung BR, Smanik PA, McGaughy VR, Fischer AH, Mazzaferri EL. An immunohistochemical study of Na+/I- symporter in human thyroid tissues and salivary gland tissues. Endocrinology. 1998; 139:4416-4419. doi: 10.1210/endo.139.10.6329.
16. Klutz K, Russ V, Willhauck MJ, Wunderlich N, Zach C, Gildehaus FJ, Goke B, Wagner E, Ogris M, Spitzweg C. Targeted radioiodine therapy of neuroblastoma tumors following systemic nonviral delivery of the sodium iodide symporter gene. Clin Cancer Res. 2009; 15:6079-6086. doi: 10.1158/1078-0432.CCR-09-0851.
17. Klutz K, Schaffert D, Willhauck MJ, Grunwald GK, Haase R, Wunderlich N, Zach C, Gildehaus FJ, Senekowitsch-Schmidtke R, Goke B, Wagner E, Ogris M, Spitzweg C. Epidermal growth factor receptor-targeted (131)I-therapy of liver cancer following systemic delivery of the sodium iodide symporter gene. Mol Ther. 2011; 19:676-685. doi: 10.1038/mt.2010.296.
18. Klutz K, Willhauck MJ, Dohmen C, Wunderlich N, Knoop K, Zach C, Senekowitsch-Schmidtke R, Gildehaus FJ, Ziegler S, Furst S, Goke B, Wagner E, Ogris M, et al. Image-guided tumor-selective radioiodine therapy of liver cancer after systemic nonviral delivery of the sodium iodide symporter gene. Hum Gene Ther. 2011; 22:1563-1574. doi: 10.1089/hum.2011.041.
19. Klutz K, Willhauck MJ, Wunderlich N, Zach C, Anton M, Senekowitsch-Schmidtke R, Goke B, Spitzweg C. Sodium iodide symporter (NIS)-mediated radionuclide ((131)I, (188)Re) therapy of liver cancer after transcriptionally targeted intratumoral in vivo NIS gene delivery. Hum Gene Ther. 2011; 22:1403-1412. doi: 10.1089/hum.2010.158.
20. Knoop K, Kolokythas M, Klutz K, Willhauck MJ, Wunderlich N, Draganovici D, Zach C, Gildehaus FJ, Boning G, Goke B, Wagner E, Nelson PJ, Spitzweg C. Image-guided, tumor stroma-targeted 131I therapy of hepatocellular cancer after systemic mesenchymal stem cell-mediated NIS gene delivery. Mol Ther. 2011; 19:1704-1713. doi: 10.1038/mt.2011.93.
21. Knoop K, Schwenk N, Dolp P, Willhauck MJ, Zischek C, Zach C, Hacker M, Goke B, Wagner E, Nelson PJ, Spitzweg C. Stromal targeting of sodium iodide symporter using mesenchymal stem cells allows enhanced imaging and therapy of hepatocellular carcinoma. Hum Gene Ther. 2013; 24:306-316. doi: 10.1089/hum.2012.104.
22. Knoop K, Schwenk N, Schmohl K, Muller A, Zach C, Cyran C, Carlsen J, Boning G, Bartenstein P, Goke B, Wagner E, Nelson PJ, Spitzweg C. Mesenchymal stem cell-mediated, tumor stroma-targeted radioiodine therapy of metastatic colon cancer using the sodium iodide symporter as theranostic gene. J Nucl Med. 2015; 56:600-606. doi: 10.2967/jnumed.114.146662.
23. Grunwald GK, Klutz K, Willhauck MJ, Schwenk N, Senekowitsch-Schmidtke R, Schwaiger M, Zach C, Goke B, Holm PS, Spitzweg C. Sodium iodide symporter (NIS)-mediated radiovirotherapy of hepatocellular cancer using a conditionally replicating adenovirus. Gene Ther. 2013; 20:625-633. doi: 10.1038/gt.2012.79.
24. Grunwald GK, Vetter A, Klutz K, Willhauck MJ, Schwenk N, Senekowitsch-Schmidtke R, Schwaiger M, Zach C, Wagner E, Goke B, Holm PS, Ogris M, Spitzweg C. Systemic image-guided liver cancer radiovirotherapy using dendrimer-coated adenovirus encoding the sodium iodide symporter as theranostic gene. J Nucl Med. 2013; 54:1450-1457. doi: 10.2967/jnumed.112.115493.
25. Grunwald GK, Vetter A, Klutz K, Willhauck MJ, Schwenk N, Senekowitsch-Schmidtke R, Schwaiger M, Zach C, Wagner E, Goke B, Holm PS, Ogris M, Spitzweg C. EGFR-Targeted Adenovirus Dendrimer Coating for Improved Systemic Delivery of the Theranostic NIS Gene. Mol Ther Nucleic Acids. 2013; 2: e131. doi: 10.1038/mtna.2013.58.
26. Urnauer S, Morys S, Krhac Levacic A, Muller AM, Schug C, Schmohl KA, Schwenk N, Zach C, Carlsen J, Bartenstein P, Wagner E, Spitzweg C. Sequence-defined cMET/HGFR-targeted Polymers as Gene Delivery Vehicles for the Theranostic Sodium Iodide Symporter (NIS) Gene. Mol Ther. 2016; 24:1395-1404. doi: 10.1038/mt.2016.95.
27. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014; 74:2913-2921. doi: 10.1158/0008-5472.can-14-0155.
28. Schober M, Javed MA, Beyer G, Le N, Vinci A, Sund M, Neesse A, Krug S. New Advances in the Treatment of Metastatic Pancreatic Cancer. Digestion. 2015; 92:175-184. doi: 10.1159/000439523.
29. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009; 324:1457-1461. doi: 10.1126/science.1171362.
30. Mahajan UM, Teller S, Sendler M, Palankar R, van den Brandt C, Schwaiger T, Kuhn JP, Ribback S, Glockl G, Evert M, Weitschies W, Hosten N, Dombrowski F, et al. Tumour-specific delivery of siRNA-coupled superparamagnetic iron oxide nanoparticles, targeted against PLK1, stops progression of pancreatic cancer. Gut. 2016; 65:1838-1849. doi: 10.1136/gutjnl-2016-311393.
31. Li Z, Zhao R, Wu X, Sun Y, Yao M, Li J, Xu Y, Gu J. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005; 19:1978-1985. doi: 10.1096/fj.05-4058com.
32. Schaffert D, Kiss M, Rodl W, Shir A, Levitzki A, Ogris M, Wagner E. Poly (I:C)-mediated tumor growth suppression in EGF-receptor overexpressing tumors using EGF-polyethylene glycol-linear polyethylenimine as carrier. Pharm Res. 2011; 28:731-741. doi: 10.1007/s11095-010-0225-4.
33. Smrekar B, Wightman L, Wolschek MF, Lichtenberger C, Ruzicka R, Ogris M, Rodl W, Kursa M, Wagner E, Kircheis R. Tissue-dependent factors affect gene delivery to tumors in vivo. Gene Ther. 2003; 10:1079-1088. doi: 10.1038/sj.gt.3301965.
34. Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011; 6:815-823. doi: 10.1038/nnano.2011.166.
35. Miller BW, Morton JP, Pinese M, Saturno G, Jamieson NB, McGhee E, Timpson P, Leach J, McGarry L, Shanks E, Bailey P, Chang D, Oien K, et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol Med. 2015; 7:1063-1076. doi: 10.15252/emmm.201404827.
36. Li J, Liu F, Gupta S, Li C. Interventional Nanotheranostics of Pancreatic Ductal Adenocarcinoma. Theranostics. 2016; 6:1393-1402. doi: 10.7150/thno.15122.
37. Grandinetti G, Smith AE, Reineke TM. Membrane and nuclear permeabilization by polymeric pDNA vehicles: efficient method for gene delivery or mechanism of cytotoxicity? Mol Pharm. 2012; 9:523-538. doi: 10.1021/mp200368p.
38. Moghimi SM, Symonds P, Murray JC, Hunter AC, Debska G, Szewczyk A. A two-stage poly (ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol Ther. 2005; 11:990-995. doi: 10.1016/j.ymthe.2005.02.010.
39. Hall A, Larsen AK, Parhamifar L, Meyle KD, Wu LP, Moghimi SM. High resolution respirometry analysis of polyethylenimine-mediated mitochondrial energy crisis and cellular stress: Mitochondrial proton leak and inhibition of the electron transport system. Biochim Biophys Acta. 2013; 1827:1213-1225. doi: 10.1016/j.bbabio.2013.07.001.
40. Chollet P, Favrot MC, Hurbin A, Coll JL. Side-effects of a systemic injection of linear polyethylenimine-DNA complexes. J Gene Med. 2002; 4:84-91. doi: 10.1002/jgm.237.
41. Wiseman JW, Goddard CA, McLelland D, Colledge WH. A comparison of linear and branched polyethylenimine (PEI) with DCChol/DOPE liposomes for gene delivery to epithelial cells in vitro and in vivo. Gene Ther. 2003; 10:1654-1662. doi: 10.1038/sj.gt.3302050.
42. Sidi AA, Ohana P, Benjamin S, Shalev M, Ransom JH, Lamm D, Hochberg A, Leibovitch I. Phase I/II marker lesion study of intravesical BC-819 DNA plasmid in H19 over expressing superficial bladder cancer refractory to bacillus Calmette-Guerin. J Urol. 2008; 180:2379-2383. doi: 10.1016/j.juro.2008.08.006.
43. Schmohl KA, Muller AM, Schwenk N, Knoop K, Rijntjes E, Kohrle J, Heuer H, Bartenstein P, Goke B, Nelson PJ, Spitzweg C. Establishment of an Effective Radioiodide Thyroid Ablation Protocol in Mice. Eur Thyroid J. 2015; 4:74-80. doi: 10.1159/000381019.
44. Lee TC, Threadgill DW. Generation and validation of mice carrying a conditional allele of the epidermal growth factor receptor. Genesis. 2009; 47:85-92. doi: 10.1002/dvg.20464.
45. Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci U S A. 2007; 104:17081-17086. doi: 10.1073/pnas.0704126104.
46. Heid I, Lubeseder-Martellato C, Sipos B, Mazur PK, Lesina M, Schmid RM, Siveke JT. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology. 2011; 141:719-730, 730.e711-717. doi: 10.1053/j.gastro.2011.04.043.
47. Russ V, Gunther M, Halama A, Ogris M, Wagner E. Oligoethylenimine-grafted polypropylenimine dendrimers as degradable and biocompatible synthetic vectors for gene delivery. J Control Release. 2008; 132:131-140. doi: 10.1016/j.jconrel.2008.09.003.
48. Spitzweg C, Zhang S, Bergert ER, Castro MR, McIver B, Heufelder AE, Tindall DJ, Young CY, Morris JC. Prostate-specific antigen (PSA) promoter-driven androgen-inducible expression of sodium iodide symporter in prostate cancer cell lines. Cancer Res. 1999; 59:2136-2141.
49. Weiss SJ, Philp NJ, Grollman EF. Iodide transport in a continuous line of cultured cells from rat thyroid. Endocrinology. 1984; 114:1090-1098. doi: 10.1210/endo-114-4-1090.
50. Willhauck MJ, Sharif Samani BR, Gildehaus FJ, Wolf I, Senekowitsch-Schmidtke R, Stark HJ, Goke B, Morris JC, Spitzweg C. Application of 188rhenium as an alternative radionuclide for treatment of prostate cancer after tumor-specific sodium iodide symporter gene expression. J Clin Endocrinol Metab. 2007; 92:4451-4458. doi: 10.1210/jc.2007-0402.
51. Spitzweg C, Baker CH, Bergert ER, O'Connor MK, Morris JC. Image-guided radioiodide therapy of medullary thyroid cancer after carcinoembryonic antigen promoter-targeted sodium iodide symporter gene expression. Hum Gene Ther. 2007; 18:916-924. doi: 10.1089/hum.2007.081.