
INTRODUCTION
Endometriosis, an estrogen-dependent chronic gynecological disease, is defined as the presence of functional endometrial stroma and glands outside the uterine cavity. The disease affects 10%-15% of woman in reproduction age [1], often associated with chronic pelvic pain and infertility [2]. There are three types of endometriotic lesions: peritoneal, ovarian endometriosis and rectovaginal, which are classified by the different location of lesions, may have different pathogeneses [3]. However, study has found that all endometriotic lesions would share the same commonality as being the repeated tissue injury and repair (ReTIAR), resulting in fibrosis [4]. Endometriosis is considered as a multifactor disease affected by hormonal, immunological, and environmental factors, while lacking a clear Mendelian pattern of inheritance. Recent molecular cytogenetic studies provide novel evidence that acquired chromosome-specific alterations may induce endometriosis [5]. But strong gene-environmental interactions might definitively influence the approaches to identify genetic variants involved [6]. So even endometriosis has long been studied, the exact cause of this disease is still unclear.
There are various theories that have been proposed to explain the development of endometriosis. The most widely accepted theory is the retrograde menstruation or transplantation theory [7]. Recently, a new branch of this theory implicates that mesothelial epithelial to mesenchymal transition (EMT) as the pathogenic mechanism [8]. A patch of mesothelial cells that has undergone EMT no longer provides the sheet-like protective barrier between the basal layer and the luminal space. Absent of the mesothelial barrier, endometrial cells could easily adhere to the underlying peritoneal stroma and establish endometriotic lesions. In both peritoneal and ovarian endometriotic lesions, epithelial markers were found downregulated in the epithelial cells, while the mesenchymal markers upregulated [9, 10]. Besides the change of cell makers, there are several other evidences indicating that EMT is essential for the development of endometriosis. For example, one of the challenge of Sampson’s retrograde hypothesis is that cells would die when they detach from extracellular matrix (ECM) or adhere to inappropriate locations, which named as anoikis. However, EMT has a main feature associated with anoikis resistance, contributing to the spread of ectopic lesions [11]. Secondly, during the development of the urogenital system, endometrium is derived from intermediate mesoderm via mesenchymal to epithelial transition (MET). As some imprint of their mesenchymal origin retained, endometrial epithelial cells may have a particularly tend to return to the original state, via EMT [12].
However, since EMT process is mainly studied in cancer field and the origin of endometriosis is controversial, there are few investigations of EMT in endometriosis. Nowadays, the related work is still stagnant on tissue level, no accurate EMT signal pathways have been studied in endometriotic cells. So far majority of studies didn’t explain which type of EMT exactly occurs in endometriosis. Based on existed literature research, we conclude that two different types of EMT may be involved in. This hypothesis urgently need further experimental evidences, and it may provide a new therapy target for this disease. Besides, two stimulating signals, hypoxia and estrogen, can through different pathways to activate the EMT process in endometriosis. Those pathways involve many cellular factors such as TGF-β and Wnt, ultimately leading to cell proliferation and migration. At last, we are trying to explain the infertility, a severe problem caused by endometriosis, through an EMT related aspect and provide a potential therapy implication. We hope this relative comprehensive review of EMT in endometriosis could appeal to more investigations, ultimately lead to radical treatments for this disease.
DIFFERENT TYPES OF EMT ARE INVOLVED IN ENDOMETRIOSIS
At a 2007 meeting on EMT in Poland and a subsequent meeting in March 2008 at Cold Spring Harbor Laboratories, EMT is classified into three different biological subtypes based on the biological context in which they occur. Type 1 EMT occurs during embryo development both in vertebrates and invertebrates, involved in the generation of tissues and organs [13]. It is an entirely normal physiological process and not associated with other abnormal function such as inflammation, fibrosis, or invasion; Type 2 EMT, often occurs in response to wound or inflammatory injury [14]. When the injury is acute, Type 2 EMT will be limited. However, under chronic injury condition, the damage and inflammatory response are persistent, Type 2 EMT would contribute to tissue fibrosis and other organ destructions; Type 3 EMT is recognized as the major cause of the tumor metastatic. With the induction of angiogenesis, Type 3 EMT can strongly promote the cancer cells to undergo a series of steps characteristic of metastatic cascade [15, 16]. Another way to classify different types of EMT is meta-analysis of multiple microarray datasets. The three previous lists of EMT-related genes are referred as PO-List [17], EM-list [18] and SC-list [19]. However, low overlap of the generic signatures generated from different investigations suggests that generic network employed by EMT could be more complex than expected [20]. In this review, we will follow the classic three types of EMT theory, which is more widely acknowledged.
EMT is a biology process that cells lose the epithelial features and instead gain properties of mesenchymal cells. This process requires a series of complex changes in cell architecture and behavior, which often driven by the various cellular signals (Figure 1). The molecular change correlated with this transition include the loss of other epithelial markers such as E-cadherin, Desmoplakin, Mucin-1, occludin, and claudin, and the gain of mesenchymal markers such as N-cadherin, smooth-muscle actin, vimentin, and fibronectin et al. All of these molecular changes are associated with the alteration of cell functions such as enhanced migration, invasiveness and resistance to apoptosis [21, 22].
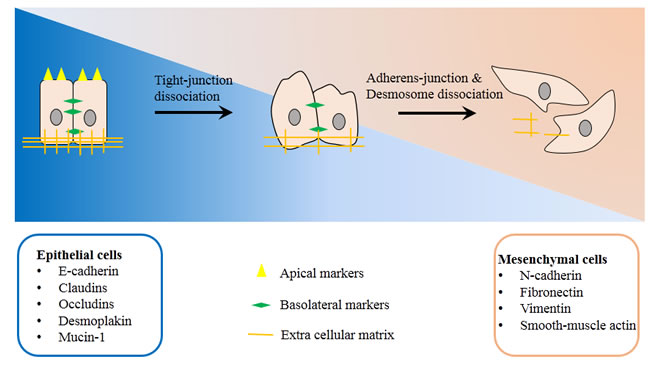
Figure 1: Basic mechanism of Epithelial-to-mesenchymal transition. Epithelial cells gradually loss cell-to-cell junctions and degrade extra cellular matrix, finally transformed into mesenchymal cells. Phenotype and functional markers of EMT include: Spindle shape to fibroblast like phenotype; losing of epithelial markers such as E-cadherin, Desmoplakin, Mucin-1, occludin, and claudin; gain of mesenchymal markers such as N-cadherin, smooth-muscle actin, vimentin, and fibronectin et al; increased invasion capacity; increased resistance to anoikis/ apoptosis.
Transcription factors such as the Snail family of zinc-finger transcription factors (Snail, Slug, and Smuc), the δEF1 family of two-handed zinc-finger factors (δEF1/ZEB1 and SIP1/ZEB2), and the basic helix–loop–helix factors Twist and E12/E47 are known as the key EMT regulators [23]. Except for Twist, these transcription factors could repress expression of E-cadherin by directly binding to the E-box sites in its promoter [24]. MicroRNAs are also tightly connected with the regulatory network of those transcriptions. For example, members of the miR-200 family maintain an epithelial status and prevent EMT through inhibition of ZEB1 and ZEB2 [25]. In turn, miR-200 members are transcriptionally repressed by ZEB factors—as well as Snail1—thus forming a double-negative loop that maintain cells in either an epithelial or mesenchymal state [26]. P53 could down-regulate Snail1 and Snail2 via induction of miR-34a/b/c. [27] Conversely, Snail1 and Snail2 (and ZEB1) transcriptionally repress miR-34a/b/c [28]. These double-negative loops regulate the transition direction of cells, and well explained the complex of EMT process. These cellular changes are almost common features in all three EMT types, which make the specified study of each type more difficult.
Though the detailed differences governing the three EMT types are not yet clear, their functional distinctions are apparent. Considering endometriosis is a chronic gynecological disease and has a relative risk deteriorating to tumor, we speculate that Type 2 and Type 3 EMT might be involved in this process. As mentioned above, endometriosis is a ReTIAR process and is histologically characterized by dense fibrous tissue surrounding the lesions [1]. This is more distinct in deep infiltrating endometriosis, and about 10%–15% of endometriosis cases formed dense scarring [29]. Besides, endometriosis has long been recognized as an inflammatory disease, for dysregulation of immune response has been noted in patients [30, 31]. Based on those evidence, we can infer that Type 2 EMT might be involved in the fibrosis of endometriosis, by participating the chronic inflammatory reaction. On the other hand, one of the most specific feature that depart the endometriosis from other benign disease, is the metastasis ability as malignancy induced by Type 3 EMT. This enhanced metastasis ability is often related with angiogenesis, which promotes endothelial functions, vascular permeability and development of experimental endometriosis [32].
As it’s not enough to focus on part of characteristics of endometriosis to design effective therapeutics, further studies need to pay attention to the different types of EMT in different phase of the endometriosis.
TWO STIMULATING SIGNALS MAY INDUCE EMT IN ENDOMETRIOSIS
Hypoxia signal
The hypoxic conditions is marked by a transcriptional response of the exposed cells that result in upregulated expression of hypoxia-inducible factors (HIFs). HIF-1 is the most characterized regulator of the cellular response to hypoxia, composed of HIF-1α and HIF-1β subunits. In contract to the constitutively expressed HIF-1β subunit, the expression and activity of the HIF-1α subunit are precisely regulated by the cellular O2 concentration [33]. In normal, eutopic and ectopic endometrial tissues, overexpression of HIF-1α and morphology changes associated with EMT were observed [34]. Hypoxic and ischemic conditions can induce the EMT of endometrial cells, not only in the healthy endometria, but also the eutopic endometria of patients with endometriosis [35]. According to the retrograde menstruation theory, we can hypothesis that endometrial epithelial cells might be adapted to specific microenvironments like hypoxia during implantation, and the enhanced invasion capacity by EMT can contribute to the development of ectopic lesions. Obviously, this process also requires angiogenesis, which is the most essential process induced by hypoxia in the endometriotic lesions. Vascular endothelial growth factor (VEGF) is strongly expressed in both in eutopic and ectopic lesions [36], and an increased level of VEGF was found in the peritoneal fluid of women with endometriosis [37]. The VEGF/NRP-1 axis may also be associated with enhanced EMT process and the NF-κB and β-catenin signaling in tumor metastasis [38]. Subsequent to HIF-1α and VEGF-dependent pathway, Reactive oxygen species (ROS) are found to stimulate EMT and increased invasion capability in several human cancer cells [39, 40]. Higher endogenous oxidative stress was found in endometriotic cells and could lead to an increase of ROS production and a drop in catalase levels [41]. In addition, lysyl oxidases isoforms (LOX and LOXL2) are also suggested as EMT mediators of hypoxia, which probably promote epithelial cell plasticity by functional interacting with certain transcriptional repressors. A study demonstrated that higher LOXs was expressed in endometriotic lesions compared with normal endometrium [42]. It should be noted that several other factors related to EMT process were up-regulated under hypoxia, including HGF, NF-κB, Tswist1 and TGF-β [43, 44]. Moreover, the Notch signaling pathway was also involved in hypoxia-induced EMT by increasing motility and invasiveness [45].
Estrogen signal
As a hormone dependent disease, endometriosis is deeply regulated by estrogen-mediated cellular signaling. Compared with normal endometrium, endometriotic lesions exhibit upregulated estradiol biosynthesis and low level of estradiol inactivation [46]. Here are two different forms of the estrogen receptor (ER), ERα and ERβ, independently encoded by two different gene, ESR1 and ESR2. Each ER isoform has a unique expression pattern in the endometriotic tissues. Indeed, in the female reproductive system, ERα is expressed primarily in the uterus, whereas ERβ is expressed primarily in the ovary [47]. In the previous studies, people believed that whether of ovarian or peritoneal locations, had higher ERβ expression rather than ERα compared with normal human endometrial cells [48], which was similarly detected in many animal models of endometriosis [49, 50]. However, a recent study shows that ERα expression was increased in ovarian endometriotic lesions compared with normal endometrium [51]. Those complicated results suggest that more detailed works should be carried out to understand the exact mechanisms of ERs action in endometriosis progression. And here we will temporally focus on the roles of both ERα and ERβ in the EMT process.
Firstly, ERα could directly bind to hepatocyte growth factor (HGF) promoter, then induces EMT in the human endometrial epithelial cells [52]. Besides, ERα could also possibly up-regulate of both Snail and Slug through activation of their promoter activities [53]. In the case of ERβ, no direct research has shown that it can regulate EMT in the endometriosis. But in other disease especially the cancer, the expression of ERβ usually negatively related to EMT process. For example, in the triple-negative breast cancer, ERβ1 could bind to and oppose the transcriptional activity of mutant p53 at the promoters of genes that regulate metastasis [54]. In basal-like breast cancer cells, the inhibition of EMT correlates with an ERβ-mediated upregulation of miR-200a/b/429 and the subsequent repression of ZEB1 and SIP1, which results in increased expression of E-cadherin [55]. However, not in all the conditions ERβ could inhibit EMT process, for Lung Adenocarcinoma as a contract example [56].
Hypoxia and Estrogen are two major stimulating signals found in endometriotic lesions, both contributing to EMT process through various pathways (Figure 2). As hypoxia condition has been found in majority cancer lesions, the studies of hypoxia in EMT are more abundant than estrogen signal. However, the high expressed estrogen is the unique feature of endometriosis, so the investigations of estrogen may provide a more effective approach to therapy. Meanwhile, these extracellular signals also could be used as effective methods to induce experimental endometriosis for study.
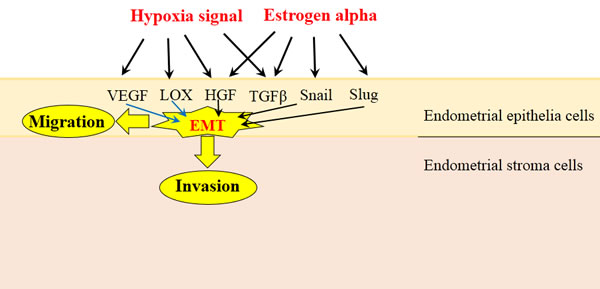
Figure 2: Hypoxia and Estrogen signals stimulate EMT in endometrial epithelial cells. Hypoxia and Estrogen are two major stimulating signals found in endometriotic lesions, both could activate various cellular factor, such as HGF, TGF-β, VEGF, LOX, Snail and Slug, then induce the EMT process in endometrial epithelial cells (black arrows). The role of in EMT only been reported in cancer cells while no study in endometrial cells, so we use blue arrows to show them. Endometrial epithelial cells undergo EMT process will gain enhanced migration and invasion ability, which lead to the form of endometriotic lesions.
Basic cellular pathways promote EMT process in endometriosis
TGF-β/Smad
TGF-β gains more attention among EMT inducers, largely because of its potency in inducing EMT in cell culture and its roles in cancer and development associated EMT. The basic TGF-β signaling pathway starts through two types of serine-threonine kinase receptors, the TGFβRI and TGFβRII. The dimerized Receptors leads to the recruitment and phosphorylation of transcription factors Smad2 and Smad3, which could trimerize with Smad4 and then the complex translocates to the nucleus, ultimately regulates related gene transcription [57]. Smad could activate the transcription factors such as Snail1, Snail2/Slug, ZEB1, ZEB2, E12/E42 and Twist1, resulting in the loss of desmosome, cell-cell tight and adherens junctions, and the gain of mesenchymal markers. However, TGF-β also induces non-Smad signaling pathways, leading to activation of Rho GTPases, MAP kinase (MAPK) pathways and the PI3 kinase-Akt-mTOR pathway [58]. These non-Smad signaling pathways often cooperate with Smad-mediated gene expression during EMT, yet also directly regulate the stabilities and activities of Smads.
Up-regulated TGF-β in peritoneum and serum/peritoneal fluid in endometriotic tissues are frequently observed in patients with endometriosis [59, 60], which suggesting that they may be essential in establishment and/or maintenance of endometriosis [61]. Functionally, TGF-β1 could inhibit natural killer cell activity in the peritoneal fluid from women with endometriosis, and promote angiogenesis and cell proliferation in ovarian endometriotic cysts [62]. TGF-β1 could activate the expression of VEGF-A in the peritoneal mesothelial cell to support endometriotic lesions vascularization [63]. In animal mode, TGFβ1-null mice developed fewer and smaller peritoneal endometriosis-like lesions compared with their wild-type counterparts [64]. However, detailed mechanisms of how increased TGF-β concentrations contribute to endometriosis remains poorly understood.
Recently, study shows that pluripotent transcription factor OCT4, initiated by TGF-β signals, could in turn regulate the TGF-βI/TGF-βRI-induced the endometriotic lesions growth by stimulating endometrial cell migration. In human endometriotic stromal cells, TGF-βI dose-dependently increased the gene and protein levels of OCT4, SNAIL and N-Cadherin and silencing of endogenous OCT4 significantly suppressed the TGF-βI-induced expressions of N-Cadherin and SNAIL [65]. This finding demonstrates that TGF-β, cooperating with OCT4, may contribute to EMT process in endometriosis.
Additionally, TGF-β has long been regarded as a major regulator of fibrosis. For example, EMT and fibroblast-to-myofibroblast transition (FMT) induced by TGF-β1/Smad3 could result in increased collagen production, cellular contractility and smooth muscle metaplasia (SMM). The SMM has been frequently found in peritoneal, ovarian, extragenital, and pleuropulmonary endometriosis [66]. Another process abundant in peritoneal fibrotic tissue is mesothelial-to-mesenchymal transition (MMT), that mesothelial cells (MCs) in the peritoneal cavity could transform into myofibroblasts under pathological conditions. In mouse model, blockage of TGF-β resulted in molecular reprogramming of markers related to the mesenchymal conversion of MCs and in a significant decrease in the severity of the peritoneal adhesions [67].
Wnt/β-catenin
Wnt/β-catenin signaling pathway is widespread in development, tissue self-renewal and various diseases including endometriosis. Β-catenin is a central intracellular transducer, which could bind the transcription factor/lymphoid enhancing-binding factor (TCF/LEF) family (TCF1, TCF3, TCF-4 and LEF) to regulate expression of hundreds of genes, including those that promote cell proliferation and survival [68]. In normal condition, the level of β-catenin in cytoplasmic is tightly controlled by GSK-3β phosphorylation, which triggers its degradation process through the ubiquitin pathway. Wnt-1 and Wnt-3 could inhibit the phosphorylation of GSK-3β thus preventing β-catenin from entering the ubiquitination pathway, thereby forming β-catenin pools [69]. The free cytoplasmic β-catenin could relocate into the nucleus to activate transcription factors LEF-1/TCFs, then induce the cells undergo EMT [70].
Many studies have revealed the association between Wnt signaling pathway and endometriosis. Significantly higher expression of WNT7A has been found in human endometriotic lesions, which may influence cell survival, promoting an improved implantation of scattered endometrial cells and hence an increase in formation of endometriosis lesions [71]. In addition, elevated secretion of Wnt2 in ectopic stromal cells could activate the growth of epithelial cells via Wnt2/β-catenin signaling pathway in a paracrine manner [72]. Estrogen could also up-regulate β-catenin expression by directly binding to the estrogen response element site on the promoter of β-catenin in endometriotic lesions [51]. Just like TGF-β, another role of Wnt/β-catenin signaling pathway in endometriosis is inducing fibrosis. Aberrant activation of the Wnt3/β-catenin was involved in mediating fibrogenesis in this disease. After the blocking of TCF/β-catenin pathway, the expression of myofibroblasts markers, such as α-SMA, collagen I and fibronectin are decreased in endometriotic stroma cells [73].
TGF-β/Smad and Wnt/β-catenin pathways may exist cooperation in EMT and fibrosis. In renal tubular epithelial cells, hydrogen Sulfide could Inhibit TGF-β1-Induced EMT via Wnt/Catenin Pathway [74]. TGF-β could mediate decreased expression of Wnt antagonist Dickkopf-1 via p38 dependent manner, highlight a key role for the interaction of both pathways in the pathogenesis of fibrotic diseases [75]. TGF-β might also stimulate Wnt signaling by other mechanisms such as the inhibition of GSK-3β aside to increase β-catenin [76]. As a marker of endometriosis, SOX-2 participates in the network of TGF-β and β-catenin [77]. SOX2 could transcriptional repress the transcriptional intermediary factor 1 γ (TIF1γ), and therefore promote TGF-β-induced EMT and cell invasion [78]. SOX2 could also reduce the expression of E-cadherin at plasma membranes, thus reducing the binding capacity of the β-catenin complexes. Therefore, the Wnt signaling components in the nucleus could recruit more β-catenin to activate the downstream targets [79]. The complex processes of EMT requires us to take a network-wide view on signal transduction (Figure 3), and further studies about the relationship between Wnt/β-catenin and other EMT pathways are needed.
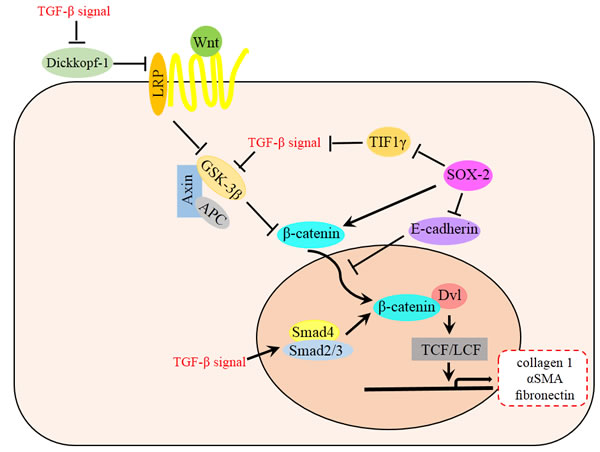
Figure 3: Relationship between Wnt and TGF-β signaling pathway in EMT. TGF-β signal is involved in many steps of Wnt signaling pathway, which ultimately accumulate β-catenin and then promote the EMT and fibrosis. TGF-β mediates decreased expression of Wnt antagonist Dickkopf-1 via p38 dependent manner, meanwhile, TGF-β stimulates Wnt signaling by inhibiting GSK-3β or promoting Smad4 and Smad2/3 complex to increase β-catenin both in cytoplasm and nucleus. TGF-β signal is also activated by SOX2 through the repression of TIF1γ. Besides, SOX2 could promote the Wnt signaling pathway by transcriptionally increasing the expression of β-catenin and enhancing the nucleus relocation process..
EMT IN ENDOMETRIA MAY LEAD TO INFERTILITY
It has been estimated that at least one third of reproductive-age women would be affected with fertility problems [80]. Altered peritoneal function resulting in impaired folliculogenesis and oocyte quality, pelvic anatomy distortion, immunologic dysfunction, and impaired implantation are part of contributing factors to reduced fertility [81]. Besides, significantly reduced implantation rates are shown in women who received embryos from endometriotic ovaries [82].
Uterine receptivity, which allows the developing embryo to implant, is a complex process involving regulation by hormones, cytokines, adhesion molecules and other factors [83]. Normally at the time of implantation, estrogen receptors are downregulated; however, women with endometriosis have an upregulation of endometrial estrogen receptors [84]. Aromatase is also aberrantly expressed in the endometrium of women with endometriosis, increasing the amount of active estradiol [85]. Conversely, an increase of progesterone compared with estrogen must occur for successful endometrial receptivity to the implanting blastocyst. However, progesterone resistance has been reported in eutopic and ectopic endometrium [1]. EMT process is often associated with progesterone resistance, and progesterone could inhibit EMT by inactivating PI3K/Akt pathway in basal phenotype breast cancers [86]. This suggests that EMT process happened in endometriotic lesions may cause a low response of endometrial cells to progesterone, ultimately result in infertility. Embryo implantation is a continuous process initiated by interaction between the blastocyst and uterine luminal epithelium. Subsequently, the embryo forms stable adhesion with epithelial cells and then makes an invasion into endometrial stroma. Although the detailed mechanisms remain incompletely, it is well established that EMT plays a key role in this process [87]. However, based on insufficient evidence, we can’t go to the conclusion that enhanced EMT in endometriosis could promote implantation. Because the expression of various molecules during embryo implantation was frequently changed in different phage, EMT is not always needed throughout the implantation. Meanwhile, the altered EMT in endometriosis is a pathological process, so it may have different ways from normal condition and lead to the failure of implantation.
CONCLUSION
Studies on EMT in the development of endometriosis represented a limited amount of publications, and these findings are mainly concentrate in recent years. However, the phenomenon of EMT has long been found in endometriotic lesions. A number of studies focus on the invasion and fibrosis in endometriosis also showed the significance of EMT. Stimulating signals such as hypoxia and estrogen activate the origination and migration of endometriotic lesions. Basic pathways of EMT, such as TGF-β and Wnt, could provide potential therapeutic approach to inhibit endometriotic cell motility and invasiveness. The detailed mechanisms of these factors in the connection between EMT and endometriosis are still unclear, so further studies are needed. Infertility as the major threaten to the life of women, is tightly regulated by EMT process during implantation. At last, we hope this relative comprehensive review of EMT in the development of endometriosis could appeal to more investigations, ultimately lead to radically novel treatments for this disease.
Abbreviations
EMT, epithelial-to-mesenchymal transition; TGF-β, TGF-beta; ReTIAR, repeat tissue injury and repair; ECM, extracellular matrix; MET, mesenchymal to epithelial transition; MSCs, Mesenchymal stem cells; VEGF, vascular endothelial growth factors; MAPK, Mitogen-activated protein kinase; FMT, fibroblast-to-myofibroblast transition; SMM, smooth muscle metaplasia; MMT, mesothelial-to-mesenchymal transition; MCs, mesothelial cells; HIFs, hypoxia-inducible factors; HGF, hepatocyte growth factor; TIF1γ, Transcriptional intermediary factor 1 γ.
Authors' contributions
Y.-M. Yang and W.-X. Yang conceived of and authored the manuscript.
ACKNOWLEDGMENTS
The authors are grateful to all the members of the Sperm Laboratory in Zhejiang University in their provision of valuable discussion. This project was supported in part by the National Natural Science Foundation of China (Nos. 31572603 and 41276151).
CONFLICTs OF INTEREST
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the review.
REFERENCES
1. Giudice LC, Kao LC. Endometriosis. Lancet. 2004; 364:1789–99.
2. Bulun SE. Endometriosis. N Engl J Med. 2009; 360:268–79.
3. Nisolle M, Donnez J. Peritoneal endometriosis, ovarian endometriosis, and adenomyotic nodules of the rectovaginal septum are three different entities. Fertil Steril. 1997; 68:585-96.
4. Zhang Q, Duan J, Olson M, Fazleabas A, Guo SW. Cellular Changes Consistent With Epithelial-Mesenchymal Transition and Fibroblast-to-Myofibroblast Transdifferentiation in the Progression of Experimental Endometriosis in Baboons. Reprod Sci. 2016; 23:1409-21.
5. Bischoff FZ, Simpson JL. Heritability and molecular genetic studies of endometriosis. Hum Reprod Update. 2000; 6:37-44.
6. Vigano P, Somigliana E, Vignali M, Busacca M, Blasio AM. Genetics of endometriosis: current status and prospects. Front Biosci. 2007; 12: 3247-55.
7. Sampson J. Peritoneal endometriosis due to dissemination of endometrial tissue into the peritoneal cavity. Am J Obstet Gynecol. 1927; 14:93-94.
8. Hans M. Albertsen, Kenneth Ward. Genes Linked to Endometriosis by GWAS Are Integral to Cytoskeleton Regulation and Suggests That Mesothelial Barrier Homeostasis Is a Factor in the Pathogenesis of Endometriosis. Reprod Sci. 2016. pii: 1933719116660847.
9. Proestling K, Birner P, Gamperl S, Nirtl N, Marton E, Yerlikaya G, Wenzl R, Streubel B, Husslein H. Enhanced epithelial to mesenchymal transition (EMT) and upregulated MYC in ectopic lesions contribute independently to endometriosis. Reprod Biol Endocrinol. 2015; 13:75.
10. Bartley J, Jülicher A, Hotz B, Mechsner S, Hotz H. Epithelial to mesenchymal transition (EMT) seems to be regulated differently in endometriosis and the endometrium. Arch Gynecol Obstet. 2014; 289:871-81.
11. Jia J, Zhang W, Liu JY, Chen G, Liu H, Zhong HY, Liu B, Cai Y, Zhang JL, Zhao YF. Epithelial mesenchymal transition is required for acquisition of anoikis resistance and metastatic potential in adenoid cystic carcinoma. PLoS One. 2012; 7:e51549.
12. Matsuzaki S, Darcha C. Epithelial to mesenchymal transition-like and mesenchymal to epithelial transition-like processes might be involved in the pathogenesis of pelvic endometriosis. Human Reproduction. 2012; 27:712-21.
13. Acloque H, Adams MS, Fishwick K, et al. Epithelial–mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009; 119:1438–1449.
14. Kalluri R, Weinberg RA. The basics of epithelial–mesenchymal transition. J Clin Invest. 2009; 119:1420–1428
15. Yang J, Weinberg RA. Epithelial–mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008; 14:818–829.
16. Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013; 27:2192–220
17. Groger CJ, Grubinger M, Waldhor T, Vierlinger K, Mikulits W. Meta-analysis of gene expression signatures defining the epithelial to mesenchymal transition during cancer progression. PloS one. 2012; 7:e51136.
18. Tan TZ, Miow QH, Miki Y, Noda T, Mori S, Huang RY, Thiery JP. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO molecular medicine. 2014; 6:1279–93.
19. Zhao M, Kong L, Liu Y, Qu H. dbEMT: an epithelial-mesenchymal transition associated gene resource. Sci Rep. 2015; 5:11459.
20. Liang L, Sun H, Zhang W. Meta-Analysis of EMT Datasets Reveals Different Types of EMT. PLoS One. 2016; 11:e0156839.
21. Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013; 342:1234850.
22. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2014; 15:178–196.
23. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009; 139:871–890
24. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype. Nat Rev Cancer 2007; 7:415–28.
25. Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop—a motor of cellular plasticity in development and cancer. EMBO Rep. 2010; 11:670–677
26. Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008; 9:582–589
27. Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE, Cha SY, Ryu JK, Yoon D, Fearon ER, Rowe RG, Lee S, Maher CA, Weiss SJ, Yook JI. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol. 2011; 195:417–433
28. Siemens H, Jackstadt R, Hünten S, Kaller M, Menssen A, Götz U, Hermeking H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle. 2011; 10:4256–4271
29. Koninckx PR, Ussia A, Adamyan L, Wattiez A, Donnez J. Deep endometriosis: definition, diagnosis, and treatment. Fertil Steril. 2012; 98:564-71.
30. Kyama CM, Debrock S, Mwenda JM, D’Hooghe TM. Potential involvement of the immune system in the development of endometriosis. Reprod Biol Endocrinol. 2003; 1:123.
31. Herington JL, Bruner-Tran KL, Lucas JA, Osteen KG.Immune interactions in endometriosis. Expert Rev Clin Immunol. 2011; 7:611-26.
32. Xu H, Zhang T, Man GC, May KE, Becker CM, Davis TN, Kung AL, Birsner AE, D’Amato RJ, Wong AW, Wang CC. Vascular endothelial growth factor C is increased in endometrium and promotes endothelial functions, vascular permeability and angiogenesis and growth of endometriosis. Angiogenesis. 2013; 16:541-51.
33. Majmundar AJ, Wong WJ, Simon MC. Hypoxia inducible factors and the response to hypoxic stress. Mol Cell 2010; 40:294–309.
34. Wu MH, Chen KC, Lin SC, Lgu CW, Tsai SJ. Aberrant expression of leptin in human endometriotic stromal cells is induced by elevated levels of hypoxia inducible factor-1alpha. Am J Pathol 2007; 170:590–598.
35. Xiong Y, Liu Y, Xiong W, Zhang L, Liu H, Du Y, Li N. Hypoxia-inducible factor 1α-induced epithelial-mesenchymal transition of endometrial epithelial cells may contribute to the development of endometriosis. Human Reprod, 2016; 31:1327-38.
36. Di Carlo C, Bonifacio M, Tommaselli GA, Bifulco G, Guerra G, Nappi C. Metalloproteinases, vascular endothelial growth factor, and angiopoietin 1 and 2 in eutopic and ectopic endometrium. Fertil Steril. 2009 Jun; 91:2315-23.
37. Mclaren J. Vascular endothelial growth factor and endometriotic angiogenesis. Human Reprod Update. 2000; 6:45-55.
38. Luo M, Hou L, Li J, Shao S, Huang S, Meng D, Liu L, Feng L, Xia P, Qin T, Zhao X. VEGF/NRP-1axis promotes progression of breast cancer via enhancement of epithelial-mesenchymal transition and activation of NF-κB and β-catenin. Cancer Lett. 2016; 373:1-11.
39. Cannito S, Novo E, Compagnone A, Valfrè di Bonzo L, Busletta C, Zamara E, Paternostro C, Povero D, Bandino A, Bozzo F, Cravanzola C, Bravoco V, Colombatto S, Parola M. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis. 2008; 29:2267-78.
40. Jung SH, Kim SM, Lee CE. Mechanism of suppressors of cytokine signaling 1 inhibition of epithelial-mesenchymal transition signaling through ROS regulation in colon cancer cells: suppression of Src leading to thioredoxin up-regulation. Oncotarget. 2016; 7:62559-62571. doi: 10.18632/oncotarget.11537.
41. Ngô C, Chéreau C, Nicco C, Weill B, Chapron C, Batteux F. Reactive oxygen species controls endometriosis progression. Am J Pathol. 2009; 175:225-34.
42. Ruiz LA, Báez-Vega PM, Ruiz A, Peterse DP, Monteiro JB, Bracero N, Beauchamp P, Fazleabas AT, Flores I. Dysregulation of Lysyl Oxidase Expression in Lesions and Endometrium of Women With Endometriosis. Reprod Sci. 2015; 22:1496-508.
43. Gort EH, Groot AJ, van der Wall E, van Diest PJ, Vooijs MA. Hypoxic regulation of metastasis via hypoxia-inducible factors. Curr Mol Med. 2008; 8:60-7.
44. Liu F, Kong X, Lv L, Gao J. TGF-β1 acts through miR-155 to down-regulate TP53INP1 in promoting epithelial-mesenchymal transition and cancer stem cell phenotypes. Cancer Lett. 2015; 359:288-98.
45. Tian Q, Xue Y, Zheng W, Sun R, Ji W, Wang X, An R. Overexpression of hypoxia-inducible factor 1α induces migration and invasion through Notch signaling. Int J Oncol. 2015; 47:728-38.
46. Bulun SE, Zeitoun KM, Takayama K, Sasano H. Estrogen biosynthesis in endometriosis: molecular basis and clinical relevance. J Mol Endocrinol. 2000; 25: 35–42.
47. Matthews J, Gustafsson JA. Estrogen signaling: a subtle balance between ER alpha and ER beta. Mol Interv 2003; 3:281–292.
48. Bulun SE, Monsavais D, Pavone ME, Dyson M, Xue Q, Attar E, Tokunaga H, Su EJ. Role of estrogen receptor-β in endometriosis. Semin Reprod Med. 2012 Jan; 30:39-45.
49. Greaves E, Cousins FL, Murray A, Esnal-Zufiaurre A, Fassbender A, Horne AW, Saunders PT. A novel mouse model of endometriosis mimics human phenotype and reveals insights into the inflammatory contribution of shed endometrium. Am J Pathol. 2014; 184:1930-9.
50. Heard ME, Simmons CD, Simmen FA, Simmen RC. Krüppel-like factor 9 deficiency in uterine endometrial cells promotes ectopic lesion establishment associated with activated notch and hedgehog signaling in a mouse model of endometriosis. Endocrinology. 2014; 155:1532–1546.
51. Zhang L, Xiong W, Xiong Y, Liu H, Liu Y. 17 β-Estradiol promotes vascular endothelial growth factor expression via the Wnt/β-catenin pathway during the pathogenesis of endometriosis. Mol Hum Reprod. 2016; 22:526-35.
52. Ono YJ, Hayashi M, Tanabe A, Hayashi A, Kanemura M, Terai Y, Ohmichi M. Estradiol-mediated hepatocyte growth factor is involved in the implantation of endometriotic cells via the mesothelial-to-mesenchymal transition in the peritoneum. Am J Physiol Endocrinol Metab. 2015; 308:E950-9.
53. Park SH, Cheung LW, Wong AS, Leung PC. Estrogen regulates Snail and Slug in the down-regulation of E-cadherin and induces metastatic potential of ovarian cancer cells through estrogen receptor alpha. Mol Endocrinol. 2008; 22:2085-98.
54. Bado I, Nikolos F, Rajapaksa G, Gustafsson JÅ, Thomas C. ERβ decreases the invasiveness of triple-negative breast cancer cells by regulating mutant p53 oncogenic function. Oncotarget. 2016; 7:13599-611. doi: 10.18632/oncotarget.7300.
55. Thomas C, Rajapaksa G, Nikolos F et al. ERbeta1 represses basal breast cancer epithelial to mesenchymal transition by destabilizing EGFR. Breast Cancer Res. 2012; 14:R148.
56. Zhao G, Nie Y, Lv M, He L, Wang T, Hou Y. ERβ-mediated estradiol enhances epithelial mesenchymal transition of lung adenocarcinoma through increasing transcription of midkine. Mol Endocrinol. 2012; 26:1304-15.
57. Wrighton K H, Lin X, Feng X H. Phospho-control of TGF-beta superfamily signaling. Cell Res. 2009; 19:8-20.
58. Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009; 19:128-39.
59. Kyama CM, Overbergh L, Debrock S, Valckx D, Vander Perre S, Meuleman C, Mihalyi A, Mwenda JM, Mathieu C, D’Hooghe TM. Increased peritoneal and endometrial gene expression of biologically relevant cytokines and growth factors during menstrual phase in women with endometriosis. Fertil Steril. 2006; 85:1667-75.
60. Pizzo A, Salmeri FM, Ardita FV, Sofo V, Tripepi M, Marsico S. Behaviour of cytokine levels in serum and peritoneal fluid of women with endometriosis. Gynecol Obstet Invest. 2002; 54: 82–87.
61. Omwandho CO, Konrad L, Halis G, Oehmke F, Tinneberg HR. Role of TGF-betas in normal human endometrium and endometriosis. Hum Reprod. 2010; 25: 101–109.
62. Loverro G, Maiorano E, Napoli A, Selvaggi L, Marra E, Perlino E. Transforming growth factor-beta 1 and insulin-like growth factor-1 expression in ovarian endometriotic cysts: a preliminary study. Int J Mol Med. 2001; 7:423-9.
63. Young VJ, Ahmad SF, Brown JK, Duncan WC, Horne AW. Peritoneal VEGF-A expression is regulated by TGF-β1 through an ID1 pathway in women with endometriosis. Sci Rep. 2015; 5:16859.
64. Hull ML, Johan MZ, HodgeWL, Robertson SA, IngmanWV. Host-derived TGFB1 deficiency suppresses lesion development in a mouse model of endometriosis. Am J Pathol. 2012; 180: 880–887.
65. Au HK, Chang JH, Wu YC, Kuo YC, Chen YH, Lee WC, Chang TS, Lan PC, Kuo HC, Lee KL, Lee MT, Tzeng CR, Huang YH. TGF-βI Regulates Cell Migration through Pluripotent Transcription Factor OCT4 in Endometriosis. PLoS One. 2015; 10:e0145256.
66. Itoga T, Matsumoto T, Takeuchi H, Yamasaki S, Sasahara N, Hoshi T, Kinoshita K. Fibrosis and smooth muscle metaplasia in rectovaginal endometriosis. Pathol Int. 2003 ; 53:371-5.
67. Sandoval P, Jiménez-Heffernan JA, Guerra-Azcona G, Pérez-Lozano ML, Rynne-Vidal Á, Albar-Vizcaíno P, Gil-Vera F, Martín P, Coronado MJ, Barcena C, Dotor J, Majano PL, Peralta AA, López-Cabrera M. Mesothelial-to-mesenchymal transition in the pathogenesis of post-surgical peritoneal adhesions. J Pathol. 2016; 239:48-59.
68. Brembeck F H, Rosário M, Birchmeier W. Balancing cell adhesion and Wnt signaling, the key role of β-catenin. Curr Opin Genet Dev. 2006; 16:51-9.
69. Espada J, Pérez-Moreno M, Braga VM, Rodriguez-Viciana P, Cano A. H-Ras Activation Promotes Cytoplasmic Accumulation and Phosphoinositide 3-Oh Kinase Association of β-Catenin in Epidermal Keratinocytes. J Cell Biol. 1999; 146:967-80.
70. Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol Int. 2002; 26:463-76.
71. Gaetje R, Holtrich U, Karn T, Cikrit E, Engels K, Rody A, Kaufmann M. Characterization of WNT7A, expression in human endometrium and endometriotic lesions. Fertil Steril. 2007; 88:1534-40.
72. Zhang H, Xue J, Li M, Zhao X, Wei D, Li C. Metformin regulates stromal-epithelial cells communication via Wnt2/β-catenin signaling in endometriosis. Mol Cell Endocrinol. 2015; 413:61-5.
73. Matsuzaki S, Darcha C. Involvement of the Wnt/β-catenin signaling pathway in the cellular and molecular mechanisms of fibrosis in endometriosis. PLoS One. 2013; 8:e76808.
74. Guo L, Peng W, Tao J, Lan Z, Hei H, Tian L, Pan W, Wang L, Zhang X. Hydrogen Sulfide Inhibits Transforming Growth Factor-β1-Induced EMT via Wnt/Catenin Pathway. PLoS One. 2016; 11:e0147018.
75. Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, Horn A, Kireva T, Beyer C, Zwerina J, Schneider H, Sadowski A, Riener MO, MacDougald OA, Distler O, Schett G, Distler JH. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun. 2012; 3:735.
76. Amini Nik S, Ebrahim RP, Van Dam K, Cassiman JJ, Tejpar S. TGF-beta modulates beta-Catenin stability and signaling in mesenchymal proliferations. Exp Cell Res. 2007; 313:2887-95.
77. Götte M, Wolf M, Staebler A, Buchweitz O, Kiesel L, Schüring AN. Aberrant expression of the pluripotency marker SOX-2 in endometriosis. Fertil Steril. 2011; 95:338-41.
78. Wang L, Yang H, Lei Z, Zhao J, Chen Y, Chen P, Li C, Zeng Y, Liu Z, Liu X, Zhang HT. Repression of TIF1γ by SOX2 promotes TGF-β-induced epithelial-mesenchymal transition in non-small-cell lung cancer. Oncogene. 2016; 35:867-77.
79. Yang N, Hui L, Wang Y, Yang H, Jiang X. Overexpression of SOX2 promotes migration, invasion, and epithelial-mesenchymal transition through the Wnt/β-catenin pathway in laryngeal cancer Hep-2 cells. Tumour Biol. 2014; 35:7965-73.
80. D’Hooghe TM, Debrock S, Hill JA, Meuleman C. Endometriosis and subfertility: is the relationship resolved. Semin Reprod Med. 2003; 21:243–254.
81. Sasson IE, Taylor HS. Stem cells and the pathogenesis of endometriosis. Ann N Y Acad Sci. 2008 ; 1127:106–115.
82. Simón C, Gutiérrez A, Vidal A, de los Santos MJ, Tarín JJ, Remohí J, Pellicer A. Outcome of patients with endometriosis in assisted reproduction: results from in-vitro fertilization and oocyte donation. Hum Reprod. 1994; 9:725–729.
83. Aghajanova L, Hamilton AE, Giudice LC. Uterine receptivity to human embryonic implantation: histology, biomarkers, and transcriptomics. Semin Cell Dev Biol. 2008;19:204–211.
84. Lessey BA, Killam AP, Metzger DA, Haney AF, Greene GL, McCarty KS Jr. Immunohistochemical analysis of human uterine estrogen and progesterone receptors throughout the menstrual cycle. J Clin Endocrinol Metab. 1988; 67:334–340
85. Attar E, Bulun SE. Aromatase and other steroidogenic genes in endometriosis: translational aspects. Hum Reprod Update. 2006; 12:49–56
86. Zuo L, Li W, You S. Progesterone reverses the mesenchymal phenotypes of basal phenotype breast cancer cells via a membrane progesterone receptor mediated pathway. Breast Cancer Res. 2010; 12:R34.
87. Simón C, Gutiérrez A, Vidal A, de los Santos MJ, Tarín JJ, Remohí J, Pellicer A. Studies using an in vitro model show evidence of involvement of epithelial-mesenchymal transition of human endometrial epithelial cells in human embryo implantation. J Biol Chem. 2012; 287:4441-50.