
INTRODUCTION
Tumor heterogeneity is an important feature of many solid malignant tumors and is mainly caused by a number of factors, such as the tumor microenvironment, mutations, changes in cellular hierarchy and epigenetic modifications. Although tumor heterogeneity is well explained by clonal evolution theory [1], a new hypothesis termed the cancer stem cell (CSC) model was recently proposed to account for this observed heterogeneity. This model suggests that tumors exhibit a hierarchical organization, including a small subpopulation of CSCs with stem cell-like characteristics and a large subgroup of tumor cells that have differentiated from CSCs [2]. This hypothesis is attracting much attention because it can well explain the chemo-resistance, metastasis and relapse of malignant tumors, and CSCs are an important research focus due to their potential clinical significance.
Although CSCs are widespread in many cancers, their presence still does not explain the functional and phenotypic heterogeneity observed in all tumors. Therefore, integration of the two models may better explain tumor formation [3]. For example, extrinsic environmental factors, together with chromosomal instability among the CSC population, may result in CSC phenotypic or functional heterogeneity. Many recent studies have addressed the impact of epigenetic modifications on CSC maintenance, among which the Polycomb group (PcG) genes are especially interesting within the CSC context.
PcG genes comprise a set of epigenetic regulators that catalyze specific histone posttranslational modifications. It has been reported that epigenetic modification by PcG proteins is critical for maintaining stem cell-like characteristics of adult stem cells as well as embryonic stem cells (ESCs) [4]. As a key protein in the PcG family, EZH2 mediates trimethylation of histone H3 lysine 27 (H3K27me3) and inhibition of downstream target genes [5]. More importantly, EZH2 has also been found to increase the proliferation of tumor cells and maintain the pluripotency of stem cells [4, 6]. Several studies have shown that EZH2 is aberrantly overexpressed in various malignant tumors, such as prostate cancer [7], breast cancer [8], ovarian cancer [9] and others [10, 11]. In addition, it is reported that EZH2 acts a critical factor in promoting tumor growth and metastasis in many malignant tumor models [12-14]. Therefore, as a new biomarker, EZH2 can be considered to be a novel target for the treatment of malignant tumors.
Recent studies have shown that EZH2 has a critical function in maintaining CSC properties and promoting CSC metastasis in various solid tumors as well as in leukemia. In addition, both EZH2 inhibition and knockdown (KD) dramatically decrease CSC tumorigenicity [15-19], supporting the notion that EZH2 may serve as a novel CSC marker and a potential target for cancer therapy. However, before these important discoveries can be translated into clinical applications, the specific function of EZH2 in maintaining CSC properties should be further elucidated.
THE STRUCTURE AND FUNCTIONS OF EZH2
As important epigenetic regulators, PcG proteins can form chromatin-modifying complexes in a cell context-dependent manner. Two important PcG proteins, PRC1 and PRC2, have been identified in mammals [20]. The PRC1 complex includes several subunits, such as PCGF, HPH, CBX, and RING1 paralog groups, whereas the PRC2 complex mainly consists of EED, SUZ12, RbAp46/48, and either EZH1 or EZH2 (Figure 1A) [21].
Human EZH2, a 751-amino acid histone-lysine methyltransferase, is organized into several domains, among which the conserved SET domain at the C-terminus functions in maintaining histone methyl transferase (HMT) activity. Other functional domains, such as CXC and ncRBD, are required for interaction with other PRC2 components and regulatory proteins (Figure 1B) [22-24].
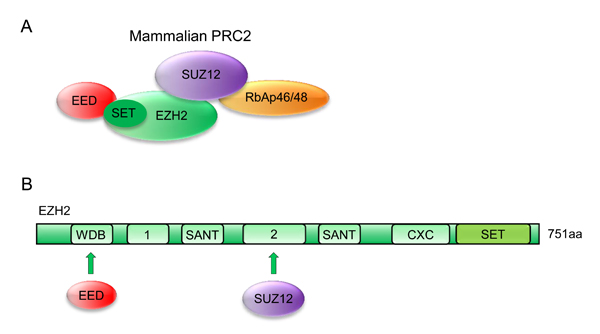
Figure 1: Possible architecture of the PRC2 complex. A. Models of core human PRC2 complexes. Subunit composition and established contacts between subunits are depicted. B. Domain organization of human EZH2 subunits. EZH2 contains a C-terminal SET domain, an adjacent cysteine-rich CXC domain, and additional conserved regions as indicated. WDB, binding domain for EED; Domain “1”, binding region for PHF1 in human cells; domain “2”, binding region for SUZ12; CXC, cysteine-rich domain; SANT, domain that allows chromatin remodeling proteins to interact with histones; SET, catalytic domain of EZH2.
EZH2 is essential for epigenetic gene silencing. After EZH2 catalyzes trimethylation of H3K27, the PRC1 protein binds to monoubiquitinated histone H2A at lysine 119 (H2AK-119ub1) and H3K27me3. This complex then mediates chromatin compaction followed by transcriptional repression (Figure 2) [25, 26].
Recently, another mechanism for PRC2-mediated stable gene inhibition was proposed. It was reported that EZH2 directly methylates the non-histone target cardiac transcription factor GATA4 at Lys299 in a PRC2-dependent manner, thereby attenuating GATA4 acetylation by p300 and resulting in GATA4 transcriptional repression and gene silencing [27]. In addition, EZH2 was shown to bind to and methylate ROR at Lys38, followed by ubiquitination of the DDB1/DCAF1/CUL4 ubiquitin ligase complex and leading to ROR target gene silencing [28]. Moreover, EZH2 has been shown to directly bind to several DNA methyltransferases (DNMTs), such as DNMT3A, DNMT3B and DNMT1, to methylate CpG sites and mediate chromatin compaction [29] (Figure 2). These studies explored a novel mechanism in which EZH2 directly methylates non-histone targets and inhibits transcriptional activity in a PRC2-dependent manner.
In addition to transcriptional repression, emerging research shows that EZH2 has a PRC2-independent function in activating downstream genes via methylation of non-histone targets or direct binding to other proteins [30-34] (Figure 2). Previous studies showed that as a transcriptional inducer, EZH2 functions independently (not as part of the PRC2 complex) in prostate cancer, breast cancer, intestinal cancer and natural killer/T-cell lymphoma (NKTL). Xu K and colleagues demonstrated that following phosphorylation at Ser21 by AKT, EZH2 directly binds to and methylates the androgen receptor (AR), thus activating its downstream genes [31]. Another study proved that EZH2 was able to directly bind to and methylate STAT3, thereby promoting the tumorigenicity of glioblastoma and prostate CSCs [34]. Similarly, Lee ST and colleagues identified that in estrogen receptor (ER)-negative basal-like breast cancer cells, EZH2 binds to a NF-κB component to form a ternary complex, leading to transcriptional activation of its downstream target genes. These processes were independent of EZH2 HMTase activity [33]. Moreover, EZH2 has also been reported to activate target genes through the ER/Wnt/β-catenin pathway [32]. Another study by Jung HY and colleagues demonstrated that EZH2 could promote transactivation of Wnt target genes by establishing a complex with TCF/β-catenin and the DNA repair protein PCNA-associated factor (PAF), which resulted in intestinal tumorigenesis [30]. EZH2 function independent of its HMTase activity was also observed in natural killer/T-cell lymphoma (NKTL). Interestingly, EZH2 overexpression in NKTL was found not to be associated with H3K27 trimethylation [35], and ectopic expression of an EZH2 mutant lacking HMTase activity in NKTL cell lines rescued the tumor growth inhibition resulting from depletion of endogenous EZH2 [35].
In conclusion, recent findings support the hypothesis that EZH2 functions as a dual-faced molecule that acts both as a transcriptional suppressor and a transcriptional co-activator depending on its interaction with other PRC2 components and the cellular context.
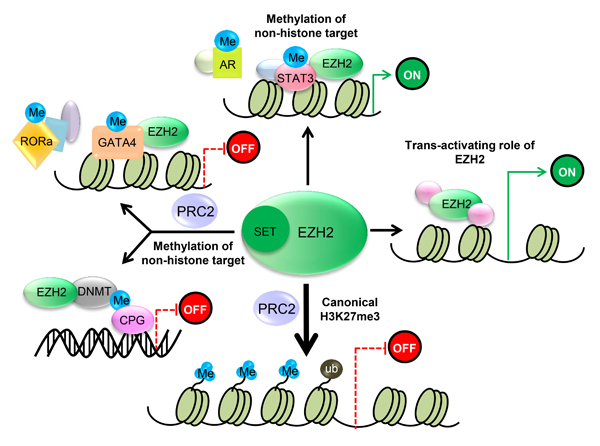
Figure 2: EZH2 regulates transcriptional activity. 1) PRC2 methylates Histone 3 on lysine 27, which contributes to transcriptional silencing. 2) EZH2 is also capable of methylating several non-histone protein substrates, which contributes to both transcriptional silencing and transcriptional activation. 3) EZH2 also has a PRC2-independent role in transcriptional activation.
EZH2 OVEREXPRESSION IN CSCs
Compared to normal tissue cells or benign tumor cells, EZH2 is overexpressed in and promotes tumor progression in various cancers [36]. Therefore, analyses of the EZH2 expression in CSCs are of research interest.
EZH2 levels have been demonstrated to be increased in CSCs in various malignant tumors, such as melanoma cancer [17], breast cancer [37, 38], ovarian cancer [16, 39], pancreatic cancer [37], skin cancer [40], colorectal cancer [18] and leukemia [41, 42] (Table 1). Despite this evidence, the link between EZH2 and CSCs has been questioned by recent work comparing EZH2 expression in differentiated cancer cells and CSCs [43]. Surprisingly, the authors found significantly lower EZH2 expression and global levels of H3K27me3 in CD44+/CD24− CSCs sorted from MCF7 cells and in side population cells sorted from HUH7 cells (Table 1). These findings were coupled to global DNA demethylation in CSCs compared with differentiated cancer cells. One possible explanation for this phenomenon is that CSCs comprise only a small subset of tumor cells, less than 5% even in CSC-enriched populations [44]. In addition, the CSC gene expression profile is highly dependent on the composition of the culture medium, and culturing CSCs in serum-containing media can induce CSC differentiation and abolish their self-renewal ability. A previous study also showed that the CSC differentiation status and epigenetic profile can be affected by the specific culture medium composition [45].
In general, testing the clinical significance of the CSC hypothesis requires a functional approach [46]. It is thus important to illuminate the function of a CSC-related gene within the context of a specific tumor using pharmacological inhibition and gene silencing. Therefore, in contrast to simply determining the level of EZH2 expression, identifying the functional importance of EZH2 in CSCs is of great significance and may pave the way for the clinical application of EZH2 inhibitors.
Table 1: Expression of EZH2 in CSCs
Cancer type |
Cell type |
CSCs |
EZH2 |
H3k27me3 |
Method |
References |
Breast cancer |
HCC193, T47D, Primary tumor |
CD44+CD24- |
Upregulated |
Upregulated |
WB; qPCR |
[37] |
Primary tumor |
Spheroids |
Upregulated |
Upregulated |
WB; qPCR |
[37] |
|
SUM149, Primary tumor |
ALDH+ |
Upregulated |
Unknown |
qPCR |
[38] |
|
MCF7 |
CD44+CD24- |
Downregulated |
Downregulated |
RT-PCR |
[43] |
|
Pancreatic cancer |
HPAC, Panc-1, Primary tumor |
EpCam+CD44+CD24+ |
Upregulated |
Upregulated |
WB; qPCR |
[37] |
Primary tumor |
Spheroids |
Upregulated |
Upregulated |
WB; qPCR |
[37] |
|
Ovarian cancer |
Primary tumor |
CD44+CD117+ |
Upregulated |
Unknown |
qPCR |
[16] |
Ascitic fluid |
Side population |
Upregulated |
Unknown |
qPCR |
[39] |
|
IGROV1 |
Side population |
Upregulated |
Unknown |
qPCR |
[39] |
|
Melanoma cancer |
WM793, A375 |
Spheroids |
Upregulated |
Upregulated |
WB |
[17] |
Skin cancer |
SCC-13 |
Spheroids |
Upregulated |
Upregulated |
WB |
[40] |
Colorectal cancer |
SW480 |
CD133+CD44+ |
Upregulated |
Unknown |
qPCR |
[18] |
SW480 |
Spheroids |
Upregulated |
Unknown |
WB |
[18] |
|
Leukemia |
CML mice cells |
Lin-c-Kit+Sca1+ |
Upregulated |
Unknown |
qPCR |
[41] |
CML patient cells |
CD34+CD38- |
Upregulated |
Upregulated |
qPCR |
[42] |
|
Hepatocarcinoma |
HUH7 |
Side population |
Downregulated |
Downregulated |
RT-PCR |
[43] |
EZH2 CONFERS CSC PROPERTIES
EZH2 and breast CSCs
Breast cancer is one of the leading causes of cancer-associated death in women. However, the detailed mechanism remains unclear. Recent research supports that EZH2 overexpression and CSC expansion can result in the progression of breast cancer [37]. Moreover, a previous study suggested that EZH2 overexpression significantly increased mammosphere formation: each sphere overexpressing EZH2 contained two or three times more cells than the control group, which proved that EZH2 can increase the self-renewal ability of breast CSCs [15]. Similarly, a study by Gonzalez ME et al. demonstrated that EZH2 KD in primary tumor cells isolated from patients with triple-negative (TN) invasive breast carcinoma, as well as in breast cancer cell lines, significantly reduced the proportion of CD44+/CD24− and ALDH1+ cells compared with controls. Moreover, reduced EZH2 expression dramatically decreased the tumorigenic ability of ALDH1+/SUM149 cells compared to control cells. However, EZH2 KD in ALDH1- populations had no significant effect on tumor onset and tumor volume [38].
EZH2 and pancreatic CSCs
One of the most lethal malignancies, pancreatic ductal adenocarcinoma (PDAC) has a five-year survival rate of less than 5% [47]. Recent findings suggest that PDAC is characterized by overexpression of EZH2, promoting self-renewal capacity of CSCs through H3K27me3. Van Vlerken LE and colleagues [48] demonstrated that EZH2 KD significantly reduced the frequency of CSCs in PDAC. In Panc-1 cells, the EpCam+CD44+CD24+ frequency declined 1.6-fold from 12.7% in control cells to 7.8% after EZH2 loss (p < 0.001). In addition, CSC frequency among HPAC cells was similarly reduced two-fold from 13.8% under control conditions to 7% after EZH2 KD (p < 0.001). Therefore, it was concluded that EZH2 is essential for maintaining the pancreatic CSC population.
EMT and chemoresistance are important properties of CSCs [49], and a recent report suggested that EZH2 overexpression results in reduced E-cadherin levels, which may result in poor prognosis of PDAC patients [50]. It was also reported that EZH2 could increase the chemotherapy resistance of PDAC cells to gemcitabine [51].
EZH2 and glioblastoma multiforme CSCs
Glioblastoma multiforme (GBM) is considered to be the most aggressive type of malignant glioma. Due to self-renewal and differentiation capacity, CSCs can lead to tumor initiation in GBM [52, 53]. Recently, Suvà ML and colleagues showed that pharmacologic inhibition of EZH2 in GBM spheres by 3-deazaneplanocin A (DZNep) at 5 μmol/L for 5 days resulted in a > 80% decrease in the clonogenic index. However, temozolomide treatment of GBM spheres at the same concentration as DZNep had no effect on CSC clonogenicity. In addition, these authors identified that a short in vitro treatment of CSCs with 5 μmol/L DZNep before transplantation dramatically improved survival of the host animal [54]. In summary, EZH2 is considered to function in maintaining the self-renewal capacity and tumorigenicity of GBM CSCs.
EZH2 and ovarian CSCs
Epithelial ovarian cancer (EOC) is one of the most common malignancies in the female reproductive system. Similar to several other human carcinomas, EZH2 overexpression is critical for the maintenance of ovarian CSC populations. Recently, Rizzo S and colleagues reported EZH2 to be overexpressed in ovarian tumor-derived side population (SP) cells, which are stem cell-like cells enriched by chemotherapy, and demonstrated that EZH2 KD results in loss of SP cells and reduced anchorage-independent growth in ovarian tumor models [39]. This evidence suggests that EZH2 expression is increased in ovarian CSCs, which may contribute to EOC chemoresistance. Using chromatin immunoprecipitation (CHIP) and gene sequencing, Li H et al. reported 60 genes directly targeted by EZH2, with ALDH1A1 as a novel target of EZH2 [55]. ALDH1A1 has previously been reported as a CSC marker in ovarian and breast cancers [56-58], and the Li et al. study revealed that EZH2 directly increased ALDH1A1 expression in ovarian cancer cells, supporting the notion that EZH2 can increase the proportion of CSCs by promoting ALDH1A1 expression.
EZH2 and prostate CSCs
Prostate cancer (PCa) accounts for the majority of cancer-associated deaths among men in the United States [47]. Recent studies have shown that as the most aggressive form of PCa, castration-resistant prostate cancer (CRPC) has a poor prognosis and high mortality, which has been in part attributed to the existence of CSCs. As in other cancer types, epigenetic alterations and microRNA (miRNA, miR) deregulation are considered important factors in prostate carcinogenesis [59]. The let-7 family has an important function in promoting PCa progression through CSC regulation. Kong D and colleagues found a lack of let-7 expression to be associated with EZH2 overexpression in human PCa tissues. In addition, enhanced let-7 expression led to decreased levels of EZH2 expression and inhibited the sphere-forming capacity and clonogenic ability of PCa cells. Moreover, the authors found that BioResponse 3,3’-diindolylmethane (BR-DIM) treatment increased expression of let-7 and decreased that of EZH2 in PCa cells, leading to repression of clonogenic and self-renewal capacity in these cells. In summary, these data indicate that reduced let-7 expression results in EZH2 overexpression, which may promote CSCs and contribute to PCa aggressiveness and recurrence [60]. Another study utilized immunohistochemical staining to examine the potential clinical significance of the levels of ALDH1 and EZH2 proteins in PCa. The results suggested that the expression level of ALDH1 is associated with tumor stage, lymphovascular invasion and extraprostatic extension, whereas that of EZH2 was correlated with the Gleason score and lymph node metastasis. Therefore, it was concluded that immunohistochemical analysis of CSC markers, such as ALDH1 and EZH2, can be applied as a predictor of tumor aggressiveness in PCa [61].
EZH2 and skin CSCs
Skin cancer is one of the most common cancers in the United States, with more than 2 million people treated for nonmelanoma (basal cell or squamous cell carcinoma (SCC)) and 76,690 new melanoma cases each year [62]. Melanoma is the most aggressive type of skin cancer and has a poor prognosis [63]; the median survival time of metastatic melanoma is only 3-11 months [63-65], partly due to the chemo-resistance of CSCs to conventional therapy. As in several other cancers, EZH2 is overexpressed in the progression of benign nevi to invasive or metastatic melanoma [66, 67], and acquired functional mutations in EZH2 account for 3% of melanomas [17]. Further investigation identified that EZH2 is essential for maintaining MCS cell survival: inhibition of EZH2 with GSK126 and EPZ-6438 or EZH2 KD in WM793 and A375 cell lines reduced sphere-forming capacity as well as MCS cell invasion and migration [17].
Similarly, Adhikary G and colleagues considered SCC-13-derived spheroids to be epidermal CSCs (ECS cells) and demonstrated that EZH2 can promote the survival, invasion and tumor formation capacity of ECS cells, with associated increases in H3K27me. They also showed that inhibition of EZH2 by GSK126 and EPZ-6438 or EZH2 KD could reduce expression and activity of EZH2, resulting in decreased ECS cell sphere formation, invasion and tumorigenic capacity. Moreover, GSK126 and EPZ-6438 reduced levels of Bmi-1 and Oct4 but did not influence those of Sox2 or K15 [68]. Additionally, research in laryngeal squamous cell carcinoma showed that EZH2 overexpression in AMC-HN-8 cells could promote sphere-forming ability, chemotherapy resistance and tumorigenic ability of CSCs [69].
EZH2 and colorectal CSCs
As one of the most common cancers, 1.23 million people worldwide are diagnosed with colorectal cancer (CRC) each year [70]. CRC stem-like cells (CCS-like cells) have recently attracted increasing attention due to their contribution to the poor prognosis of cancer patients [71, 72]. Chen JF and colleagues [18] reported EZH2 to be indispensable for CCS-like cell maintenance. These authors first identified that EZH2 KD cells exhibit significantly reduced mammosphere growth, as analyzed by sphere size (~75%), with fewer mammospheres (~50%) compared to WT or control cells. They also found the CD133+/CD44+ subpopulation of SW480 cells to decrease dramatically, from 42.8 ± 0.8% of WT cells and 43.3 ± 1.1% of control cells to 20.5 ± 0.5% of EZH2 KD cells. Additionally, they reported reduced expression of stem cell-associated genes such as Nanog and Sox2 in EZH2 KD cells and further showed that the tumors generated by EZH2 KD cells were smaller than those generated by the control group. In summary, these results demonstrate that silencing EZH2 reduces CCS-like cell properties.
EZH2 and leukemia stem cells
The existence of CSCs, which were first reported in acute myeloid leukemia (AML) [73], is considered as a reasonable explanation for tumor initiation and propagation. EZH2 was recently identified as promoting leukemia stem cell (LSC) tumorigenesis by suppressing cell differentiation and maintaining stem cell properties [19]. It was also reported that EZH2 KD prevented initiation and maintenance of chronic myelogenous leukemia (CML) and survival of CML stem cells. Moreover, EZH2 was defined as a selective vulnerability of CML stem cells, irrespective of BCR-ABL1 mutational status [41]. Another study demonstrated that PRC2 is misregulated in CSCs in the chronic phase of CML, which is largely due to the extensive reprogramming of H3K27me3 targets in CML stem cells. In addition, treatment with either EZH2 siRNA or tyrosine kinase inhibitor (TKI) alone increased expression of H3K27me3 targets, and combined treatment enhanced these effects and decreased the proportion of CSCs in CML compared to treatment with TKI alone [42]. A study on mixed-lineage leukemia (MLL) showed that EZH2 KD or inhibition by DZNep reduced LSC frequency and repressed MLL proliferation. Further analysis showed that p16 KD could expand LSCs and inhibit prognosis improvement of DZNep-treated mice. Moreover, CHIP assays revealed that EZH2 is enriched around the transcriptional start site of the p16 gene in MLL⁄ENL and in Hoxa9⁄Meis1-transduced cells [74]. In conclusion, these data suggest that EZH2 can be regarded as a potential therapeutic target for MLL.
MECHANISMS OF EZH2 ACTION IN REGULATING CSCs
BMP signaling
The BMP-mediated PS-SMAD1/5/8 pathway is a well-known cytokine-mediated pathway regulating normal neural stem cell (NSC) differentiation. Glioblastoma CSCs display some similarities with early embryonic NSCs; for example, they both lack BMPR1B expression and have a temporary response to BMP ligands [75]. Lee J and colleagues found that BMPR1B hypermethylation in 0308 CSCs may result in a lack of signal transduction responses to the CNTF and BMP pathways [76]. They also proved that EZH2 KD can increase expression of BMPR1B in these cells. Further investigation confirmed that EZH2 KD in 0308 CSCs dramatically decreased methylation of CpG nucleotides in the BMPR1B promoter region, indicating that deficiency in BMPR1B expression can be primarily attributed to PRC2-mediated transcriptional repression via methylation of CpG nucleotides in the BMPR1B promoter [75]. In conclusion, these data support that EZH2-mediated BMPR1B repression is essential for maintaining CSC properties in GBM, especially with regard to inhibiting differentiation (Figure 3).
AKT/EZH2/STAT3 signaling
STAT3 signaling is regarded as a target for cancer treatment due to its important function in a variety of malignant tumors [77, 78]. Activation of STAT3 involves a series of events [79], and recent research identified that STAT3 downstream genes are overexpressed in the mesenchymal GBM subtype, supporting that STAT3 signaling has a key function in controlling mesenchymal transformation in glioma [80, 81]. Another study reported that EZH2 S21D overexpression induces STAT3 methylation and promotes STAT3 activity in GSCs compared to non-CSCs [34]. Further analysis found that inhibition of AKT signaling could mediate EZH2 phosphorylation, which in turn resulted in STAT3 inactivation. These results indicate that interactions between EZH2 and STAT3 in GSCs are regulated by upstream PI3K/AKT signaling. Therefore, it is concluded that EZH2 participates in PRC2-independent transcriptional activation by directly binding to STAT3, which results in GSC expansion and GBM progression. Similarly, Wen S and colleagues [82] indicated that EZH2-STAT3 signals influence prostate tumor progression by directly methylating the promoters of stem cell-related genes, such as Sox-2, Oct-4 and Nanog. In addition, these authors found that ASC-J9®, a newly identified AR degradation enhancer, could mediate inhibition of STAT3 methylation and phosphorylation in prostate CSCs by suppressing the function of EZH2. Therefore, ASC-J9®, as well as the EZH2 inhibitor GSK-126, may be effective at suppressing PCa metastasis by targeting EZH2-STAT3 signals (Figure 3). Together, the evidence to date suggests that inhibition of EZH2 suppresses the functions of STAT3 signals involved in CSC maintenance, supporting that EZH2 can be regarded as a target for cancer therapy.
Wnt/β-catenin signaling
β-Catenin activation is important in maintaining the self-renewal capacity and survival of CSCs in various tumors, such as leukemia, skin cancer and gastrointestinal cancer [83-85]. As the upstream regulator of β-catenin, the wingless pathway is also reported to maintain the properties of Drosophila ovarian stem cells [86]. Moreover, in CD117+ ovarian CSCs, β-catenin has been identified as the gene downstream of the stem cell factor receptor [87]. The Wnt/β-catenin pathway is vital to CCS-like cell maintenance [88]. Recently, gene expression data from 433 human CRC specimens in The Cancer Genome Atlas (TCGA) database were used to analyze EZH2 expression levels relative to those of randomly chosen target genes in the Wnt/β-catenin pathway. The authors found that expression of a Wnt signature, based on combined mRNA expression of all 12 Wnt/β-catenin pathway target genes, to be positively correlated with EZH2 mRNA expression. They also found that EZH2 KD could decrease the expression levels of β-catenin and downstream target genes such as vimentin and c-Myc in SW480 and SW620 cells. In addition, β-catenin, c-Myc and vimentin levels were increased following EZH2 overexpression, with significant increases in sphere formation and the CD44+ population. Collectively, these findings demonstrate that EZH2 is indispensable for activating Wnt/β-catenin signaling to maintain CCS-like cell properties. In an attempt to determine the mechanism, another study showed that levels of P21cip1, a cell cycle regulator that can arrest the cell cycle at the G1/S phase [89], were increased following EZH2 knockdown in CD133+/CD44+ SW480 cells, whereas the levels of β-catenin, vimentin and c-Myc expression were reduced. Similarly, treatment with EPZ-6438, a specific inhibitor of EZH2 [90], increased p21cip1 expression and inactivated the Wnt/β-catenin pathway. These findings support the notion that EZH2 knockdown inactivates Wnt/β-catenin pathway by increasing p21cip1expression, resulting in G1/S-phase arrest. Similarly, EZH2 overexpression-mediated repression of DNA damage repair was found to result in RAF1 gene amplifications in breast CSCs, which promoted breast CSC expansion by activating p-ERK-β-catenin signaling. This study also revealed that AZD6244, a RAF1-ERK signaling inhibitor, could improve the prognosis of breast cancer by eliminating breast CSCs [15] (Figure 3).
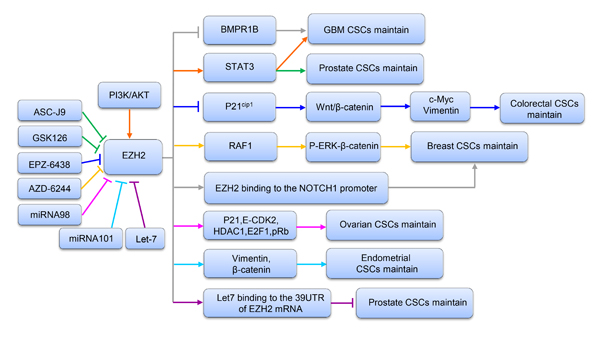
Figure 3: A schematic diagram illustrating the mechanism of EZH2 in maintaining CSCs in various tumors. Upstream and downstream genes involved in the process are shown.
Notch signaling
Notch signaling is involved in a series of biological events, such as cell proliferation, differentiation, apoptosis and embryogenesis [91]. A previous report indicated that enhanced expression of Notch pathway components is essential for maintaining self-renewal and chemoresistance in CSCs and SP cells [92]. A recent study suggested that increased EZH2 expression could increase both NOTCH1 expression and signaling. Moreover, NOTCH1 inhibition prevented EZH2-mediated CSC expansion in breast cancer. Further study revealed that in TN breast cancer, EZH2 directly binds to the NOTCH1 promoter to active NOTCH1 signaling (Figure 3), independent of PRC2-mediated H3K27me3 [38].
EZH2 and CSC-related genes
The epigenetic mechanism of gene repression by EZH2 is involved in CSC-associated features such as differentiation, migration, invasion and chemoresistance [15, 17, 18, 38, 39, 51, 54, 60]. The function of EZH2 in malignant tumors may due to its transcriptional repression of differentiation-related and antimetastatic genes. It has been reported that CSCs can maintain their pool by suppressing cell differentiation genes, such as p16 and p19, and that these cells exhibit other characteristics, such as decreased E-cadherin expression, that can induce metastasis [93]. A recent study identified that EZH2 can mediate p16, p19, and E-cadherin silencing through H3K27me3 [62].
c-Myc, which is critical for maintaining self-renewal and survival in glioma CSCs [94, 95], is also regarded as a key molecule involved in genetic reprogramming [96]. Suvà ML and colleagues analyzed global gene expression profile changes in primary cultures of GBM CSCs (named BT-CSCs) following treatment with DZNep for five days. As the top DZNep-deregulated gene, c-myc was chosen for further investigation. Compared with untreated cells, the clonogenic ability was decreased by 40% in c-myc-infected BT-CSCs after treatment with DZNep, an 80% decreased compared to empty vector-infected BT-CSCs. These findings suggest that c-myc partially rescues the decreased clonogenic capacity of BT-CSCs treated with DZNep. Additionally, EZH2 KD resulted in reductions in c-myc expression and depletion of c-myc transcripts by approximately 80%, further supporting that c-myc is a potential downstream target of EZH2 in the regulation of BT-CSCs [54].
EZH2 and CSC-related microRNAs
MicroRNAs (miRNAs) are defined as short RNA molecules of 21-23 nt that block or interfere with mRNA translation to negatively regulate gene expression [97-99]. Previous studies report that specific miRNAs control CSCs in various tumors by activating or suppressing target genes [97, 99].
Liu T and colleagues showed that miRNA-98 could decrease the EZH2 expression level and inhibit the proliferation of ovarian CSCs. Moreover, EZH2 KD decreased expression of the cyclin E-CDK2 and cell cycle protein p21, as well as E2F1, HDAC1, and pRb. These authors concluded that EZH2-specific microRNA-98 could inhibit the cell cycle in ovarian CSCs by regulating the CDK-pRb-E2F pathway [16] (Figure 3).
Previous studies reported that miRNA-101 suppresses the proliferation, migration and invasion of tumor cells in various cancer types [100-104]. Konno Y and colleagues used computational analysis and microarray screening to predict three direct targets of miRNA-101, namely, FOS, EZH2 and MCL-1. Moreover, a negative correlation between the expression levels of miR-101 and its targeted genes, including FOS, EZH2 and MCL-1, was found in EC samples. In addition, knockdown of FOS, EZH2 and MCL-1 expression repressed the proliferation, migration, invasion and CSC-like phenotypes of EC cells. These results provide the first evidence that miR-101 governs multiple malignant phenotypes including proliferation, migration, invasion and cancer stemness in aggressive EC cells. This at least partly occurs through reduced EZH2 expression, followed by increased levels of p21, Bax, TIMP-3 and epithelial markers and inhibited expression of mesenchymal markers and target genes of Wnt/β-catenin signaling (Figure 3) [105].
Members of the let-7 family inhibit the progression and recurrence of several cancers by negatively regulating CSCs [106, 107]. Kong D et al. used Target Scan software to predict EZH2 as a direct target of the let-7 family. Further study proved that the let-7 family can bind to the 3’ untranslated region (UTR) of the EZH2 mRNA to decrease EZH2 expression, leading to repression of CSC self-renewal ability in prostate cancer [60] (Figure 3).
TARGETING EZH2 FOR CANCER THERAPY
Significant experimental evidence clearly demonstrates that EZH2 participates in a series of biological events in many cancer types, including tumor initiation, progression and metastasis. Therefore, EZH2 is regarded as a potential anticancer target in current epigenetic strategies. Several EZH2 inhibitors are undergoing preclinical studies or phase 1 clinical trial, further supporting the potential clinical significance of EZH2.
Enzymatic inhibition of EZH2
DZNep, a 3-deazaadenosine analog that can increase the expression level of S-adenosyl-l-homocysteine hydrolase (SAH) and inhibit S-adenosylmethionine (SAM)-dependent histone lysine methyltransferase activity, is a widely used EZH2 inhibitor (Table 2) [108]. Although a non-specific EZH2 inhibitor, DZNep is still able to inhibit EZH2 expression and its transcriptional repression function. The antitumor activity of DZNep in thyroid cancer is well established [109], and it is reported that DZNep significantly inhibits the sphere-forming and tumorigenic abilities of ovarian and glioblastoma CSCs [39, 54]. Nonetheless, the non-specific inhibitory effect of DZNep is a huge obstacle for its clinical application because it may also affect other SAM-dependent processes. In addition, there are remaining questions regarding its short plasma half-life and toxicity profile in animal models [110]. Therefore, it is imperative to develop DZNep analogs and select EZH2 inhibitors with increased antitumor effects and reduced toxicity. A recent study identified that the DZNep analog D9 could decrease the proliferation and tumorigenesis of CSCs, with antitumor effects in AML via inhibition of several oncogenic pathways [111]. Therefore, due to its anti-cancer effects, D9 is regarded as a potential drug candidate.
Recently, several potent selective inhibitors, including EPZ005687, EPZ-6438, EPZ-011989, EI1, GSK126, GSK343 and UNC1999 (Table 2), have been identified through high-throughput screening. All of these compounds act as SAM competitive inhibitors.
EPZ005687 exhibits more than 500-fold selectivity toward EZH2 compared with other methyltransferases. This compound can also inhibit H3K27me3 in various cancer cells harboring wild-type, tyrosine Y641- or alanine A677-mutant EZH2 in a dose-dependent manner [112].
Compared to EPZ005687, EPZ-6438 has a strong inhibitory effect and better oral efficacy [113]. Treatment with EPZ-6438 dose-dependently decreased H3K27me3 levels and improved prognosis in EZH2-mutant non-Hodgkin lymphoma (NHL) mouse models [90]. Currently, EPZ-6438 is being applied in phase 1/2 clinical trials of refractory B-cell lymphoma and advanced solid tumors (NCT01897571). In this study, the researchers observed partial or complete response in 9 out of 15 NHL patients, including complete response in a malignant rhabdomyoma patient and partial response in a patient with EZH2 mutation (EZH2Y646H); these results show the potential drug effects of EPZ-6438. Two other clinical trials (NCT02395601 and NCT02082977) of SMARCB1-deficient tumors and EZH2-mutant B cell NHL are underway [36]. Additionally, a recent study reported that another molecule, EPZ-011989, sharing a similar structure with EPZ-6438 could expand the inhibition range of EZH2 and improve pharmacodynamic and pharmacokinetic qualities in a mouse B cell lymphoma model [114].
El1, another SAM-competitive inhibitor of EZH2, exhibits more than 10,000-fold selectivity toward both wild-type and mutant EZH2 compared with other methyltransferases [115]. It is reported that El1 modulates H3K27me2 and H3K27me3 levels without altering EZH2 expression. El1 also exhibited an antitumor effect only in SMARCB1-mutant rhabdoid tumors and EZH2-mutant diffuse large B cell lymphoma (DLBCL) [115].
As the most potent inhibitor of EZH2, GSK126 shows greater than 1,000-fold selectivity for EZH2 compared with other methyltransferases containing SET or non-SET domains and 150-fold selectivity compared to EZH1 [116, 117]. One study showed that GSK126 was able to inhibit proliferation of EZH2 mutants in DLBCL cell lines as well as in EZH2-mutant DLBCL xenografts. Therefore, GSK126 has an important function in improving the poor prognosis of tumors with EZH2 overexpression. GSK343, as a derivative of GSK126, has been shown to exhibit antitumor activity in EOC cells [118].
UNC1999, a GSK126 analog, exhibits high selectivity for both wild-type and Y641-mutant EZH2, with good oral efficacy. Additionally, it was reported that UNC1999 reduced levels of H3K27me3 and displayed an antitumor effect in EZH2 Y641N-mutant DLBCL cells [119].
Non-enzymatic inhibition of EZH2
In addition to enzymatic inhibition, some natural compounds have also been proven to be effective in repressing the expression level and function of EZH2 in several malignant tumor types (Table 2). It is reported that curcumin, a natural component of turmeric, can reduce EZH2 expression through the MAPK pathway rather than by increasing protein degradation, which could inhibit tumor cell proliferation [120]. Another compound, epigallocatechin-3-gallate (EGCG), is an important component of dietary omega-3 polyunsaturated fatty acids and green tea. It was reported that EGCG reduced EZH2 expression levels by increasing proteasomal degradation [121, 122]. In addition, sulforaphane (SFN), a natural tumor-preventing compound extracted from broccoli and other cruciferous vegetables [123] was shown to induce apoptosis in melanoma tumor cells and have significant therapeutic potential in skin cancer [124-126]. Previous reports identified that SFN could decrease expression of EZH2 through a proteasome-dependent mechanism [122, 127]. Recently, Tiffen J and colleagues reported that SFN reduced EZH2 expression and H3K27me3 marks in the melanoma cancer cell line A375, though these effects were not reversed by lactacystin, a proteasome inhibitor. Instead, the authors identified that SFN treatment decreased the level of EZH2 by inhibiting its transcription [128].
Table 2: EZH2 inhibitors and their status in clinical development
Compound |
Mechanism |
Specificity to EZH2 |
Clinical Status |
References |
|
Enzymatic inhibition of EZH2 |
DZNep |
SAH hydrolase inhibitor of methyltransferases |
No |
Preclinical |
[108] |
D9 |
SAH hydrolase inhibitor of methyltransferases |
No |
Preclinical |
[111] |
|
EPZ005687 |
SAM-competitive inhibitor of PRC2 |
Yes |
Preclinical |
[112] |
|
EPZ-6438 |
SAM-competitive inhibitor of PRC2 |
Yes |
Phase 1/2 |
||
EPZ-011989 |
SAM-competitive inhibitor of PRC2 |
Yes |
Preclinical |
[114] |
|
El1 |
SAM-competitive inhibitor of PRC2 |
Yes |
Preclinical |
[115] |
|
GSK126 |
SAM-competitive inhibitor of PRC2 |
Yes |
Phase 1 |
||
GSK343 |
SAM-competitive inhibitor of PRC2 |
Yes |
Preclinical |
[118] |
|
UNC1999 |
SAM-competitive inhibitor of PRC2 |
Yes |
Preclinical |
[119] |
|
Non-enzymatic inhibition of EZH2 |
Curcumin |
MAPK pathway |
No |
Preclinical |
[120] |
EGCG |
Promotion of EZH2 proteasomal degradation |
No |
Preclinical |
||
SFN |
Inhibitor of Ezh2 transcription |
No |
Preclinical |
CONCLUSIONS, QUESTIONS AND FUTURE DIRECTIONS
In summary, EZH2 is required to maintain CSC self-renewal capacity and tumorigenic ability and confers resistance to chemotherapy and radiotherapy. Moreover, we discuss a variety of agents targeting EZH2 that may improve cancer treatment. However, EZH2 is not always highly expressed in CSCs. For instance, the level of EZH2 is low in CSCs in hepatocarcinoma and breast cancer [43]. Thus, EZH2 expression and function should be investigated in each individual type of tumor. Moreover, there are no universal markers to date that can be used for CSC detection in all cancer types, and putative CSC markers have not been identified for every type of cancer [129]. This situation leads to ambiguity in defining CSCs and may lead to variable results regarding EZH2 expression in CSCs in different types of cancer. It important to note that low EZH2 in CSCs and the presence of compensatory regulation that overcomes EZH2 inhibition will affect the efficacy of EZH2-targeting treatments. Overall, EZH2-targeted therapy is likely to be less cytotoxic when applied as a single agent. Such treatment may be beneficial when combined with other treatments such as sensitivity enhancers in adjuvant settings.
In addition, interaction between EZH2 and various factors is extremely complex. The function of EZH2 occurring through PRC2 depends on the existence of other components of the complex, such as SUZ12 and EED [21]. Moreover, interplay between PRC1 and PRC2 can improve recruitment and activity of PRC2 at target sites of chromatin, which is also regulated by other types of histone modifications such as phosphorylation and acetylation, which function antagonistically with PRC2 [130]. lncRNAs, such as HOTAIR [131], Xist [132] and Meg3 [133], are also involved in controlling PRC2 function. Regarding the PRC2-independent action of EZH2, it can directly methylate non-histone targets such as GATA4, ROR and STAT3, bind to DNA methyltransferases to methylate CpGs and form transcriptional complexes with other proteins to regulate gene transcription, protein function and degradation. Although limited, emerging evidence suggests that EZH2 can regulate CSCs via both PRC2-dependent and PRC2-independent mechanisms (Figure 4). Regardless, the molecular mechanisms by which EZH2 maintains CSC properties require further investigation.
In conclusion, regardless of whether it functions as a classical oncogene or a CSC-specific regulator, novel anti-cancer therapeutic strategies will emerge from further research on the molecular mechanisms of EZH2 action in CSC regulation.
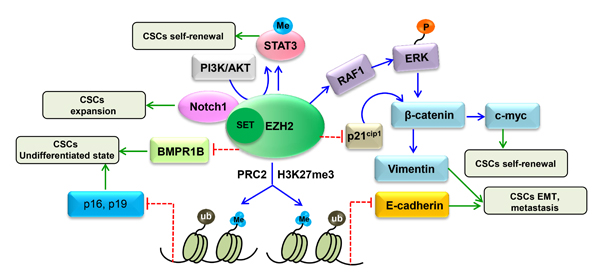
Figure 4: Proposed mechanism involving the regulation of CSC properties by EZH2. EZH2 regulates CSCs through multiple pathways, including BMP signaling, WNT/β-catenin signaling, AKT/EZH2/STAT3 signaling and Notch signaling. In addition, EZH2 mediates p16, p19 and E-cadherin silencing through H3K27me3 to maintain CSC properties. EZH2 also directly methylates STAT3 and inhibits BMPR1B through a PRC2-independent pathway to regulate CSCs.
Abbreviations
AML, acute myeloid leukemia; AR, androgen receptor; BR-DIM, 3, 39-diindolylmethane by Bio Response, Boulder, CO; CHIP, chromatin immunoprecipitation; CML, chronic myelogenous leukemia; CRC, colorectal cancer; CRPC, castration-resistant prostate cancer; CSCs, cancer stem cells; ECSs, epidermal cancer stem cells; EGCG, sepigallocatechin-3-gallate; EMT, epithelial-mesenchymal transition; EOC, epithelial ovarian cancer; EPCs, early precursor cells; ER, estrogen receptor; ESCs, embryonic stem cells; EZH2, enhancer of zeste homolog 2; GBM, glioblastoma multiforme; H3K27me3, trimethylation of histone H3 lysine 27; KD, knock down; MCSs, melanoma cancer stem cells; MLL, mixed-lineage leukemia; NHL, non-Hodgkin lymphoma; NKTL, natural killer/T-cell lymphoma; NSCs, neural stem cells; PCa, prostate cancer; PDAC, pancreatic ductal adenocarcinoma; PcG, Polycomb group; PRC2, Polycomb repressive complex 2; SAH, S-adenosyl-l-homocysteine hydrolase; SCC, squamous cell carcinoma; SFN, sulforaphane, 1-isothiocyanato-4-(methylsulfinyl) butane; SP, side population; TCGA, The Cancer Genome Atlas; TKI, tyrosine kinase inhibitor.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (81472443, 81572572).
CONFLICTS OF INTEREST
There is no conflict of interest regarding this work.
REFERENCES
1. Shlush LI, Hershkovitz D. Clonal evolution models of tumor heterogeneity. Am Soc Clin Oncol Educ Book. 2015: e662-5.
2. Singh AK, Arya RK, Maheshwari S, Singh A, Meena S, Pandey P, Dormond O, Datta D. Tumor heterogeneity and cancer stem cell paradigm: updates in concept, controversies and clinical relevance. Int J Cancer. 2015; 136: 1991-2000.
3. Odoux C, Fohrer H, Hoppo T, Guzik L, Stolz DB, Lewis DW, Gollin SM, Gamblin TC, Geller DA, Lagasse E. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. 2008; 68: 6932-41.
4. Richly H, Aloia L, Di Croce L. Roles of the Polycomb group proteins in stem cells and cancer. Cell Death Dis. 2011; 2: e204.
5. Tsang DP, Cheng AS. Epigenetic regulation of signaling pathways in cancer: role of the histone methyltransferase EZH2. J Gastroenterol Hepatol. 2011; 26: 19-27.
6. Huang BY, Pan XY, Li ZX, Wang ZC, Yu YS, Dou ZH. Polycomb group proteins and their roles in regulating stem cell development. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2012; 34: 281-5.
7. Melling N, Thomsen E, Tsourlakis MC, Kluth M, Hube-Magg C, Minner S, Koop C, Graefen M, Heinzer H, Wittmer C, Sauter G, Wilczak W, Huland H, et al. Overexpression of enhancer of zeste homolog 2 (EZH2) characterizes an aggressive subset of prostate cancers and predicts patient prognosis independently from pre- and postoperatively assessed clinicopathological parameters. Carcinogenesis. 2015; 36: 1333-40.
8. Guo S, Li X, Rohr J, Wang Y, Ma S, Chen P, Wang Z. EZH2 overexpression in different immunophenotypes of breast carcinoma and association with clinicopathologic features. Diagn Pathol. 2016; 11: 41.
9. Hu S, Yu L, Li Z, Shen Y, Wang J, Cai J, Xiao L, Wang Z. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol Ther. 2010; 10: 788-95.
10. Zingg D, Debbache J, Schaefer SM, Tuncer E, Frommel SC, Cheng P, Arenas-Ramirez N, Haeusel J, Zhang Y, Bonalli M, McCabe MT, Creasy CL, Levesque MP, et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun. 2015; 6: 6051.
11. Geng J, Li X, Zhou Z, Wu CL, Dai M, Bai X. EZH2 promotes tumor progression via regulating VEGF-A/AKT signaling in non-small cell lung cancer. Cancer Lett. 2015; 359: 275-87.
12. Yang F, Lv LZ, Cai QC, Jiang Y. Potential roles of EZH2, Bmi-1 and miR-203 in cell proliferation and invasion in hepatocellular carcinoma cell line Hep3B. World J Gastroenterol. 2015; 21: 13268-76.
13. Eskander RN, Ji T, Huynh B, Wardeh R, Randall LM, Hoang B. Inhibition of enhancer of zeste homolog 2 (EZH2) expression is associated with decreased tumor cell proliferation, migration, and invasion in endometrial cancer cell lines. Int J Gynecol Cancer. 2013; 23: 997-1005.
14. Sun M, Liu XH, Lu KH, Nie FQ, Xia R, Kong R, Yang JS, Xu TP, Liu YW, Zou YF, Lu BB, Yin R, Zhang EB, et al. EZH2-mediated epigenetic suppression of long noncoding RNA SPRY4-IT1 promotes NSCLC cell proliferation and metastasis by affecting the epithelial-mesenchymal transition. Cell Death Dis. 2014; 5: e1298.
15. Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, Woodward WA, Hsu JM, Hortobagyi GN, Hung MC. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell. 2011; 19: 86-100.
16. Liu T, Hou L, Huang Y. EZH2-specific microRNA-98 inhibits human ovarian cancer stem cell proliferation via regulating the pRb-E2F pathway. Tumour Biol. 2014; 35: 7239-47.
17. Tiffen J, Gallagher SJ, Hersey P. EZH2: an emerging role in melanoma biology and strategies for targeted therapy. Pigment Cell Melanoma Res. 2015; 28: 21-30.
18. Chen JF, Luo X, Xiang LS, Li HT, Zha L, Li N, He JM, Xie GF, Xie X, Liang HJ. EZH2 promotes colorectal cancer stem-like cell expansion by activating p21cip1-Wnt/beta-catenin signaling. Oncotarget. 2016; 7: 41540-58. doi: 10.18632/oncotarget.9236.
19. Lund K, Adams PD, Copland M. EZH2 in normal and malignant hematopoiesis. Leukemia. 2014; 28: 44-9.
20. Di Croce L, Helin K. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol. 2013; 20: 1147-55.
21. Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010; 7: 299-313.
22. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011; 469: 343-9.
23. Nekrasov M, Klymenko T, Fraterman S, Papp B, Oktaba K, Kocher T, Cohen A, Stunnenberg HG, Wilm M, Muller J. Pcl-PRC2 is needed to generate high levels of H3-K27 trimethylation at Polycomb target genes. Embo j. 2007; 26: 4078-88.
24. Walker E, Chang WY, Hunkapiller J, Cagney G, Garcha K, Torchia J, Krogan NJ, Reiter JF, Stanford WL. Polycomb-like 2 associates with PRC2 and regulates transcriptional networks during mouse embryonic stem cell self-renewal and differentiation. Cell Stem Cell. 2010; 6: 153-66.
25. Francis NJ, Kingston RE, Woodcock CL. Chromatin compaction by a polycomb group protein complex. Science. 2004; 306: 1574-7.
26. Zhou W, Zhu P, Wang J, Pascual G, Ohgi KA, Lozach J, Glass CK, Rosenfeld MG. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell. 2008; 29: 69-80.
27. He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P, Wang G, Marquez VE, Orkin SH, Pu WT. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev. 2012; 26: 37-42.
28. Lee JM, Lee JS, Kim H, Kim K, Park H, Kim JY, Lee SH, Kim IS, Kim J, Lee M, Chung CH, Seo SB, Yoon JB, et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol Cell. 2012; 48: 572-86.
29. Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006; 439: 871-4.
30. Jung HY, Jun S, Lee M, Kim HC, Wang X, Ji H, McCrea PD, Park JI. PAF and EZH2 induce Wnt/beta-catenin signaling hyperactivation. Mol Cell. 2013; 52: 193-205.
31. Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, Xu H, Cato L, Thornton JE, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012; 338: 1465-9.
32. Shi B, Liang J, Yang X, Wang Y, Zhao Y, Wu H, Sun L, Zhang Y, Chen Y, Li R, Zhang Y, Hong M, Shang Y. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol. 2007; 27: 5105-19.
33. Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, Liou YC, Yu Q. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol Cell. 2011; 43: 798-810.
34. Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, Nakano I, Lee C, Joo KM, Rich JN, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013; 23: 839-52.
35. Yan J, Ng SB, Tay JL, Lin B, Koh TL, Tan J, Selvarajan V, Liu SC, Bi C, Wang S, Choo SN, Shimizu N, Huang G, et al. EZH2 overexpression in natural killer/T-cell lymphoma confers growth advantage independently of histone methyltransferase activity. Blood. 2013; 121: 4512-20.
36. Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016; 22: 128-34.
37. van Vlerken LE, Kiefer CM, Morehouse C, Li Y, Groves C, Wilson SD, Yao Y, Hollingsworth RE, Hurt EM. EZH2 is required for breast and pancreatic cancer stem cell maintenance and can be used as a functional cancer stem cell reporter. Stem Cells Transl Med. 2013; 2: 43-52.
38. Gonzalez ME, Moore HM, Li X, Toy KA, Huang W, Sabel MS, Kidwell KM, Kleer CG. EZH2 expands breast stem cells through activation of NOTCH1 signaling. Proc Natl Acad Sci U S A. 2014; 111: 3098-103.
39. Rizzo S, Hersey JM, Mellor P, Dai W, Santos-Silva A, Liber D, Luk L, Titley I, Carden CP, Box G, Hudson DL, Kaye SB, Brown R. Ovarian cancer stem cell-like side populations are enriched following chemotherapy and overexpress EZH2. Mol Cancer Ther. 2011; 10: 325-35.
40. Adhikary G, Grun D, Kerr C, Balasubramanian S, Rorke EA, Vemuri M, Boucher S, Bickenbach JR, Hornyak T, Xu W, Fisher ML, Eckert RL. Identification of a population of epidermal squamous cell carcinoma cells with enhanced potential for tumor formation. PLoS One. 2013; 8: e84324.
41. Xie H, Peng C, Huang J, Li BE, Kim W, Smith EC, Fujiwara Y, Qi J, Cheloni G, Das PP, Nguyen M, Li S, Bradner JE, et al. Chronic Myelogenous Leukemia- Initiating Cells Require Polycomb Group Protein EZH2. Cancer Discov. 2016; 6: 1237-47.
42. Scott MT, Korfi K, Saffrey P, Hopcroft LE, Kinstrie R, Pellicano F, Guenther C, Gallipoli P, Cruz M, Dunn K, Jorgensen HG, Cassels JE, Hamilton A, et al. Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discov. 2016; 6: 1248-57.
43. Yasuda H, Soejima K, Watanabe H, Kawada I, Nakachi I, Yoda S, Nakayama S, Satomi R, Ikemura S, Terai H, Sato T, Suzuki S, Matsuzaki Y, et al. Distinct epigenetic regulation of tumor suppressor genes in putative cancer stem cells of solid tumors. Int J Oncol. 2010; 37: 1537-46.
44. Richard V, Nair MG, Santhosh Kumar TR, Pillai MR. Side population cells as prototype of chemoresistant, tumor-initiating cells. Biomed Res Int. 2013; 2013: 517237.
45. Hurt EM, Kawasaki BT, Klarmann GJ, Thomas SB, Farrar WL. CD44+ CD24(-) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br J Cancer. 2008; 98: 756-65.
46. Dick JE. Looking ahead in cancer stem cell research. Nat Biotechnol. 2009; 27: 44-6.
47. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66: 7-30.
48. Avan A, Crea F, Paolicchi E, Funel N, Galvani E, Marquez VE, Honeywell RJ, Danesi R, Peters GJ, Giovannetti E. Molecular mechanisms involved in the synergistic interaction of the EZH2 inhibitor 3-deazaneplanocin A with gemcitabine in pancreatic cancer cells. Mol Cancer Ther. 2012; 11: 1735-46.
49. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133: 704-15.
50. Toll AD, Dasgupta A, Potoczek M, Yeo CJ, Kleer CG, Brody JR, Witkiewicz AK. Implications of enhancer of zeste homologue 2 expression in pancreatic ductal adenocarcinoma. Hum Pathol. 2010; 41: 1205-9.
51. Ougolkov AV, Bilim VN, Billadeau DD. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin Cancer Res. 2008; 14: 6790-6.
52. Zhang X, Zhang W, Mao XG, Zhen HN, Cao WD, Hu SJ. Targeting role of glioma stem cells for glioblastoma multiforme. Curr Med Chem. 2013; 20: 1974-84.
53. Romaguera-Ros M, Peris-Celda M, Oliver-De La Cruz J, Carrion-Navarro J, Perez-Garcia A, Garcia-Verdugo JM, Ayuso-Sacido A. Cancer-initiating enriched cell lines from human glioblastoma: preparing for drug discovery assays. Stem Cell Rev. 2012; 8: 288-98.
54. Suva ML, Riggi N, Janiszewska M, Radovanovic I, Provero P, Stehle JC, Baumer K, Le Bitoux MA, Marino D, Cironi L, Marquez VE, Clement V, Stamenkovic I. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 2009; 69: 9211-8.
55. Li H, Bitler BG, Vathipadiekal V, Maradeo ME, Slifker M, Creasy CL, Tummino PJ, Cairns P, Birrer MJ, Zhang R. ALDH1A1 is a novel EZH2 target gene in epithelial ovarian cancer identified by genome-wide approaches. Cancer Prev Res (Phila). 2012; 5: 484-91.
56. Li H, Ma F, Wang H, Lin C, Fan Y, Zhang X, Qian H, Xu B. Stem cell marker aldehyde dehydrogenase 1 (ALDH1)-expressing cells are enriched in triple-negative breast cancer. Int J Biol Markers. 2013; 28: e357-64.
57. Kida K, Ishikawa T, Yamada A, Shimada K, Narui K, Sugae S, Shimizu D, Tanabe M, Sasaki T, Ichikawa Y, Endo I. Effect of ALDH1 on prognosis and chemoresistance by breast cancer subtype. Breast Cancer Res Treat. 2016; 156: 261-9.
58. Huang R, Li X, Holm R, Trope CG, Nesland JM, Suo Z. The expression of aldehyde dehydrogenase 1 (ALDH1) in ovarian carcinomas and its clinicopathological associations: a retrospective study. BMC Cancer. 2015; 15: 502.
59. Schulz WA, Hoffmann MJ. Epigenetic mechanisms in the biology of prostate cancer. Semin Cancer Biol. 2009; 19: 172-80.
60. Kong D, Heath E, Chen W, Cher ML, Powell I, Heilbrun L, Li Y, Ali S, Sethi S, Hassan O, Hwang C, Gupta N, Chitale D, et al. Loss of let-7 up-regulates EZH2 in prostate cancer consistent with the acquisition of cancer stem cell signatures that are attenuated by BR-DIM. PLoS One. 2012; 7: e33729.
61. Matsika A, Srinivasan B, Day C, Mader SA, Kiernan DM, Broomfield A, Fu J, Hooper JD, Kench JG, Samaratunga H. Cancer stem cell markers in prostate cancer: an immunohistochemical study of ALDH1, SOX2 and EZH2. Pathology. 2015; 47: 622-8.
62. Deb G, Singh AK, Gupta S. EZH2: not EZHY (easy) to deal. Mol Cancer Res. 2014; 12: 639-53.
63. Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE, Hahn WC, Meyerson M, Garraway LA. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011; 29: 3085-96.
64. Spagnolo F, Ghiorzo P, Orgiano L, Pastorino L, Picasso V, Tornari E, Ottaviano V, Queirolo P. BRAF-mutant melanoma: treatment approaches, resistance mechanisms, and diagnostic strategies. Onco Targets Ther. 2015; 8: 157-68.
65. Ilieva KM, Correa I, Josephs DH, Karagiannis P, Egbuniwe IU, Cafferkey MJ, Spicer JF, Harries M, Nestle FO, Lacy KE, Karagiannis SN. Effects of BRAF mutations and BRAF inhibition on immune responses to melanoma. Mol Cancer Ther. 2014; 13: 2769-83.
66. Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, Salvesen HB, Otte AP, Akslen LA. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006; 24: 268-73.
67. McHugh JB, Fullen DR, Ma L, Kleer CG, Su LD. Expression of polycomb group protein EZH2 in nevi and melanoma. J Cutan Pathol. 2007; 34: 597-600.
68. Adhikary G, Grun D, Balasubramanian S, Kerr C, Huang JM, Eckert RL. Survival of skin cancer stem cells requires the Ezh2 polycomb group protein. Carcinogenesis. 2015; 36: 800-10.
69. Huang J, Zhou L, Chen H, Wu C, Duo Z, Zhang Y. EZH2 is overexpressed in laryngeal squamous cell carcinoma and enhances the stem-like properties of AMC-HN-8 cells. Oncol Lett. 2016; 12: 837-46.
70. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010; 127: 2893-917.
71. Merlos-Suarez A, Barriga FM, Jung P, Iglesias M, Cespedes MV, Rossell D, Sevillano M, Hernando-Momblona X, da Silva-Diz V, Munoz P, Clevers H, Sancho E, Mangues R, et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell. 2011; 8: 511-24.
72. Pang R, Law WL, Chu AC, Poon JT, Lam CS, Chow AK, Ng L, Cheung LW, Lan XR, Lan HY, Tan VP, Yau TC, Poon RT, et al. A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell. 2010; 6: 603-15.
73. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997; 3: 730-7.
74. Ueda K, Yoshimi A, Kagoya Y, Nishikawa S, Marquez VE, Nakagawa M, Kurokawa M. Inhibition of histone methyltransferase EZH2 depletes leukemia stem cell of mixed lineage leukemia fusion leukemia through upregulation of p16. Cancer Sci. 2014; 105: 512-9.
75. Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, Kotliarova S, Kotliarov Y, Walling J, Ahn S, Kim M, Totonchy M, Cusack T, et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell. 2008; 13: 69-80.
76. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006; 9: 391-403.
77. Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs. 2009; 18: 45-56.
78. Guryanova OA, Wu Q, Cheng L, Lathia JD, Huang Z, Yang J, MacSwords J, Eyler CE, McLendon RE, Heddleston JM, Shou W, Hambardzumyan D, Lee J, et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell. 2011; 19: 498-511.
79. Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell Res. 2008; 18: 443-51.
80. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010; 17: 98-110.
81. Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, Lasorella A, Aldape K, Califano A, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010; 463: 318-25.
82. Wen S, Tian J, Niu Y, Li L, Yeh S, Chang C. ASC-J9((R)), and not Casodex or Enzalutamide, suppresses prostate cancer stem/progenitor cell invasion via altering the EZH2-STAT3 signals. Cancer Lett. 2016; 376: 377-86.
83. Zhou Y, Wang Y, Wen J, Zhao H, Dong X, Zhang Z, Wang S, Shen L. Aquaporin 3 promotes the stem-like properties of gastric cancer cells via Wnt/GSK-3beta/beta-catenin pathway. Oncotarget. 2016; 7: 16529-41. doi: 10.18632/oncotarget.7664.
84. Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010; 327: 1650-3.
85. Hoffmeyer K, Raggioli A, Rudloff S, Anton R, Hierholzer A, Del Valle I, Hein K, Vogt R, Kemler R. Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science. 2012; 336: 1549-54.
86. Song X, Xie T. Wingless signaling regulates the maintenance of ovarian somatic stem cells in Drosophila. Development. 2003; 130: 3259-68.
87. Chau WK, Ip CK, Mak AS, Lai HC, Wong AS. c-Kit mediates chemoresistance and tumor-initiating capacity of ovarian cancer cells through activation of Wnt/beta-catenin-ATP-binding cassette G2 signaling. Oncogene. 2013; 32: 2767-81.
88. Holland JD, Klaus A, Garratt AN, Birchmeier W. Wnt signaling in stem and cancer stem cells. Curr Opin Cell Biol. 2013; 25: 254-64.
89. Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005; 65: 3980-5.
90. Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, Wigle TJ, Klaus CR, Allain CJ, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014; 13: 842-54.
91. Conner SD. Regulation of Notch Signaling Through Intracellular Transport. Int Rev Cell Mol Biol. 2016; 323: 107-27.
92. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011; 8: 97-106.
93. Crea F, Paolicchi E, Marquez VE, Danesi R. Polycomb genes and cancer: time for clinical application? Crit Rev Oncol Hematol. 2012; 83: 184-93.
94. Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, Stommel JM, Dunn KL, Wiedemeyer R, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008; 455: 1129-33.
95. Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, Hjelmeland AB, Rich JN. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One. 2008; 3: e3769.
96. Gifford CA, Meissner A. Epigenetic obstacles encountered by transcription factors: reprogramming against all odds. Curr Opin Genet Dev. 2012; 22: 409-15.
97. Qin W, Ren Q, Liu T, Huang Y, Wang J. MicroRNA-155 is a novel suppressor of ovarian cancer-initiating cells that targets CLDN1. FEBS Lett. 2013; 587: 1434-9.
98. Li M, Li J, Ding X, He M, Cheng SY. microRNA and cancer. Aaps j. 2010; 12: 309-17.
99. Cheng W, Liu T, Wan X, Gao Y, Wang H. MicroRNA-199a targets CD44 to suppress the tumorigenicity and multidrug resistance of ovarian cancer-initiating cells. Febs j. 2012; 279: 2047-59.
100. Strillacci A, Valerii MC, Sansone P, Caggiano C, Sgromo A, Vittori L, Fiorentino M, Poggioli G, Rizzello F, Campieri M, Spisni E. Loss of miR-101 expression promotes Wnt/beta-catenin signalling pathway activation and malignancy in colon cancer cells. J Pathol. 2013; 229: 379-89.
101. Semaan A, Qazi AM, Seward S, Chamala S, Bryant CS, Kumar S, Morris R, Steffes CP, Bouwman DL, Munkarah AR, Weaver DW, Gruber SA, Batchu RB. MicroRNA-101 inhibits growth of epithelial ovarian cancer by relieving chromatin-mediated transcriptional repression of p21(waf(1)/cip(1)). Pharm Res. 2011; 28: 3079-90.
102. Ren G, Baritaki S, Marathe H, Feng J, Park S, Beach S, Bazeley PS, Beshir AB, Fenteany G, Mehra R, Daignault S, Al-Mulla F, Keller E, et al. Polycomb protein EZH2 regulates tumor invasion via the transcriptional repression of the metastasis suppressor RKIP in breast and prostate cancer. Cancer Res. 2012; 72: 3091-104.
103. He XP, Shao Y, Li XL, Xu W, Chen GS, Sun HH, Xu HC, Xu X, Tang D, Zheng XF, Xue YP, Huang GC, Sun WH. Downregulation of miR-101 in gastric cancer correlates with cyclooxygenase-2 overexpression and tumor growth. Febs j. 2012; 279: 4201-12.
104. Cho HM, Jeon HS, Lee SY, Jeong KJ, Park SY, Lee HY, Lee JU, Kim JH, Kwon SJ, Choi E, Na MJ, Kang J, Son JW. microRNA-101 inhibits lung cancer invasion through the regulation of enhancer of zeste homolog 2. Exp Ther Med. 2011; 2: 963-7.
105. Konno Y, Dong P, Xiong Y, Suzuki F, Lu J, Cai M, Watari H, Mitamura T, Hosaka M, Hanley SJ, Kudo M, Sakuragi N. MicroRNA-101 targets EZH2, MCL-1 and FOS to suppress proliferation, invasion and stem cell-like phenotype of aggressive endometrial cancer cells. Oncotarget. 2014; 5: 6049-62. doi: 10.18632/oncotarget.2157.
106. Kong D, Banerjee S, Ahmad A, Li Y, Wang Z, Sethi S, Sarkar FH. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS One. 2010; 5: e12445.
107. Jin B, Wang W, Meng XX, Du G, Li J, Zhang SZ, Zhou BH, Fu ZH. Let-7 inhibits self-renewal of hepatocellular cancer stem-like cells through regulating the epithelial-mesenchymal transition and the Wnt signaling pathway. BMC Cancer. 2016; 16: 863.
108. Glazer RI, Hartman KD, Knode MC, Richard MM, Chiang PK, Tseng CK, Marquez VE. 3-Deazaneplanocin: a new and potent inhibitor of S-adenosylhomocysteine hydrolase and its effects on human promyelocytic leukemia cell line HL-60. Biochem Biophys Res Commun. 1986; 135: 688-94.
109. Cui B, Yang Q, Guan H, Shi B, Hou P, Ji M. PRIMA-1, a mutant p53 reactivator, restores the sensitivity of TP53 mutant-type thyroid cancer cells to the histone methylation inhibitor 3-Deazaneplanocin A. J Clin Endocrinol Metab. 2014; 99: E962-70.
110. Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE, Jones PA. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009; 8: 1579-88.
111. Jiang X, Lim CZ, Li Z, Lee PL, Yatim SM, Guan P, Li J, Zhou J, Pan J, Chng WJ, Chai CL, Yu Q. Functional Characterization of D9, a Novel Deazaneplanocin A (DZNep) Analog, in Targeting Acute Myeloid Leukemia (AML). PLoS One. 2015; 10: e0122983.
112. Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, Scott MP, Jin L, Smith JJ, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012; 8: 890-6.
113. Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013; 110: 7922-7.
114. Campbell JE, Kuntz KW, Knutson SK, Warholic NM, Keilhack H, Wigle TJ, Raimondi A, Klaus CR, Rioux N, Yokoi A, Kawano S, Minoshima Y, Choi HW, et al. EPZ011989, A Potent, Orally-Available EZH2 Inhibitor with Robust in Vivo Activity. ACS Med Chem Lett. 2015; 6: 491-5.
115. Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, Gu J, Ardayfio O, Zhang JH, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012; 109: 21360-5.
116. Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley JA, Graves AP, Scherzer DA, Shu A, et al. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med Chem Lett. 2012; 3: 1091-6.
117. McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A 3rd, Diaz E, LaFrance LV, Mellinger M, Duquenne C, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012; 492: 108-12.
118. Amatangelo MD, Garipov A, Li H, Conejo-Garcia JR, Speicher DW, Zhang R. Three-dimensional culture sensitizes epithelial ovarian cancer cells to EZH2 methyltransferase inhibition. Cell Cycle. 2013; 12: 2113-9.
119. Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, Liu F, Gao C, Huang XP, Kuznetsova E, Rougie M, Jiang A, Pattenden SG, et al. An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013; 8: 1324-34.
120. Hua WF, Fu YS, Liao YJ, Xia WJ, Chen YC, Zeng YX, Kung HF, Xie D. Curcumin induces down-regulation of EZH2 expression through the MAPK pathway in MDA-MB-435 human breast cancer cells. Eur J Pharmacol. 2010; 637: 16-21.
121. Dimri M, Bommi PV, Sahasrabuddhe AA, Khandekar JD, Dimri GP. Dietary omega-3 polyunsaturated fatty acids suppress expression of EZH2 in breast cancer cells. Carcinogenesis. 2010; 31: 489-95.
122. Choudhury SR, Balasubramanian S, Chew YC, Han B, Marquez VE, Eckert RL. (-)-Epigallocatechin-3-gallate and DZNep reduce polycomb protein level via a proteasome-dependent mechanism in skin cancer cells. Carcinogenesis. 2011; 32: 1525-32.
123. Clarke JD, Dashwood RH, Ho E. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008; 269: 291-304.
124. Rudolf K, Cervinka M, Rudolf E. Sulforaphane-induced apoptosis involves p53 and p38 in melanoma cells. Apoptosis. 2014; 19: 734-47.
125. Hamsa TP, Thejass P, Kuttan G. Induction of apoptosis by sulforaphane in highly metastatic B16F-10 melanoma cells. Drug Chem Toxicol. 2011; 34: 332-40.
126. Chinembiri TN, du Plessis LH, Gerber M, Hamman JH, du Plessis J. Review of natural compounds for potential skin cancer treatment. Molecules. 2014; 19: 11679-721.
127. Balasubramanian S, Chew YC, Eckert RL. Sulforaphane suppresses polycomb group protein level via a proteasome-dependent mechanism in skin cancer cells. Mol Pharmacol. 2011; 80: 870-8.
128. Fisher ML, Adhikary G, Grun D, Kaetzel DM, Eckert RL. The Ezh2 polycomb group protein drives an aggressive phenotype in melanoma cancer stem cells and is a target of diet derived sulforaphane. Mol Carcinog. 2016; 55: 2024-36.
129. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012; 10: 717-28.
130. Lu H, Li G, Zhou C, Jin W, Qian X, Wang Z, Pan H, Jin H, Wang X. Regulation and role of post-translational modifications of enhancer of zeste homologue 2 in cancer development. Am J Cancer Res. 2016; 6: 2737-54.
131. Liu YW, Sun M, Xia R, Zhang EB, Liu XH, Zhang ZH, Xu TP, De W, Liu BR, Wang ZX. LincHOTAIR epigenetically silences miR34a by binding to PRC2 to promote the epithelial-to-mesenchymal transition in human gastric cancer. Cell Death Dis. 2015; 6: e1802.
132. Sarma K, Cifuentes-Rojas C, Ergun A, Del Rosario A, Jeon Y, White F, Sadreyev R, Lee JT. ATRX directs binding of PRC2 to Xist RNA and Polycomb targets. Cell. 2014; 159: 869-83.
133. Kaneko S, Bonasio R, Saldana-Meyer R, Yoshida T, Son J, Nishino K, Umezawa A, Reinberg D. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol Cell. 2014; 53: 290-300.