
INTRODUCTION
Adenoid cystic carcinoma (ACC) is a rare type of cancer, most commonly originating from the salivary glands, with an indolent behavior but a high propensity to metastasize [1]. Surgery with wide resection is the mainstay of treatment for localized ACC. A watch and wait approach is appropriate if a patient has metastases, especially if confined to the lungs with minimal symptoms [2].
Several anti-cancer agents have been studied to treat metastatic ACC, however none have shown a robust response rate, with stable disease (SD) being the most common reported outcome. SD duration over 6 months is a common metric used in ACC trials, varying among different agents, with reported ranges of 30–60% (reviewed in [3]). Currently, there is no Food and Drug Administration (FDA) approved agent for the treatment of ACC.
Suberoylanilide hydroxamic acid (SAHA), also known as vorinostat, is a small molecule inhibitor of histone deacetylase (HDAC). Two patients with ACC on a previously published National Cancer Institute (NCI) trial evaluating vorinostat in patients with advanced cancers and liver dysfunction experienced significant radiological and/or clinical improvement [4]. Additionally, recent next generation sequencing (NGS) studies of ACC have demonstrated a low mutation rate with few recurring single gene mutations converging on regulators of chromatin remodeling [5, 6].
Based on these encouraging findings, we undertook this multi-institutional phase 2 trial of vorinostat in patients with locally advanced or metastatic ACC.
RESULTS
Baseline demographics and patient characteristics
Thirty patients were enrolled over 22 months. As shown in Table 1, 19 patients (63%) were female and 24 (80%) were Caucasian. The median age was 53 years (range 21–73), 21 had a performance status of 1 and 19 were chemo-naïve. Twenty-eight had metastatic disease and 2 patients had locally advanced disease. While not an eligibility criterion, 27 out of 30 patients had radiographic disease progression before study entry.
Table 1: Baseline demographics and disease characteristics (N = 30)
Characteristics |
No. of Patients |
% |
|---|---|---|
Race |
||
White |
24 |
80 |
African-American |
3 |
10 |
Asian |
2 |
6.7 |
Middle Eastern |
1 |
3.3 |
Sex |
||
Male |
11 |
37 |
Female |
19 |
63 |
ECOG PS |
||
0 |
7 |
23 |
1 |
21 |
70 |
2 |
2 |
7 |
Age (years) |
||
Median (range) |
53 (21–73) |
|
Disease Site |
||
Metastatic |
28 |
93 |
Locally Advanced |
2 |
7 |
Prior Chemotherapy |
||
Chemo-naive |
19 |
63 |
ECOG PS- Eastern Cooperative Oncology Group performance status
Efficacy
Partial responses by Response Evaluation Criteria in Solid Tumors (RECIST) were observed in two patients. These responses were not immediate. One patient had a decrease in the size of lung nodules achieving a partial response (PR) at cycle 8 and the other patient experienced reduction in size and number of liver and lung lesions achieving a PR by cycle 10 (Figure 1). One of the patients with PR had surgery and radiation as prior therapies and the other patient with PR also received surgery, radiation and one systemic chemotherapy prior to enrolling in this trial. While stable disease (SD) was the best response in 27 patients, 20 patients demonstrated a decrease in the size of their tumors (Figure 2). Another patient had improvement in ascites from cycle 6 through cycle 20 (minor response). The overall response rate (RR) was 7% with a clinical benefit rate (PR+SD) of 97% (Table 2). Anecdotal improvement in symptoms was also noted in 3 patients despite SD by RECIST. Three patients received extensive therapy to 56, 62 and 66 cycles.
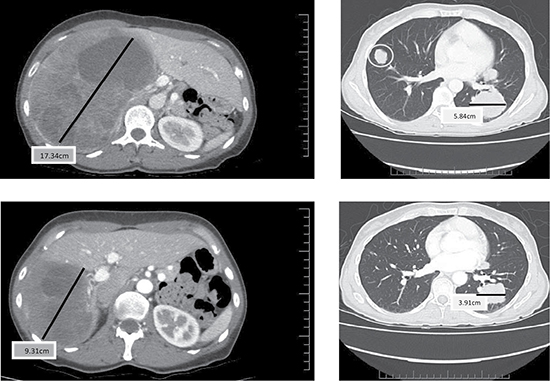
Figure 1: Baseline (top) and Cycle 13 (bottom) computer tomography (CT) scan (chest and abdomen) from a patient who had a partial response and a duration of 7.2 months.
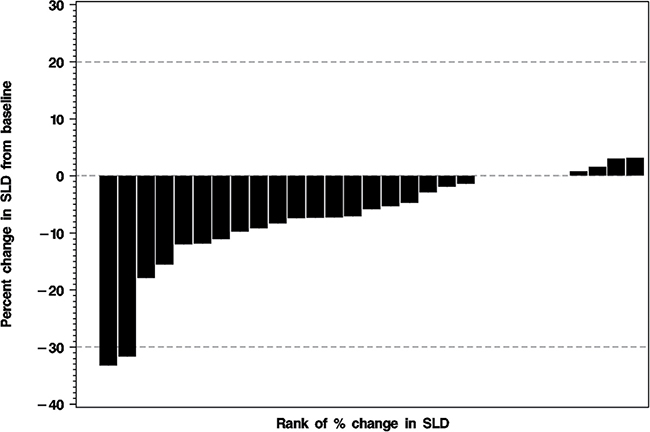
Figure 2: Waterfall plot of the percent change from baseline to best response in the sum of the longest diameters (SLD) of the index lesions for 29 treated patients. For one treated patient the follow-up CT was not available. The horizontal dashed lines indicate the thresholds for partial response (PR: ≥ 30% decrease in SLD) and for progressive disease (PD: ≥ 20% increase in SLD).
Table 2: Response rate (among N = 30 treated patients)
Response Category |
No. of Patients (%) |
95% Confidence Interval |
|---|---|---|
Partial Response (PR) |
2/30 (7%) |
2%–21% |
Stable Disease (SD) |
27/30 (90%) |
74%–97% |
Clinical Benefit (PR+SD) |
29/30 (97%) |
83%–99% |
Progressive Disease |
1/30 (4%) |
1%–17% |
Toxicity
The most frequent grade 3 toxicities were lymphopenia (7/30 = 23%), hypertension and fatigue (3/30 = 10% each), headache, oral pain and thromboembolic event (2/30 = 7% each) (Table 3). Grade 4 bronchopulmonary hemorrhage and hypoxia occurred in 3% (1 patient each), but neither was drug-related. Eleven patients were dose reduced, 10 patients were reduced by one dose level to 300mg daily and 1 patient to 300 mg once a day for 5 days with 2 days off (reduction of two dose levels). Three patients discontinued treatment due to toxicity.
Table 3: Grade 3 or 4 toxicities experienced by at least 1 patient (N = 30 treated patients)
Type of toxicity a |
Worst grade experienced |
||||
|---|---|---|---|---|---|
0 |
1 |
2 |
3 |
4 |
|
Bronchopulmonary hemorrhage |
0 |
0 |
0 |
0 |
1 |
Hypoxia |
0 |
0 |
0 |
0 |
1 |
Lymphopenia |
13 |
7 |
3 |
7 |
0 |
Hypertension |
15 |
5 |
7 |
3 |
0 |
Fatigue |
9 |
12 |
6 |
3 |
0 |
Headache |
21 |
4 |
3 |
2 |
0 |
Oral pain |
27 |
0 |
1 |
2 |
0 |
Thrombo-embolic event |
26 |
2 |
0 |
2 |
0 |
aThere were 14 other miscellaneous toxicities whose worst Grade was 3, and they occurred in only 1 patient: appendicitis, menorrhagia, atelectasis, bronchial obstruction, pneumonia, lung infection, wound complication, thrombocytopenia, diarrhea, cataract, oral mucositis, hypophosphatemia, increased lymphocyte count, and non-cardiac chest pain.
Time to event end-points
Time to tumor response (TTR) was 7.7 and 10.0 months in the 2 patients who achieved PR. The duration of their response was 53 and 7.2 months, respectively, with the former patient still receiving study drug. Prior to study entry, 27 (90%) of the 30 patients had documented progressive disease on their most recent scans. The median follow-up among the censored patients was 8.0 months for stable disease duration (SDD), 10.0 months for progression free survival (PFS), and 11.5 months for overall survival (OS). The 6 month rates for SDD, PFS, and OS were 75%, 72%, and 94%, respectively. The 12 month rates for SDD, PFS and OS were 45%, 46%, and 88%, respectively. (Supplementary Table 1- Supplementary Appendix). The median PFS and SDD were both 11.4 months, and the median OS has not been reached. The estimated SDD and PFS distributions are shown in the Supplementary Appendix (Supplementary Figures 1 and 2, respectively).
Exploratory studies
We hypothesized that the variation in clinical response to vorinostat (“responders” or “non-responders”- Table 4) is associated with the presence or absence of driver mutations including those targeting chromatin remodeling genes involved in the regulation of the cancer epigenome. To test our hypothesis, we used both targeted and whole exome sequencing to survey the mutational landscape of each tumor.
Table 4: Selected responses to vorinostat and mutations identified
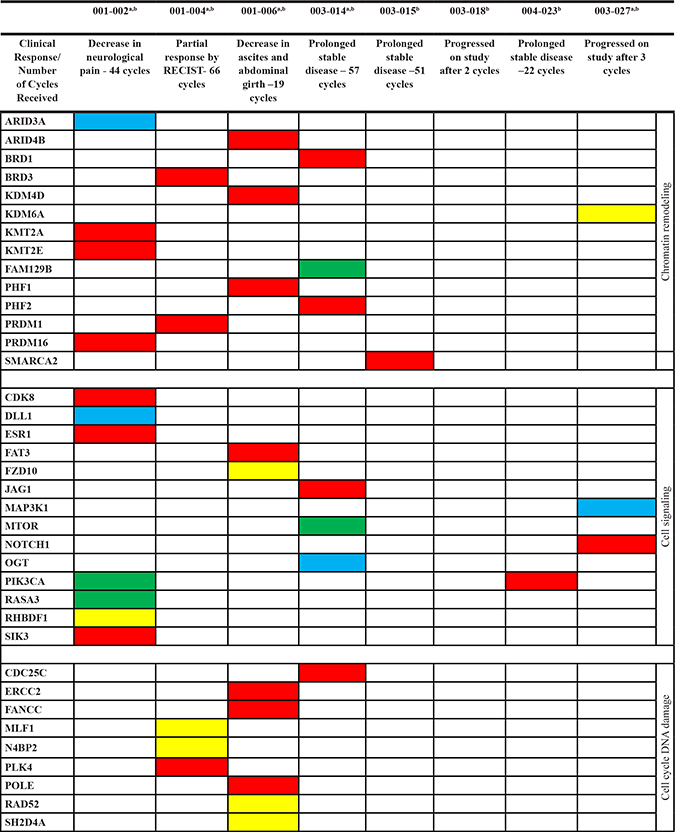
RECIST- Response Evaluation Criteria in Solid Tumors; Yellow- Nonsense; Red- Missense; Blue- Frame shift indel; Green- Essential splice, a- Exome, b- 10-gene panel.
Exploratory studies results
"Responders"
We sequenced the exomes from 4 of these patients (001–002, 001–004, 001–006, 003–014). In each case we found somatic mutations of interest after filtering. These and 2 additional responder cases (003–015, 004–023) were screened with our 10 gene panel. A striking observation was the lack of recurring gene specific mutations in these 4 cases (Table 4). However, recurring cell pathways and processes were targeted.
Patient 001–002
There were a series of unique somatic variants detected in this tumor. Notably these converged on chromatin remodeling, transcriptional regulation, and cell signaling. The DNA binding factor ARID3A has been implicated in RB1/E2F mediated control of cell cycle progression while KMT2E and KMT2A belong to the myeloid/lymphoid or mixed-lineage leukemia (MLL) class of epigenetic writers (Supplementary Figure 3- Supplementary Appendix). In addition, mutations targeted regulators of RAS (RASA3), EGFR (RHBDF1), and estrogen (ESR1) and NOTCH (DLL1) signaling.
Patient 001–004
Variants in genes associated with chromatin remodeling and DNA repair were detected. These included BRCA1Q356R, BRCA2N372H, and EZH2D185H. However, each of these variants appears in dbSNP, has a population frequency of > 5%, and has not been associated with an increased cancer risk [7]. We also detected ATMR720H in the tumor tissue. However, the targeted amino acids in both wild type and this somatic variant have positive charge side chains. Thus, the functional effects of this variant on ATM activity are not clear. In contrast the MLF1L120* and N4BP2K76* non sense variants have low or absent reported allele frequencies. MLF1 (Myeloid leukemia factor 1) is a transcription factor that prevents cells from exiting the cell cycle through suppression of CDKN1B/p27Kip1 levels and activation of TP53 [8]. Mutations in TP53 have been reported in ACC [6]. However, we did not observe any TP53 mutations in the current study. Thus the MLF1L120* mutation may provide a novel mechanism to target TP53 mediated repair and cell cycle checkpoint functions in ACC. N4BP2 (NEDD4 binding protein 2) has 5′-polynucleotide kinase and nicking endonuclease activity and may play a role in DNA repair or recombination [9].
Patient 001–006
We identified two additional novel ACC mutations (SH2D4AR237*, FZD10G345*) that have previously been reported in colon and gastric cancer [10–13]. SH2D4A inhibits estrogen-induced cell proliferation by competing with phospholipase C, gamma 2 (PLCG) for binding to ESR1, blocking the effect of estrogen on PLCG and repressing estrogen-induced proliferation [12]. It may play a role in T-cell development and function. Both of these unique SH2D4A and FZD10 mutations were validated in our targeted resequencing analyses. The Drosophila frizzled polarity gene homolog 10 (FZD10) is a member of the G protein coupled receptor (GPCR) superfamily, exhibiting characteristics of a WNT receptor [13]. We also detected variants in chromatin remodeler genes including ARID4BT84R and PHF1E128Q. The latter gene encodes a Polycomb group protein that is a component of a histone H3 lysine-27 (H3K27)-specific methyltransferase complex, and functions in transcriptional repression of homeotic genes [14]. The protein is also recruited to double-strand breaks, and reduced protein levels results in X-ray sensitivity and increased homologous recombination [15, 16]. Recurring chromosomal aberrations involving PHF1 may be a cause of endometrial stromal tumors [17, 18]. In addition, we detected variants in the DNA damage checkpoint regulators ATRPhe926Leu and RAD52Y415*.
Patient 003–014
The mutations in this tumor included 3 predicted high impact mutations in MTOR a regulator of stress response, OGT a glycosyltransferase that modifies a broad range of targets including H2B, AKT1, EZH2, PFKL, KMT2E/MLL5, MAPT/TAU and HCFC1, and FAM129B a negative regulator of apoptosis [19]. Additional mutations included the cell cycle regulator CDC25C, the chromatin regulators PHF2 and BRD1, and the NOTCH1 ligand JAG1. The latter is a transcriptional target of MYB [20].
Patients 003–015 and 004–023
Both patients were screened with our 10 gene panel. We detected a somatic variant in SMARCA2 in 003–015 adding to the list of unique mutations in chromatin regulators. In addition, we detected a PIK3CA mutation in 004–023. Mutations in both these genes have been previously reported in ACC [5, 6].
"Non-responders"
We obtained WES data from one of four non-responders (003–027). Targeted resequencing provided additional data for a second non- responder (003–018) and validation of the KDM6A mutation detected in the whole exome results (Table 4).
Patient 003–027
A striking finding was the presence of a mutation in the NOTCH1 receptor (Supplementary Figure 3). This gain of function mutation destabilizes the heterodimerization domain of the receptor. It results in ligand-independent cleavage of Notch1 at site S2 and subsequent receptor activation. This mutation is a recurrent driver of T cell ALL [21, 22]. To our knowledge, this is the first report of the association of this recurring mutation in ACC with resistance to vorinostat. We also detected a nonsense mutation in the lysine demethylase KDM6A. Mutations in this lysine demethylase, also known as UTX, have been reported in several cancers and are one of several classes of mutations that are believed to converge on chromatin remodeling in ACC [11, 23]. Pathogenic KDM6A variants disrupt histone structure [23]. This latter mutation was unique to non-responder 003–027 and was validated by targeted resequencing with our 10-gene panel.
Patient 003–018
The exome data for the tumor sample failed overall quality metrics including coverage and percent target bases. Targeted resequencing was of sufficient quality for this tumor normal pair however we did not identify any somatic variants in the 10-gene panel.
DISCUSSION
ACC is a rare neoplasm with an initial indolent pace. However, once it becomes metastatic, it runs an inexorable, albeit slow growing course with poor and short-lived responses to available treatments [2, 24].
In our study we saw 2 PRs by RECIST out of 30 patients treated with vorinostat. Both responses occurred late into treatment after 8 and 10 cycles. A third patient had a minor response with reduction in ascites, and eliminated the need for therapeutic paracentesis. Anedoctal improvement in symptoms was also observed in an additional 3 patients (decreased pain, improvement in shortness of breath, improvement in eye movement, and improvement in fatigue) with SD by RECIST. Of note, 20 patients demonstrated a decrease in the size of their tumors. Additionally, 2 patients with ACC were treated with vorinostat in a prior NCI liver dysfunction trial and experienced significant clinical and radiological improvement. (decrease in size of liver lesions and normalization of liver enzymes from moderate liver dysfunction) [4]. In the patients from the liver dysfunction trial, the clinical improvement (less fatigue and pain, weight gain) also started early (after 2–3 cycles) and preceded any radiologic change by several cycles, similar to what we observed in our current trial [4].
SD duration (SDD) has been described in several trials for patients with ACC [25–31]. It is unclear if SD represents a marker of drug activity or simply the indolent behavior of this tumor. However, most ACC studies report SDD of 6 months or more as an endpoint, which may be clinically relevant, especially in view of tumors that were progressing prior to study entry (reviewed in [32]). In our study, although not required as part of eligibility criteria; 90% (27/30) of the patients had radiological evidence of disease progression prior to enrollment. We observed a 6-month rate of SD of 75%, which is comparable to recent systemic therapeutic interventions studied in this disease [27–29, 33].
Currently, response to treatment in most solid tumors is evaluated by measuring the sum of the longest diameter of target lesions according to the RECIST criteria, which has several limitations [34]. RECIST does not take into account tumor growth dynamics or rate and cannot fully capture response and progression in certain tumor types such as gastrointestinal stromal tumors (GISTs), where which incorporates tumor densities in assessing responses, have been more widely used have been more widely used [35]. Additionally, in GIST, RECIST underestimates PR rates when compared to CHOI criteria [36]. A recent study of sorafenib in patients with salivary gland tumors (both ACC and non-ACC) used both RECIST and CHOI criteria to assess responses. It revealed 6 PRs according to RECIST, and 10 PRs according to CHOI (only 2 were concordant with RECIST) [33]. Also, RECIST may not be the best tool to evaluate tumor response to targeted agents or immune therapy [37–40]. The concept of volumetric tumor growth likely is a better method to assess response in such instances [41]. Volumetric imaging analysis can predict clinical response earlier than RECIST in some cancers and it is also more sensitive than changes in unidimensional diameters [41]. Considering the late responses observed in some of the patients in the current study, along with early symptomatic improvement, a possible explanation could be that RECIST criteria may not be the best method to monitor ACC patients for response.
Patient reported outcomes (PRO) questionnaires evaluate the impact on patient’s functioning and well-being that could likely be caused by the disease and/or its treatment without modification or interpretation by the observer [42]. Given the slow growing but unrelenting progression of ACC, improvement in PRO is a reasonable measure of clinical benefit. Future trials in patients with ACC should incorporate such questionnaires as endpoints.
Another possibility is that the late responses we observed could be due to an immuno-stimulatory effect of vorinostat, which may take longer to occur and not be evident radiographically as early as with some other therapeutic interventions. Mounting evidence suggests a role for HDACi in modulating the immune system and enhancing efficacy of immunotherapeutic strategies. HDACi have been shown to alter the activation and function of macrophage and dendritic cells [43]; regulate cytokine production [44, 45] and upregulate major histocompatibility class I and II molecules [44, 46].
A feature of the ACC mutational landscape is a low mutational burden with a paucity of common recurring driver mutations typically seen in solid tumors [5, 6]. For example, TP53 mutations, one of the most frequent somatic lesions in solid tumors, have been detected in only 5% of ACC cases studied [6]. Nevertheless, this represents the second highest incidence of reported somatic variants in ACC. Notably none were detected by either WES or targeted resequencing in the present study. Recent studies suggest that the disparate low frequency mutations in ACC tumors appear to converge on specific pathways notably DNA repair, chromatin regulation, and NOTCH signaling. The exception to this inter-tumor genomic diversity is a translocation targeting c-Myb and NFIB, creating a fusion gene in > 40% of ACC tumors [47]. Except for low impact variants in UTRs we did not detect any evidence for alterations (mutations or copy number aberrant intervals) associated with this frequent event in our ACC cohort. However, our experimental approach, whole exome sequences for 5 tumors and targeted resequencing that included 3 additional samples, was designed for problematic formalin-fixed, paraffin-embedded (FFPE) samples and was not optimal for detection of genomic lesions associated with translocations.
The whole exome data for the patients in this study highlight the role of DNA repair and chromatin structure regulation in ACC. Mutations, including somatic non sense mutations, targeting key mediators of repair, including RAD52, ATR, and POLQ, were detected in multiple patients. Although none of these mutations were recurring, they converge on well-characterized steps in DNA repair and replication. In addition, we detected mutations targeting different cell signaling pathways including estrogen, NOTCH and the Wnt pathway, as well as PIK3CA/AKT signaling. In each case, unique mutations converged on the pathway. These observations are in agreement with recent whole exome data of ACC [5, 6]. In addition, the validated mutation in FZD10 provides further evidence for a role of aberrant Wnt signaling in ACC [48]. The most prominent set of mutations was present in multiple chromatin regulating genes (Table 4). These included lysine methyltransferases, bromodomain containing proteins, and members of the SWI/SNF chromatin regulator family. Variants in SMARCA2 were detected in patient 003–015 however they were either conserved or resulted in an amino acid substitution with the same polarity.
The presence of the KDM6A non sense mutation in patient 003–027 is consistent with studies suggesting that disruption of the epigenome is a driver in a subset of ACC [49, 50]. Mutations that arise in this region of the protein have been described previously in multiple tumor types. The functional impact of these KDM6A mutations was evaluated via a well-characterized assay for trimethylation at lysine 27 of histone H3 (H3K27me3). Notably abrogation of demethylase activity was observed in cells overexpressing mutant KDM6A but not in those overexpressing wild-type KDM6A. Moreover, whereas wild-type KDM6A suppressed growth, mutants either lost the ability to suppress growth or, in some cases, augmented it (dominant phenotype). Strikingly patient 003–027, whose tumor had a KDM6AR1272X high impact mutation, progressed after only 3 cycles of vorinostat. Future clinical trials should incorporate sequencing of this and other histone demethylases as a correlative for responses to vorinostat and other epigenetic targeting agents. The presence of well-characterized activating mutations in NOTCH1 supports the role of this pathway in ACC and its potential therapeutic targeting. Notably, mutations targeting NOTCH signaling were present in both responders and non-responders (Supplementary Figure 3, Table 4). Our study adds to the list of mutations reported in ACC and provides further support for the role of mutations targeting chromatin-remodeling genes in this disease.
Our small exploratory study confirms the presence of mutations targeting epigenome regulation in ACC, as 10 out of 11 patients had mutations in chromatin remodeling genes. Notably, a well characterized mutation in the histone demethylase KDM6A/UTX was present in a patient who did not respond to vorinostat. This is in contrast to the presence of mutations in histone methyltransferases (KMT2E and KMT2A), bromodomain containing proteins (BRD1 and BRD3), and members of the SWI/SNF chromatin regulator family (SMARCA2) in responders. However, this non-responder patient (003–027) had an activating NOTCH1 mutation, suggesting that it could be the driving mutation in this case, as activating NOTCH1 mutations have been shown to confer a worse prognosis [51]. The presence of a well-characterized activating mutation in NOTCH1 confirms the role of this signaling pathway in ACC and may provide a therapeutic window in future studies. To advance these observations will require a more standardized tissue collection and processing protocol for correlative studies. Ongoing improvements in NGS technologies will make it feasible to recover more data from currently limited samples. In addition, the data from our current study and recently published NGS data provide the basis for well-designed gene panels that should include KDM6A/UTX and related histone demethylases for targeted resequencing
We acknowledge the limitations of our study. First, while not all patients had documented progression of disease at study entry, nonetheless, almost all (90%) of the patients did. Second, we did not have a quality of life questionnaire in this trial, which would have helped to capture and describe the clinical improvements observed. Third, we did not have funding to incorporate other imaging modalities, (such as PET/CT), or to utilize other measurement criteria (such as volumetric assessment), which would have helped evaluate the utility of a different imaging criteria in ACC. Fourth, due to the small number of samples analyzed, we could not unequivocally identify mutations that were predictors of response or resistance to vorinostat.
In conclusion, the findings from this study are encouraging. Vorinostat was relatively well tolerated and patients remained on drug treatment for significant periods of time. The clinical benefit rate (97%) was very high, as was the 6-month stable disease rate (75%). Despite a small number of PRs overall, these responses were durable and associated with improvement in symptoms. We found mutations in chromatin remodeling genes in most samples analyzed. Perhaps the fact that ACC has a low rate of somatic mutations, coupled with the pleotropic effects of vorinostat as a modulator of epigenetics, can partly explain this drug’s antitumor activity in this disease. In the future, it will be important to assess responses to vorinostat and other HDACi in patients with ACC relative to aberrant epigenetic regulation with tumor gene sequencing. In summary, despite this being a statistically negative trial, we observed clinical improvement in symptoms in several patients, along with two partial responses and a significant decrease in ascites in another patient. We find this study encouraging and will pursue further prospective clinical trials evaluating HDAC inhibitors perhaps in combination with other agents, for the treatment of ACC we find this study encouraging and will pursue further prospective clinical trials evaluating HDAC inhibitors perhaps in combination with other agents, for the treatment of ACC. Such future trials should include additional tumor measurement criteria, such as volumetric tumor measurement and quality of life questionnaires, along with systematic molecular analysis and tumor sequencing.
MATERIALS AND METHODS
Eligibility criteria
Key eligibility criteria included: histologically or cytologically confirmed locally advanced, recurrent or metastatic ACC; age ≥ 18 years; measurable disease per RECIST v1.1; Eastern Cooperative Oncology Group (ECOG) performance status 0 to 2; life expectancy ≥ 12 weeks. Any number of prior chemotherapy regimens was allowed but not required. Laboratory parameters included adequate organ and marrow function defined as: leucocytes ≥ 3,000/μL, absolute neutrophil count ≥ 1,500/μL, platelets ≥ 100,000/μL, total bilirubin within institutional normal limits (WNL), liver enzymes ≤ 2.5X upper limit of normal, and creatinine WNL, or creatinine clearance ≥ 60 mL/min. Patients with previous brain metastases were eligible if they were treated and stable for ≥ 1 month with no requirement for steroids. All patients provided informed consent before treatment. The study complied with local institutional review board guidelines. Baseline formalin fixed paraffin embedded FFPE slides or blocks needed to be available for correlative studies.
Study design and treatment
This was a multicenter, international, single-arm, phase 2 study. Patients received oral vorinostat 400 mg once a day continuously until disease progression, death, withdrawal of consent, or unacceptable adverse events. Each cycle was defined as 28 days. Imaging was obtained at baseline and repeated every 8 weeks for the first 6 cycles. After 6 months on study, imaging was extended to every 12 weeks. Study visits occurred every 2 weeks for the first 2 cycles, then every 4 weeks thereafter. Safety assessments included physical examinations, AE assessment, and laboratory measurements. AEs were graded according to the Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. Vorinostat was distributed by Cancer Therapy Evaluation Program (CTEP) of the National Cancer Institute under a collaborative agreement with Merck & Co. Inc.
Objectives and endpoints
The primary objective was to evaluate the efficacy as defined by response rate (RR) of vorinostat in patients with ACC. Secondary objectives were to evaluate time to tumor response, response duration (RD), stable disease duration, progression free survival, overall survival and to characterize safety and tolerability of vorinostat in ACC patients. For RD, SDD, and PFS, if progression had not occurred, then their duration was censored as of the date of the most recent tumor assessment. For patients still alive, their OS duration was censored as of the most recent date that the patients’ vital status had been confirmed.
Statistical methods
Design
Complete + partial (CR+PR) rate was the primary statistical endpoint. We used a 2-stage Simon optimal design with p0 = 5%, p1 = 20%, alpha = 0.15, power = 0.90, and PET = 0.540. Stage 1 required 12 patients. At least 1 responder in Stage 1 would be needed to justify continuing to Stage 2, which would require 17 more patients. If < 3 responders, we would conclude that vorinostat has insufficient efficacy to justify further study.
Analysis
95% confidence interval (CI) estimates were calculated via Wilson’s method. TTE endpoints were estimated using the Kaplan-Meier (K-M) method. Due to the small numbers of events, point estimates of survival statistics were estimated more conservatively using linear interpolation among successive event times on the K-M curves, and a slightly lower confidence level (90%) was used when determining CI’s. A waterfall plot was used to display the degree of response (percent change in tumor burden).
Exploratory studies methods
Whole exome sequencing
The DNA from FFPE tumor tissue and from normal lymphocytes were extracted with QIAamp® FFPE Tissue Kit and QIAamp® DNA Blood Mini Kit respectively according to the supplier’s instructions. All double strand DNAs were quantified using a QuBit® fluorometer. In order to evaluate the clinical samples from the patients with differential responses to vorinostat WES of 5 available tumor normal pairs with sufficient tumor DNA (patients 001–002, 001–004, 001–006, 003–014, 003–027) from the FFPE tissue samples was done using Agilent_V5_PlusUTR_hs37d5_Baits. The sequencing was done through the TGen Collaborative Sequencing Center (for patients 001–004 and 001–006) and the Mayo Clinic Medical Genome Facility (MGF) at a mean depth of 60x coverage according to established protocols [52]. Observed variants were filtered based on a series of criteria including NextProt Feature Strength, Maximum Population Allele Frequency, and inclusion in the Catalogue of Somatic Mutations in Cancer (COSMIC) database [7]. The goal was to prioritize those variants that likely disrupt protein function, identify mutations targeting known cancer related genes and pathways, and filter out polymorphisms detected in patient matched normal samples and reported in dbSNP and other population based studies. Of significant interest were those somatic variants that introduced non-synonymous variants within well-annotated cancer associated genes.
Targeted resequencing
In addition to WES, we developed a 10 gene panel to validate mutations and to screen samples with sub optimal levels of DNA for this study. We selected 4 genes from our initial WES results for patients 001–004 and 001–006, and 6 additional genes from previously published ACC data for our 10 gene panel [5, 6]. Candidate driver aberrations targeting chromatin remodeling (SMARCA2, KDM6A, CREBBP), DNA repair and checkpoints (TP53, MLF1) and cell signaling (PIK3CA, SH2D4A, FZD10) were prioritized based on their biological functions and potential clinical utility. The final 10-gene ACC panel consisted of SH2D4A, FZD10, TP53, PIK3CA, PTEN, SMARCA2, KDM6A, CREBBP, MLF1, and N4BP2. Primers were designed for each of the 10 genes with coverage for all exons and untranslated sequencings (UTRs) using the Ion AmpliseqTM Designer https://www.ampliseq.com/browse.action. Our targeted resequencing 10 gene FFPE specific input panel has 87.65% coverage with 644 amplicons ranging from 125–175 base pairs in length, for a total of 62.09 kb of sequence. This design provides validation of the 4 novel mutations identified in the initial whole exome data and increases the probability of identifying additional mutations that converge on each of the 10 genes in the panel. For exploratory purposes, patients were qualified as either “responders” (7/11 patients) or “non-responders” (4/11 patients) based on whether they had a PR by RECIST (n = 2), prolonged SD, clinical benefit to vorinostat or progressed quickly on study. To investigate the role of somatic mutations in responses to vorinostat we assembled archival FFPE blocks from 2 patients and FFPE slides from an additional 9 patients treated with vorinostat. We obtained patient matched normal blood samples as controls. In total we screened both tumor and normal tissues from each of 8 patients (from 6 “responders” and 2 “non-responders”), including the 5 cases that were analyzed by WES with this panel using the Ion Torrent platform. In the remaining 3 cases there was insufficient material for either WES or target resequencing. A full description of the methods can be found in the Supplementary File.
Authors’ contributions
Conception and design of work: PG, LH, MB, RP, PL. Data Collection: PG, SK, AH, LS, AS, JC, MP, LC, AC, PL. Data analysis and interpretation: PG, LH, MB, DS, EL, SM, PL. Drafting the article: PG, LH, MB, PL, DS. Critical revision of the article: All authors. Final approval of the version to be published: All authors.
CONFLICTS OF INTEREST
All authors except LS and JC have no conflicts of interest. LS has research funding (for clinical trials) from Merck and is on Merck advisory board. JC has research funding (institutional for clinical trials) from Merck.
FUNDING
Supported by 5U01CA062487/19 and U01 grant (5U01CA132123). Partially funded by the Adenoid Cystic Carcinoma Research Foundation (ACCRF) and by NIH Cancer Center Support Grant CA-22453.
REFERENCES
1. Ellington CL, Goodman M, Kono SA, Grist W, Wadsworth T, Chen AY, Owonikoko T, Ramalingam S, Shin DM, Khuri FR, Beitler JJ, Saba NF. Adenoid cystic carcinoma of the head and neck: Incidence and survival trends based on 1973–2007 Surveillance, Epidemiology, and End Results data. Cancer. 2012; 118:4444–4451.
2. Spiro RH. Distant metastasis in adenoid cystic carcinoma of salivary origin. American journal of surgery. 1997; 174:495–498.
3. Chae YK, Chung SY, Davis AA, Carneiro BA, Chandra S, Kaplan J, Kalyan A, Giles FJ. Adenoid cystic carcinoma: current therapy and potential therapeutic advances based on genomic profiling. Oncotarget. 2015; 6:37117–37134. doi: 10.18632/oncotarget.5076.
4. Ramalingam SS, Kummar S, Sarantopoulos J, Shibata S, LoRusso P, Yerk M, Holleran J, Lin Y, Beumer JH, Harvey RD, Ivy SP, Belani CP, Egorin MJ. Phase I study of vorinostat in patients with advanced solid tumors and hepatic dysfunction: a National Cancer Institute Organ Dysfunction Working Group study. J Clin Oncol. 2010; 28:4507–4512.
5. Stephens PJ, Davies HR, Mitani Y, Van Loo P, Shlien A, Tarpey PS, Papaemmanuil E, Cheverton A, Bignell GR, Butler AP, Gamble J, Gamble S, Hardy C, et al. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest. 2013; 123:2965–2968.
6. Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, Ramaswami D, Walsh LA, Eng S, Huse JT, Zhang J, Dolgalev I, Huberman K, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013; 45:791–798.
7. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015; 43:D805–811.
8. Yoneda-Kato N, Tomoda K, Umehara M, Arata Y, Kato JY. Myeloid leukemia factor 1 regulates p53 by suppressing COP1 via COP9 signalosome subunit 3. EMBO J. 2005; 24:1739–1749.
9. Watanabe N, Wachi S, Fujita T. Identification and characterization of BCL-3-binding protein: implications for transcription and DNA repair or recombination. J Biol Chem. 2003; 278:26102–26110.
10. Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, Chan TL, Kan Z, Chan AS, Tsui WY, Lee SP, Ho SL, Chan AK, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011; 43:1219–1223.
11. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–337.
12. Li T, Li W, Lu J, Liu H, Li Y, Zhao Y. SH2D4A regulates cell proliferation via the ERalpha/PLC-gamma/PKC pathway. BMB Rep. 2009; 42:516–522.
13. Koike J, Takagi A, Miwa T, Hirai M, Terada M, Katoh M. Molecular cloning of Frizzled-10, a novel member of the Frizzled gene family. Biochem Biophys Res Commun. 1999; 262:39–43.
14. Cao R, Wang H, He J, Erdjument-Bromage H, Tempst P, Zhang Y. Role of hPHF1 in H3K27 methylation and Hox gene silencing. Mol Cell Biol. 2008; 28:1862–1872.
15. Yang Y, Wang C, Zhang P, Gao K, Wang D, Yu H, Zhang T, Jiang S, Hexige S, Hong Z, Yasui A, Liu JO, Huang H, et al. Polycomb group protein PHF1 regulates p53-dependent cell growth arrest and apoptosis. J Biol Chem. 2013; 288:529–539.
16. Hong Z, Jiang J, Lan L, Nakajima S, Kanno S, Koseki H, Yasui A. A polycomb group protein, PHF1, is involved in the response to DNA double-strand breaks in human cell. Nucleic Acids Res. 2008; 36:2939–2947.
17. Micci F, Panagopoulos I, Bjerkehagen B, Heim S. Consistent rearrangement of chromosomal band 6p21 with generation of fusion genes JAZF1/PHF1 and EPC1/PHF1 in endometrial stromal sarcoma. Cancer Res. 2006; 66:107–112.
18. Panagopoulos I, Micci F, Thorsen J, Gorunova L, Eibak AM, Bjerkehagen B, Davidson B, Heim S. Novel fusion of MYST/Esa1-associated factor 6 and PHF1 in endometrial stromal sarcoma. PLoS One. 2012; 7:e39354.
19. Chen S, Evans HG, Evans DR. FAM129B/MINERVA, a novel adherens junction-associated protein, suppresses apoptosis in HeLa cells. J Biol Chem. 2011; 286:10201–10209.
20. Drier Y, Cotton MJ, Williamson KE, Gillespie SM, Ryan RJ, Kluk MJ, Carey CD, Rodig SJ, Sholl LM, Afrogheh AH, Faquin WC, Queimado L, Qi J, et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet. 2016; 48:265–272.
21. Malecki MJ, Sanchez-Irizarry C, Mitchell JL, Histen G, Xu ML, Aster JC, Blacklow SC. Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol Cell Biol. 2006; 26:4642–4651.
22. Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, Leigh IM, Collisson EA, Gordon PB, Jakkula L, Pennypacker S, Zou Y, Sharma M, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proced Natl Acad Sci USA. 2011; 108:17761–17766.
23. van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, Edkins S, Hardy C, O'Meara S, Teague J, Butler A, Hinton J, Latimer C, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009; 41:521–523.
24. Sanghvi S, Patel NR, Patel CR, Kalyoussef E, Baredes S, Eloy JA. Sinonasal adenoid cystic carcinoma: comprehensive analysis of incidence and survival from 1973 to 2009. Laryngoscope. 2013; 123:1592–1597.
25. Hotte SJ, Winquist EW, Lamont E, MacKenzie M, Vokes E, Chen EX, Brown S, Pond GR, Murgo A, Siu LL. Imatinib mesylate in patients with adenoid cystic cancers of the salivary glands expressing c-kit: a Princess Margaret Hospital phase II consortium study. J Clin Oncol. 2005; 23:585–590.
26. Agulnik M, Cohen EW, Cohen RB, Chen EX, Vokes EE, Hotte SJ, Winquist E, Laurie S, Hayes DN, Dancey JE, Brown S, Pond GR, Lorimer I, et al. Phase II study of lapatinib in recurrent or metastatic epidermal growth factor receptor and/or erbB2 expressing adenoid cystic carcinoma and non adenoid cystic carcinoma malignant tumors of the salivary glands. J Clin Oncol. 2007; 25:3978–3984.
27. Thomson DJ, Silva P, Denton K, Bonington S, Mak SK, Swindell R, Homer J, Sykes AJ, Lee LW, Yap BK, Slevin NJ. Phase II trial of sorafenib in advanced salivary adenoid cystic carcinoma of the head and neck. Head Neck. 2015; 37:182–187.
28. Wong SJ, Karrison T, Hayes DN, Kies MS, Cullen KJ, Tanvetyanon T, Argiris A, Takebe N, Lim D, Saba NF, Worden FP, Gilbert J, Lenz HJ, et al. Phase II trial of dasatinib for recurrent or metastatic c-KIT expressing adenoid cystic carcinoma and for nonadenoid cystic malignant salivary tumors. Ann Oncol. 2016; 27:318–323.
29. Dillon PM, Petroni GR, Horton BJ, Moskaluk CA, Fracasso PM, Douvas MG, Varhegyi N, Zaja-Milatovic S, Thomas CY. A Phase II study of Dovitinib in Patients With Recurrent or Metastatic Adenoid Cystic Carcinoma. Clin Cancer Res April 4. [Epub ahead of print].
30. Keam B, Kim SB, Shin SH, Cho BC, Lee KW, Kim MK, Yun HJ, Lee SH, Yoon DH, Bang YJ. Phase 2 study of dovitinib in patients with metastatic or unresectable adenoid cystic carcinoma. Cancer. 2015; 121:2612–2617.
31. Ho AL, Dunn L, Sherman EJ, Fury MG, Baxi SS, Chandramohan R, Dogan S, Morris LG, Cullen GD, Haque S, Sima CS, Ni A, Antonescu CR, et al. A phase II study of axitinib (AG-013736) in patients with incurable adenoid cystic carcinoma. Ann Oncol. 2016; 27:1902–1908.
32. Laurie SA, Ho AL, Fury MG, Sherman E, Pfister DG. Systemic therapy in the management of metastatic or locally recurrent adenoid cystic carcinoma of the salivary glands: a systematic review. Lancet Oncol. 2011; 12:815–824.
33. Locati LD, Perrone F, Cortelazzi B, Bergamini C, Bossi P, Civelli E, Morosi C, Lo Vullo S, Imbimbo M, Quattrone P, Dagrada GP, Granata R, Resteghini C, et al. A phase II study of sorafenib in recurrent and/or metastatic salivary gland carcinomas: Translational analyses and clinical impact. Eur J Cancer. 2016; 69:158–165.
34. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009; 45:228–247.
35. Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR, Chen LL, Podoloff DA, Benjamin RS. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007; 25:1753–1759.
36. Dudeck O, Zeile M, Reichardt P, Pink D. Comparison of RECIST and Choi criteria for computed tomographic response evaluation in patients with advanced gastrointestinal stromal tumor treated with sunitinib. Ann Oncol. 2011; 22:1828–1833.
37. Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbe C, Maio M, Binder M, Bohnsack O, Nichol G, Humphrey R, Hodi FS. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clini Cancer Res. 2009; 15:7412–7420.
38. Nishino M, Jagannathan JP, Krajewski KM, O'Regan K, Hatabu H, Shapiro G, Ramaiya NH. Personalized tumor response assessment in the era of molecular medicine: cancer-specific and therapy-specific response criteria to complement pitfalls of RECIST. Am J Roentgenol. 2012; 198:737–745.
39. Nishino M, Jackman DM, Hatabu H, Janne PA, Johnson BE, Van den Abbeele AD. Imaging of lung cancer in the era of molecular medicine. Acad Radiol. 2011; 18:424–436.
40. Nishino M, Dahlberg SE, Cardarella S, Jackman DM, Rabin MS, Ramaiya NH, Hatabu H, Janne PA, Johnson BE. Volumetric tumor growth in advanced non-small cell lung cancer patients with EGFR mutations during EGFR-tyrosine kinase inhibitor therapy: developing criteria to continue therapy beyond RECIST progression. Cancer. 2013; 119:3761–3768.
41. Mozley PD, Schwartz LH, Bendtsen C, Zhao B, Petrick N, Buckler AJ. Change in lung tumor volume as a biomarker of treatment response: a critical review of the evidence. Ann Oncol. 2010; 21:1751–1755.
42. Lipscomb J, Gotay CC, Snyder CF. Patient-reported outcomes in cancer: a review of recent research and policy initiatives. CA Cancer J Clin. 2007; 57:278–300.
43. Brogdon JL, Xu Y, Szabo SJ, An S, Buxton F, Cohen D, Huang Q. Histone deacetylase activities are required for innate immune cell control of Th1 but not Th2 effector cell function. Blood. 2007; 109:1123–1130.
44. West AC, Christiansen AJ, Smyth MJ, Johnstone RW. The combination of histone deacetylase inhibitors with immune-stimulating antibodies has potent anti-cancer effects. Oncoimmunology. 2012; 1:377–379.
45. Reddy P, Maeda Y, Hotary K, Liu C, Reznikov LL, Dinarello CA, Ferrara JL. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proc Natl Acad Sci USA. 2004; 101:3921–3926.
46. Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, Keiser N, Santaniello F, Tomasi TB. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000; 165:7017–7024.
47. Persson M, Andren Y, Mark J, Horlings HM, Persson F, Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci USA. 2009; 106:18740–18744.
48. Queimado L, Lopes CS, Reis AM. WIF1, an inhibitor of the Wnt pathway, is rearranged in salivary gland tumors. Genes Chromosomes Cancer. 2007; 46:215–225.
49. Shao C, Sun W, Tan M, Glazer CA, Bhan S, Zhong X, Fakhry C, Sharma R, Westra WH, Hoque MO, Moskaluk CA, Sidransky D, Califano JA, et al. Integrated, genome-wide screening for hypomethylated oncogenes in salivary gland adenoid cystic carcinoma. Clin Cancer Res. 2011; 17:4320–4330.
50. Bell A, Bell D, Weber RS, El-Naggar AK. CpG island methylation profiling in human salivary gland adenoid cystic carcinoma. Cancer. 2011; 117:2898–2909.
51. Ferrarotto R, Mitani Y, Diao L, Guijarro I, Wang J, Zweidler-McKay P, Bell D, William WN Jr, Glisson BS, Wick MJ, Kapoun AM, Patnaik A, Eckhardt G, et al. Activating NOTCH1 Mutations Define a Distinct Subgroup of Patients With Adenoid Cystic Carcinoma Who Have Poor Prognosis, Propensity to Bone and Liver Metastasis, and Potential Responsiveness to Notch1 Inhibitors. J Clin Oncol. 2016; 35:352–360.
52. Borad MJ, Champion MD, Egan JB, Liang WS, Fonseca R, Bryce AH, McCullough AE, Barrett MT, Hunt K, Patel MD, Young SW, Collins JM, Silva AC, et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet. 2014; 10:e1004135.