
INTRODUCTION
Acute lymphoblastic leukemia (ALL) is one of the most common pediatric cancers [1], and leukemogenesis has been considered to be impacted by both environmental and genetic factors [2]. Through a series of independent genome-wide association studies (GWAS) in ethnic diverse populations, several risk loci for ALL susceptibility have been identified (e.g., ARID5B, IKZF1, CEBPE, PIP4K2A, CDKN2A, GATA3) [3–10], and validated by subsequent replication studies [11–14]. However, most of these GWAS signals are located in non-coding region of the related genes, except CDKN2A [9]. Nevertheless, some ALL-related single nucleotide polymorphisms (SNPs) are noted to be located in the regulatory region, and impact on gene expression (e.g., SNPs of PIP4K2A, and GATA3 loci [5, 8, 15]), indicating their possible epigenetic regulation. Notably, ALL-related GATA3 SNPs (e.g., rs3824662, located in intron3) locate in its enhancer region, with higher GATA3 expressed in risk allele carriers of EBV virus transformed lymphoblastoid cell lines (LCL), which suggests their causal mechanisms in leukemogenesis [8]. Moreover, GATA3 SNPs are associated with ALL susceptibility with varied odds radio (OR) in terms of different clinical characteristics, and mostly impacted by subtypes (i.e., Ph-like B cell lineage ALL) [8, 10], indicating the specific role of GATA3 in different cell type.
As a well-known transcription factor, GATA3 can bind to specific motif (e.g., consensus DNA sequence WGATAR, W = A/T and R =A/G), and is capable to function in determination of cell identity of hematopoietic system, mammary gland, and etc [16, 17], especially emerging as a critical regulator of both innate and adaptive immunity. GATA3 expression is associated with cell-type specification, and plays an important role on the development and functions of multiple immune cell types, including T cells and B cells [17, 18]. Actually, function of GATA3 has been firstly characterized in T cell, and is essential for Th1-Th2 commitment with higher expression level in Th2 cells [19], as a transcriptional regulator through direct action at many critical factors (e.g., cytokines, signaling molecules) [18]. Also, GATA3 plays an important role on T cells maintenance, and is required for distinct aspects of T cell activation and proliferation in cell type-specific manner [17]. Through large efforts with experimental analyses, multiple upstream regulators and downstream targets of GATA3 have been characterized in T cells [20]. For instance, interleukin 4 can promote GATA3 expression through STAT6 signal [17, 19]. Also, GATA3 is involved in multiple pathways independent of IL4-STAT6 signaling, including Notch and Wnt pathways [21–23], which are essential for T cell development.
Moreover, Knocking-out of Gata3 in mouse results in embryonic lethal between E11 and E12, displaying massive internal bleeding, gross aberrations in fetal liver hematopoiesis, and etc [24]. Importantly, aberrant GATA3 expression or mutations can impact on its downstream genes, thus induce dysfunctions including tumorigenesis, such as breast cancer [25, 26]. For instance, loss of Gata3 in adult mice leads to an expansion of undifferentiated luminal cells and basement-membrane detachment, which may promote tumor dissemination [27], while rescue of Gata3 expression reduces both tumorigenicity and metastatic potential of breast cancer cells [28, 29]. In human cancers, frequent loss-of-function of GATA3 alteration and copy number deletions were observed in breast cancer and T cell leukemia/lymphoma recently [25, 30].
Recent studies indicate that GATA3 can actively suppress B cells development [17, 31, 32], and deficiency of this gene results in development failure of T cells but not B cells in conditional hematopoietic knockout mouse model [33, 34], raising the possibility that GATA3 was involved in cell-type specific regulatory network. However, despite of studies on association of GATA3 SNPs with B-ALL susceptibility, function of GATA3 in leukemogenesis for B lineage cells was poorly understood. It will be time and effect consuming to figure out the GATA3-involved regulatory network in B lineage ALL (B-ALL) with the traditional methods, especially for those unreported genes. Fortunately, array based characterization of transcriptional profiles have been conducted in multiple independent B-ALL patient cohorts. With the public resource, we conducted transcriptional wide screening in this study to effectively find the genes those are significantly related to GATA3 expression, and built the regulatory network. Subsequent validations were also carried out for some of the candidates in ALL cell lines to evaluate the reliability of this procedure.
RESULTS
The top GWAS SNP for ALL susceptibility is located in the enhancer region of GATA3 in a tissue-type specific manner
The function of GATA3 has been largely revealed as a transcription factor and highly expressed in multiple tissues including breast, bladder, blood, skins (Supplementary Figure 1). Significant expression changes between tumor and control normal tissues were also observed in multiple types of cancers according to the dataset of The Cancer Genome Atlas (TCGA) (Supplementary Figure 2). However, opposite directions were also noticed with higher expression level in tumors (e.g., bladder cancer, cervical squamous cell carcinoma) or in normal tissues (e.g., kidney cancer) (Supplementary Figure 2), indicating the important and heterogeneity role of GATA3 in tumorigenesis for different types of cancer. Therefore, it is important to find the regulatory network of GATA3 in each type of cancer separately, including B-ALL.
Because the top SNP (i.e., rs3824662) for ALL susceptibility in GATA3 is located in its intron region, epigenetic signals were thus analyzed with the public resource (e.g., ENCODE and ROADMAP database). Interestingly, a strong enhancer close to rs3824662 was observed in a tissue-type specific manner, and blood and breast exhibit strong signals (Figure 1A), which is consistent with their higher expression level among different tissue types (Supplementary Figure 1). Additionally, differences were also observed among hematopoietic cell types. For instance, CD34 positive cells have relatively weaker DNAase hypersensitivity signal around rs3824662 compared to other type of hematopoietic cells, indicating the varied role of rs3824662 on GATA3 regulation in different development stage of hematopoietic cells (Figure 1B). Additionally, risk allele of rs3824662 is significantly related to higher expression level of GATA3 in LCLs from diverse ethnicities, (P = 0.009 after adjust for ethnicity) (Figure 1C), indicating overexpression of GATA3 may increase the risk of leukemogenesis through SNP-induced epigenetic regulation.

Figure 1: Epigenetic regulation of GATA3. (A) Epigenetic elements around the top GWAS GATA3 SNP (i.e., rs3824662) in different tissue types. Different epigenetic elements were labeled as different colors as annotation indicated, and the tissue type information was listed on the right with “Blood” and “Breast” highlighted (B) DNase hypersensitivity signals around rs3824662 in different types of blood cells. Strength of the binding for each transcriptional factor was illustrated according to the (C) Genotype-expression association between rs3824662 and GATA3 expression in LCLs, P = 0.009.
Multiple genes are significantly associated with GATA3 expression in B-ALL
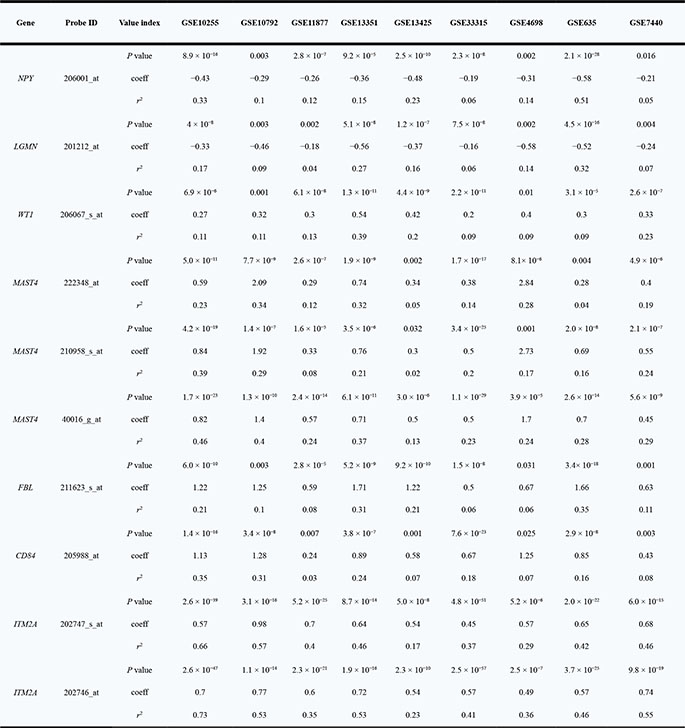
Expression array data from nine independent ALL patient cohorts were downloaded from the public resource (Table 1). Association of GATA3 expression with all the rest genes were estimated by using linear regression model. To find the potential expression related genes and build the co-expression network of GATA3 in B-ALL, a series of filter steps were applied for candidate selection, including strict P value cutoff, r2, and consistent direction for association coefficient (Figure 2). Interestingly, only 5 out of 142 genes (or 5 out of 178 array probes) were filtered out because of the inconsistency direction among cohorts, indirectly proving the high reliability of the selected candidates. Totally 83 and 54 genes were positively and negatively related to GATA3 expression, respectively (Supplementary Table 1). Due to the large sample size and availability of clinical information, data from GSE33315 was used for further analyses (with 173 probes for 137 genes have available expression information). Expression level of GATA3 in B-ALL is significantly higher than that in CD19 positive cells, and similar as that in CD34 positive cells from healthy people (Supplementary Figure 3). The highest GATA3 expression was observed in B-others subtype, possibly because Ph-like ALL was included in such subtype. Interestingly, these GATA3-related genes are tend to be clustered in ALL subtypes in heatmap, indicating their different roles on leukemia subtypes (Figure 3A). Among these candidates, some genes have already been reported as upstream regulators (e.g., SATB1 [21]) or downstream targets (e.g., ITM2A [20, 35]) in T cells (Supplementary Table 1), exhibiting the ubiquitous GATA3-related network in different cell types as well as the reliability of our screening procedure. The candidates was also listed, which are significantly related to GATA3 expression in all patient cohorts with P ≤ 2 × 10−6 and r2 ≥ 0.1 in at least 5 cohorts (Table 2). Interestingly, STAT4, which is involved in JAK/STAT pathway, has been found as one of the strongest candidates. Considering that GATA3 SNP is more related to Ph-like ALL, which is enriched in JAK pathway alteration, GATA3 may be involved in B-ALL leukemogenesis through inducing STAT4 overexpression and activating the JAK/STAT pathway. Additional, we also found another novel target (i.e., ETV6), alteration of which is frequently observed in leukemia in germline [36] or somatic level. Next, we conducted pathway analyses by using online tools (e.g., DAVID Functional Annotation Tools), and found that two gene sets were significantly enriched in GATA3-related genes (i.e., “Cyclin” and “RNA polymerase II regulatory region sequence specific DNA binding “ Supplementary Table 2), suggesting GATA3 may impact cell cycle and involved in complicated transcriptional regulation to induce leukemogenesis. Additionally, protein-protein interaction network of these candidates was also illustrated with STRING, IntAct and BioGRID to indicate the known interactions (Figure 3B), those genes that were not illustrated may be considered as novel members in GATA3 regulatory network specific in B-ALL.
Table 1: Summary information for the B-ALL microarray datasets
Year |
Author[*] |
Dataset ID |
Age group |
Analyses |
|---|---|---|---|---|
2008 |
Bhojwani D et al. [44] |
GSE7440 |
pediatric |
|
2010 |
Kang H et al. [45] |
GSE11877 |
pediatric |
|
2009 |
Bungaro S et al. [46] |
GSE10792 |
pediatric |
|
2009 |
den Boer ML et al. [47] |
GSE13351 |
pediatric |
Discovery |
2009 |
den Boer ML et al. [47] |
GSE13425 |
pediatric |
|
2004 |
Holleman A et al. [48] |
GSE635 |
pediatric |
|
2008 |
Sorich MJ et al. [49] |
GSE10255 |
pediatric |
|
2006 |
Kirschner-Schwabe R et al. [50] |
GSE4698 |
pediatric |
|
2012 |
Zhang J et al. [51] |
GSE33315 |
pediatric |
|
2009 |
Haferlach T et al. [52] |
GSE13204 |
all ages |
Validation |
*number in the brackets represent the references in the manuscript.
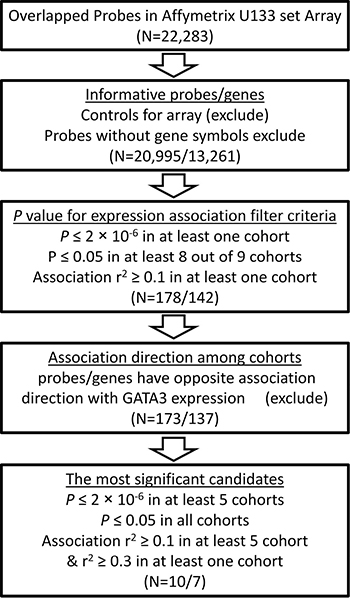
Figure 2: Flow chart for GATA3-related genes screening pipeline.
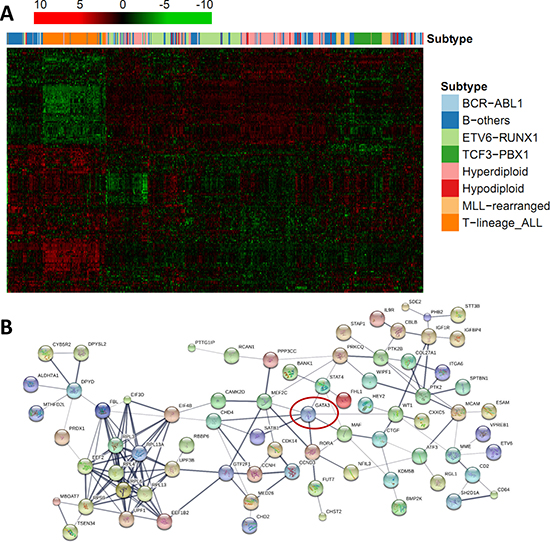
Figure 3: Regulatory statement of GATA3 and its related candidates in B-ALL. (A) Expression clustering illustration of GATA3-related 137 candidates, with B-ALL subtypes was labeled above with different colors indicated. (B) Protein-protein interaction network of GATA3-related genes. Line thickness indicates the strength of data support, and nodes that disconnected with the main network were hide.
Table 2: Strongest GATA3-related candidates in different datasets
GATA3-related genes exhibit tissue and subtype specific association
Since the clusters of the GATA3-related gene closely match B-ALL subtypes (described above), the role of GATA3 in different subtypes of B-ALL was checked in the largest pediatric B-ALL cohort (GSE33315) by analyzing each subtype separately (Supplementary Table 3). Most of candidates were only significant association with GATA3 expression in some of the subtypes, partially because of the small sample size in some subtypes such as BCR-ABL and MLL rearrangement subtypes. To exclude the impact of sample size, we next analyzed the subtypes with at least 90 patients (i.e., ETV6-RUNX1, Hyperdiploid, and B-other subtype), only 36 out of 136 genes are significantly associated with GATA3 expression in all three subtypes. All of them have the same direction except PHB2 (Figure 4), which is positively related to GATA3 expression in ETV6-RUNX1 and B-other subtypes but negatively related to that in hyperdiploid subtype (Figure 4). For the seven strongest candidates described above, ITM2A, and MAST4 exhibit statistically significant in three subtypes with varied coefficient value, and the rest 5 genes only exhibit significance in one or two subtypes (Figure 4, and Supplementary Table 3), suggesting the different regulatory network of GATA3 in each subtypes.
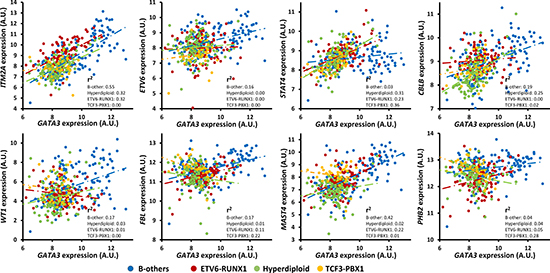
Figure 4: Expression association status of GATA3 with some of the important candidates in different subtypes of B-ALL (i.e., B-others, ETV6-RUNX1, Hyperdiploid, and TCF3-PBX1) in the largest available pediatric B-ALL cohort (i.e., GSE33315) with the P values listed in Table 1 and Supplementary Table 3.
In another hand, we also checked the association status in a dataset containing ALL, acute myeloid leukemia (AML), chronic lymphoblastic leukemia (CLL), and chronic myeloid leukemia (CML) patients in all stage of ages at diagnosis (i.e., GSE13204). Not surprisingly, most of the candidate genes (98.5%, 135/137) reached statistical significance in B-ALL, and all of them have the same direction with the previous results. However, the consistent rate dropped to 36.5% (50/137), 75.9% (104/137), 64.9% (89/137), and 40.1% (55/137) in T-ALL (N = 174), CLL (N = 448), AML (N = 542), and CML (N = 76), respectively. Among the rest filtered genes, we noticed that 20% (10/50 in T-ALL), 25.9% (27/104 in CLL), 43.8% (39/89 in AML), and 16.3% (9/55, in CML) were even in the opposite association direction with GATA3 to that in ALL (Supplementary Table 4). We next evaluated the candidate genes in breast cancer, on which GATA3 also plays an important role according to the reports. Among the available gene expression information (157 genes in 1,992 patients), only 19% genes (30/157) exhibit P < 0.05 and r2 > 0.1. In addition, 50% (15/30) of the rested candidates have the opposite association direction with GATA3 to that in ALL (Figure 5 and Supplementary Table 5). Taking STAT4 as an example, which is positively related to GATA3 expression in healthy bone marrow (P = 5.7 × 10−21, and r2 = 0.7), the association got weak gradually in CLL (P = 4.7 × 10−78, and r2 = 0.54), CML (P = 1.2 × 10−10, and r2 = 0.42), AML (P = 1.2 × 10−23, and r2 = 0.17), B-ALL (P = 1.7 × 10−23, and r2 = 0.16), and T-ALL (P = 0.16, and r2 = 0.005) (Supplementary Table 4), and even negatively related to GATA3 expression in breast cancer (P = 1.2 × 10−66, and r2 = 0.14) (Supplementary Table 5). For ETV6, expression of this gene is positively associated with GATA3 in B-ALL only, and with the opposite direction in all other types of leukemia, breast cancer as well as the healthy bone marrow, indicating its specific role on B-ALL leukemogenesis with GATA3 regulation. In conclusion, there are large differences in GATA3-related genes and corresponding regulatory network in varied tissues and subtypes.
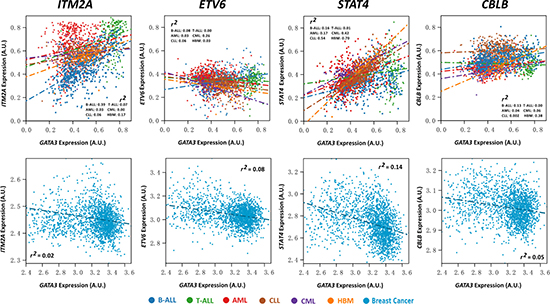
Figure 5: Expression association status of GATA3 with ITM2A, ETV6, STAT4, and CBLB in different types of leukemia, including B-ALL (N = 576), T-ALL (N = 174), AML (N = 542), CLL (N = 448), CML (N = 76), and healthy bone marrow (HBM, N = 74) based on GSE13204, and breast cancer based on EGAS00000000083 (N = 1,992).
Multiple leukemia or cancer related genes are associated with GATA3 expression in cell lines
Although we have found the candidates that are significantly associated with GATA3 expression, and build regulatory network based on the known resources, it is also important to figure out the detail relationship between GATA3 and these genes. We assumed these candidates can be upstream regulators or downstream targets of GATA3 through direct or indirect interactions. Therefore, we retrieved the expression data of the candidates from Nalm6 cells with GATA3 overexpression or empty vector control. Available expression information were got for 43 genes, which had present expression in control or/and GATA3 overexpression cells. Not surprisingly, 27 out of 43 candidates were significantly changed after GATA3 overexpression (e.g., ETV6 and WT1), with the same association direction as described above (Figure 6A and Supplementary Table 6). For those were not significant changed genes, we considered them as potential upstream of GATA3, such as SATB1, which has been reported as regulator of GATA3 in T cell. Additionally, we also picked some of the strong candidates (e.g., ITM2A) for analyses with shRNA system in other leukemia cell lines for validation. Cells with GATA3 knocking down exhibited consistent changes as well (Figure 6B), indicating the reliability of our analyses.
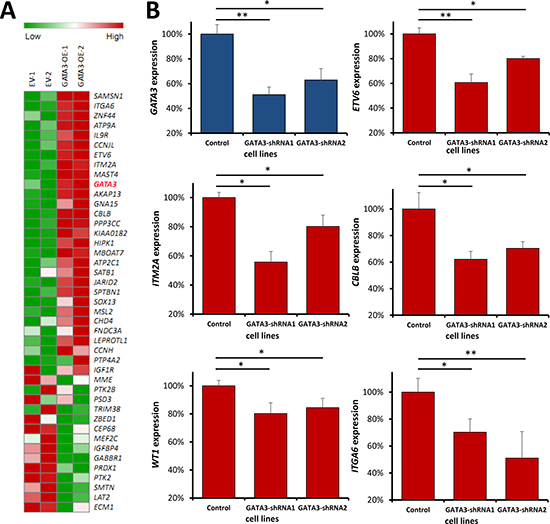
Figure 6: Impact of GATA3 expression changes on the candidates in leukemia cells. Expression changes of GATA3-related candidates in GATA3 overexpression and GATA3 down-regulated cells. (A) Heatmap for the candidate gene expression in GATA3 overexpression cells. The most significant genes were listed on the top (positive related) and the bottom (negative related) (B) The expression changes of ETV6, ITM2A, WT1, ITGA6 and CBLB were detected in GATA3 down-regulated cells. (* and ** indicate P < 0.05 and P < 0.01, respectively).
Loss of GATA3 binding motif induced by SNP can impact association of CBLB with GATA3 expression
We next checked whether the expression of the candidates can be impacted by SNPs, which alter the GATA3 binding affinity through breaking the conserved “GATA” motif. Interestingly, CBLB, which is the potential downstream target of GATA3 in leukemia as well as LCLs according to our results, contains one SNP (i.e., rs4894953) in its enhancer region. rs4894953 and its flanking nucleotide acids form a sequence of “GA(T/C)A”, in which GATA3 is more likely to bind to this motif in individuals with T allele at this SNP. Therefore, we conducted genotype-specific expression association analyses in LCLs, which comprehensive information for both SNP genotypes and gene expression were available. We separated the individuals in terms of genotypes of rs4894953, and checked the association of GATA3 expression with CBLB. Interestingly, although significant association of these two genes can be detected in both C/C (P = 0.0008) and T/T (P = 0.003) genotype groups, large difference was observed in terms of r2, (i.e., r2 = 0.06 and 0.39 in C/C and T/T genotype groups, respectively) (Figure 7A). Additionally, we also checked the available epigenetic signal in LCLs, and noticed that the DNase I hypersensitivity signal is stronger in GM19238 (T/T at rs4894953) than that in GM12878 (C/C at rs4894953) around the SNP (Figure 7B). These results indicated that the expression of GATA3-related candidates can be strongly impacted by SNPs those locate in “GATA” motif, and further suggested the reliability of the candidates we screened out.
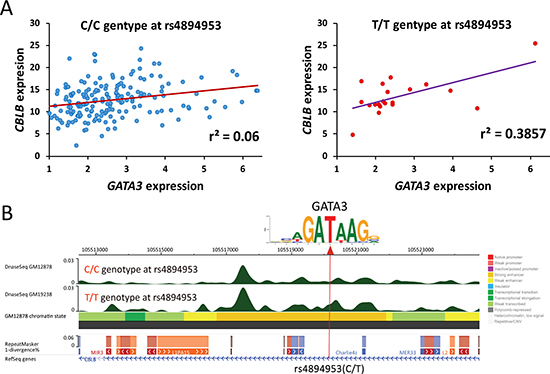
Figure 7: Impact of SNP genotypes on expression association between GATA3 and its downstream target. (A) Expression association status between GATA3 and CBLB in terms of C/C and T/T genotypes at rs4894953. (B) Impact of different allele on GATA3 binding motif and the epigenomic signals in LCL with C/C and T/T genotypes at rs4894953.
DISCUSSION
Due to the varied roles of GATA3 on different tissue types, it is important but also time/effect-consuming to find the regulatory network of GATA3 in each type of cancer separately. We assumed that the genes involved in the same regulatory network will be related in terms of expression level among patients, and the transcription factor and its direct target will exhibit the most significant association. Therefore, it will be easy and effective to screen the GATA3-related genes through whole transcriptome-wide association by using the public available microarray datasets. Interestingly, multiple GWASs revealed strong association of GATA3 SNP with ALL susceptibility, especially in Ph-like subtypes, and the risk allele of the top GWAS SNP is related to higher expression of GATA3. Therefore, the mechanism of how GATA3 involved in B lineage leukemogenesis can be studied on its upstream and downstream signals in leukemia cells from B-ALL patients. Finally, we found 137 genes that are potential involved in GATA3-related regulatory network with nine independent pediatric ALL patient cohorts, and got validated in another leukemia cohort containing all ages of B-ALL patients. Interestingly, all of strongest candidates are significantly associated with GATA3 expression in B others, which could reflect the risk allele enrichment of GATA3 SNP in Ph-like ALL subtype. However, due to the different association status of GATA3 with its related gene in different subtypes of B-ALL, analyses within a certain subtype should be done if the information can be got from more larger size of patient cohorts. Notably, some of the candidates have been reported to be upstream regulator (e.g., SATB1 [21]) or downstream targets (e.g., ITM2A [20, 35]) of GATA3 in other types of cells, indicating the reliability of our methods. Besides, our results provided a clue for further studies on how GATA3 acted in leukemogenesis.
Interestingly, some of the well-known cancer related genes were found, such as STAT4. Actually, STAT4 is involved in JAK/STAT pathway, and constitutive activation of JAK-STAT was recognized as being associated with malignancy, including leukemia [37]. Moreover, GATA3 has stronger effects in Ph-like ALL, in which gain-of-function of JAK mutations is enriched, raising the possibility that GATA3 increases the leukemogenesis risk through activating JAK-STAT signaling. Consistently, STAT4 expression decreases in leukemia cells treated with shRNA against GATA3, which was also confirmed in previous reports [38]. Paradoxically, STAT4 is negatively related to GATA3 expression in breast cancer, probably because of the tissue specific role of GATA3. Actually, we noticed that lots of GATA3-related genes in B-ALL lost their association with GATA3 expression in breast cancer, or even exhibit the opposite association directions (e.g., STAT4), providing the possible explanation of the opposite role of GATA3 in different cancer types (e.g., potential tumor suppressor in breast cancer but oncogene in leukemia). Not surprisingly, similarity of association status and regulatory network may increase in more related cell types. For instance, number of GATA3-related genes, which exhibit same significant association direction with that in pediatric B-ALL, is the largest in all ages of B-ALL, and gradually decreases in B lineage chronic lymphocytic leukemia, myeloid leukemia, and breast cancer.
As described above, the candidates can be upstream regulator or downstream targets of GATA3 with direct or indirect binding. In T cells, ITM2A is a direct target of GATA3 [35], ITGA6 can be indirectly down regulated by GATA3 via microRNA-29b [39], whereas SATB1 acts as upstream regulator and positively regulates GATA3. Therefore, knocking-down GATA3 can largely reduce expression of ITM2A, slightly for ITGA6 but not for SATB1, which can be validated in cellular experiments, indicating the ubiquitous association of some candidates among different cell types. Moreover, candidates can be impacted by SNPs in “GATA” motif in their regulatory elements, as direct downstream targets of GATA3, such as rs4894953, located in the enhancer region of CBLB, appears in the C allele, then expression of CBLB is down-regulated compared with T allele as the result of losing GATA3 binding site characterized in CBLB. Some of the other candidates have been linked to GATA3 through PPI prediction, and the validations for well-known cancer related genes should be first priority to reveal the mechanism of GATA3 induced leukemogenesis. In another hand, risk allele GATA3 SNPs are associated with higher risk of B-ALL relapse as well, suggesting higher expression of GATA3 will result in poor treatment outcomes. Recently, GATA3 overexpression has been reported to be associated with poor overall survival in Peripheral T-cell lymphoma [40], but a favorite prognostic factor for breast cancer. We assumed that the paradox might be explained by the GATA3 related candidates with opposite directions.
Importantly, pipeline we developed can be expanded to screen the regulatory network of other important genes in different cancer types, especially for those transcription factors. In this study, we used a very strict criteria to screen the strongest candidates, which can induce high rate of false negative. When this pipeline will be used in other studies, multiple factors should be considered to balance the false negative and false positive, including sample size, number of available cohorts, heterogeneity of the patients, and etc. Moreover, experimental validations are always needed for the final determination. Notably, this method can’t be used to find out the gene-related candidates through other mechanisms, such as protein-protein interaction or post-transcriptional/post-translational modifications.
In conclusion, we have used a series of public available microarray datasets, and developed an effective pipeline to find 173 GATA3-related genes in B-ALL. With the bioinformatics analyses and cellular experiment validations, multiple potential GATA3 related genes (e.g., ETV6) and signaling pathways (JAK/STAT and cell cycle pathways) were determined in ubiquitous or B-ALL specific manner. We conclude that risk allele of GATA3 SNP induces overexpression of GATA3, and subsequently impacts on the regulatory network of GATA3 to increase the susceptibility for B-ALL leukemogenesis.
MATERIALS AND METHODS
Epigenetics regulation illustration and genotype-expression association analyses
Online tools (i.e., Epigenome Browser [41]) was used to illustrate the epigenetic element around SNPs of GATA3 and CBLB by introducing Roadmap and ENCODE information from multiple tissue and cell types. Expression level of GATA3 gene was obtained from public RNA-seq data resource of Lymphoblastoid cell lines [42], and genotypes of rs3824662 was obtained from the 1000 genome project website (http://grch37.ensembl.org/index.html). As described before, Genotype-expression association was assessed through a linear regression model for the available individuals (N = 441) [43].
Expression microarray datasets searching and association analyses
Expression level of all genes in B-ALL patients were obtained from Gene Expression Omnibus (GSE7440 [44], GSE11877 [45], GSE10792 [46], GSE13351 [47], GSE13425 [47], GSE635 [48], GSE10255 [49], GSE4698 [50], GSE33315 [51], and GSE13204 [52]). Association of GATA3 expression of all the rest genes were estimated by using linear regression model, and multiple criteria were applied for candidates screening, including P value, r2, association directions, and etc. Expression information of the candidates for breast cancer was retrieved from a large patient cohort from The European Genome-phenome Archive database (EGAS00000000083) [53], and was also conducted to association analyses with GATA3 expression with linear regression model as well.
All the GATA3-related genes were imported into the STRING, IntAct and BioGRID for protein-protein interaction network construction [54], and DAVID for pathway analyses [55].
Plasmids of shRNA cloning, lentivirus production and stable cells constructions
Pairs of shRNA oligonucleotides for GATA3 were annealed and ligated into the pLKO-TRC vector with AgeI and EcoRI digested and gel-purified. The constructed plasmids were verified by Sanger sequencing. Sequence information of shRNAs against our interested candidates were obtained from online information (http://www.sigmaaldrich.com/, Supplementary Table 7). Lentivirus was prepared with calcium phosphate-mediated transfection of 293T cells, which were cultured with 10% FBS contained DMEM medium. Lentivitral vectors were cotransfected with the helper vectors pCAGkGP1R, pCAG4-RTR2 and pCAG-VSV-G, and lentiviruses were purified by 0.45 um syringe filters. 697 cells were seeded into 6-well plates at a density of 1–2 million and infected with purified lentivirus particles. Polybrene (3 ul of 5 mg/ml stock solution) was added to the cells, followed by 3 ml of lentivirus solution. Cells were spin infected in 6-well plates for 1 h at 2000 rpm at 30°C. After cells and lentivirus co-incubated for 18 h at 37°C, the supernatant was removed by centrifugation and aspiration. Next, cells were resuspended in fresh 10% FBS contained RPMI medium, and incubated at 37°C for 72 h. Next, the knockdown stable cells were selected from infected cells with appropriate puromycin concentrations.
RNA isolation and real-time PCR
RNA extractions for stable cells were performed with Animal Total RNA Isolation Kit (Foregene, RE-03013) according to the manual protocol and reverse transcribed into cDNA with PrimeScript™ RT reagent Kit with gDNA Eraser (TAKARA, RR047A). Real-time PCR was performed with PowerUp™ SYBR® Green Master Mix (Applied Biosystems™, A25776) to estimate the knockdown efficacy of shRNA as well as the selected gene expression, and primer sequence information is listed in Supplementary Table 8.
Abbreviations
ALL: Acute lymphoblastic leukemia; TCGA: The Cancer Genome Atlas; GWAS: genome-wide association studies; LCL: Lymphoblastoid cell line; AML: acute myeloid leukemia; CLL: chronic lymphoblastic leukemia; CML: chronic myeloid leukemia; GEO: Gene Expression Omnibus.
Authors’ contributions
QQH, FL carried out the data gathering and cellular experiments, SYZ and DYZ carried out data analyses, YZ, XYZ, XYX and YXY carried out figure/table preparations, HSY, LMW, XW, YPZ, ZGM, LO, YLW, and HZ provide material input and helped revising the manuscript, LY, BL, HX, and YS supervised the experimental design, data analyses, and manuscript writing, all authors read and approved the final manuscript.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (No. 81400120, No. 81522028, No. 81673452), and National Key Research Development Program (No. 2016YFC0905000), and Heng Xu are supported by the grant from “the Recruitment Program of Global Young Experts” (known as “the Thousand Young Talents Plan”).
CONFLICTS OF INTEREST
None.
REFERENCES
1. Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006; 354:166–78.
2. Buffler PA, Kwan ML, Reynolds P, Urayama KY. Environmental and genetic risk factors for childhood leukemia: appraising the evidence. Cancer Invest. 2005; 23:60–75.
3. Trevino LR, Yang W, French D, Hunger SP, Carroll WL, Devidas M, Willman C, Neale G, Downing J, Raimondi SC, Pui CH, Evans WE, Relling MV. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009; 41:1001–5.
4. Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, Kinsey SE, Lightfoot T, Roman E, Irving JA, Allan JM, Tomlinson IP, Taylor M, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009; 41:1006–10.
5. Xu H, Yang W, Perez-Andreu V, Devidas M, Fan Y, Cheng C, Pei D, Scheet P, Burchard EG, Eng C, HuntsmanS, Torgerson DG, Dean M, et al. Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst. 2013; 105:733–42.
6. Migliorini G, Fiege B, Hosking FJ, Ma Y, Kumar R, Sherborne AL, da Silva Filho MI, Vijayakrishnan J, Koehler R, Thomsen H, Irving JA, Allan JM, Lightfoot T, et al. Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood. 2013; 122:3298–307.
7. Sherborne AL, Hosking FJ, Prasad RB, Kumar R, Koehler R, Vijayakrishnan J, Papaemmanuil E, Bartram CR, Stanulla M, Schrappe M, Gast A, Dobbins SE, Ma Y, et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet. 2010; 42:492–4.
8. Perez-Andreu V, Roberts KG, Harvey RC, Yang W, Cheng C, Pei D, Xu H, Gastier-Foster J, E S, Lim JY, Chen IM, Fan Y, Devidas M, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet. 2013; 45:1494–8.
9. Xu H, Zhang H, Yang W, Yadav R, Morrison AC, Qian M, Devidas M, Liu Y, Perez-Andreu V, Zhao X, Gastier-Foster JM, Lupo PJ, Neale G, et al. Inherited coding variants at the CDKN2A locus influence susceptibility to acute lymphoblastic leukaemia in children. Nat Commun. 2015; 6:7553.
10. Perez-Andreu V, Roberts KG, Xu H, Smith C, Zhang H, Yang W, Harvey RC, Payne-Turner D, Devidas M, Cheng IM, Carroll WL, Heerema NA, Carroll AJ, et al. A genome-wide association study of susceptibility to acute lymphoblastic leukemia in adolescents and young adults. Blood. 2015; 125:680–6.
11. Prasad RB, Hosking FJ, Vijayakrishnan J, Papaemmanuil E, Koehler R, Greaves M, Sheridan E, Gast A, Kinsey SE, Lightfoot T, Roman E, Taylor M, Pritchard-Jones K, et al. Verification of the susceptibility loci on 7p12.2, 10q21.2, and 14q11.2 in precursor B-cell acute lymphoblastic leukemia of childhood. Blood. 2010; 115:1765–7.
12. Yang W, Trevino LR, Yang JJ, Scheet P, Pui CH, Evans WE, Relling MV. ARID5B SNP rs10821936 is associated with risk of childhood acute lymphoblastic leukemia in blacks and contributes to racial differences in leukemia incidence. Leukemia. 2010; 24:894–6.
13. Xu H, Cheng C, Devidas M, Pei D, Fan Y, Yang W, Neale G, Scheet P, Burchard EG, Torgerson DG, Eng C, Dean M, Antillon F, et al. ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment outcome of childhood acute lymphoblastic leukemia. J Clin Oncol. 2012; 30:751–7.
14. Moriyama T, Relling MV, Yang JJ. Inherited genetic variation in childhood acute lymphoblastic leukemia. Blood. 2015; 125:3988–95.
15. Liao F, Yin D, Zhang Y, Hou Q, Zheng Z, Yang L, Shu Y, Xu H, Li Y. Association Between PIP4K2A Polymorphisms and Acute Lymphoblastic Leukemia Susceptibility. Medicine (Baltimore). 2016; 95: e3542.
16. Rothenberg EV. Transcriptional drivers of the T-cell lineage program. Curr Opin Immunol. 2012; 24:132–8.
17. Wan YY. GATA3: a master of many trades in immune regulation. Trends Immunol. 2014; 35:233–42.
18. Jenner RG, Townsend MJ, Jackson I, Sun K, Bouwman RD, Young RA, Glimcher LH, Lord GM. The transcription factors T-bet and GATA-3 control alternative pathways of T-cell differentiation through a shared set of target genes. Proc Natl Acad Sci U S A. 2009; 106:17876–81.
19. Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997; 89:587–96.
20. Wei G, Abraham BJ, Yagi R, Jothi R, Cui K, Sharma S, Narlikar L, Northrup DL, Tang Q, Paul WE, Zhu J, Zhao K. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity. 2011; 35:299–311.
21. Notani D, Gottimukkala KP, Jayani RS, Limaye AS, Damle MV, Mehta S, Purbey PK, Joseph J, Galande S. Global regulator SATB1 recruits beta-catenin and regulates T(H)2 differentiation in Wnt-dependent manner. PLoS Biol. 2010; 8: e1000296.
22. Fang TC, Yashiro-Ohtani Y, Del Bianco C, Knoblock DM, Blacklow SC, Pear WS. Notch directly regulates Gata3 expression during T helper 2 cell differentiation. Immunity. 2007; 27:100–10.
23. Amsen D, Antov A, Jankovic D, Sher A, Radtke F, Souabni A, Busslinger M, McCright B, Gridley T, Flavell RA. Direct regulation of Gata3 expression determines the T helper differentiation potential of Notch. Immunity. 2007; 27:89–99.
24. Pandolfi PP, Roth ME, Karis A, Leonard MW, DzierzakE, Grosveld FG, Engel JD, Lindenbaum MH. Targeted disruption of the GATA3 gene causes severe abnormalities in the nervous system and in fetal liver haematopoiesis. Nat Genet. 1995; 11:40–4.
25. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012; 486:405–9.
26. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012; 486:353–60.
27. Kouros-Mehr H, Kim JW, Bechis SK, Werb Z. GATA-3 and the regulation of the mammary luminal cell fate. Curr Opin Cell Biol. 2008; 20:164–70.
28. Chu IM, Michalowski AM, Hoenerhoff M, Szauter KM, Luger D, Sato M, Flanders K, Oshima A, Csiszar K, Green JE. GATA3 inhibits lysyl oxidase-mediated metastases of human basal triple-negative breast cancer cells. Oncogene. 2012; 31:2017–27.
29. Yan W, Cao QJ, Arenas RB, Bentley B, Shao R. GATA3 inhibits breast cancer metastasis through the reversal of epithelial-mesenchymal transition. J Biol Chem. 2010; 285:14042–51.
30. Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, Totoki Y, Chiba K, Sato-Otsubo A, Nagae G, Ishii R, Muto S, Kotani S, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015; 47:1304–15.
31. Banerjee A, Northrup D, Boukarabila H, Jacobsen SE, Allman D. Transcriptional repression of Gata3 is essential for early B cell commitment. Immunity. 2013; 38:930–42.
32. Garcia-Ojeda ME, Klein Wolterink RG, Lemaitre F, Richard-Le Goff O, Hasan M, Hendriks RW, Cumano A, Di Santo JP. GATA-3 promotes T-cell specification by repressing B-cell potential in pro-T cells in mice. Blood. 2013; 121:1749–59.
33. Ku CJ, Hosoya T, Maillard I, Engel JD. GATA-3 regulates hematopoietic stem cell maintenance and cell-cycle entry. Blood. 2012; 119:2242–51.
34. Hosoya T, Kuroha T, Moriguchi T, Cummings D, Maillard I, Lim KC, Engel JD. GATA-3 is required for early T lineage progenitor development. J Exp Med. 2009; 206:2987–3000.
35. Tai TS, Pai SY, Ho IC. Itm2a, a target gene of GATA-3, plays a minimal role in regulating the development and function of T cells. PLoS One. 2014; 9: e96535.
36. Moriyama T, Metzger ML, Wu G, Nishii R, Qian M, Devidas M, Yang W, Cheng C, Cao X, Quinn E, Raimondi S, Gastier-Foster JM, Raetz E, et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol. 2015; 16:1659–66.
37. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015; 66:311–28.
38. Stanelle J, Doring C, Hansmann ML, Kuppers R. Mechanisms of aberrant GATA3 expression in classical Hodgkin lymphoma and its consequences for the cytokine profile of Hodgkin and Reed/Sternberg cells. Blood. 2010; 116:4202–11.
39. Chou J, Lin JH, Brenot A, Kim JW, Provot S, Werb Z. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat Cell Biol. 2013; 15:201–13.
40. Iqbal J, Wright G, Wang C, Rosenwald A, Gascoyne RD, Weisenburger DD, Greiner TC, Smith L, Guo S, Wilcox RA, Teh BT, Lim ST, Tan SY, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014; 123:2915–23.
41. Zhou X, Maricque B, Xie M, Li D, Sundaram V, Martin EA, Koebbe BC, Nielsen C, Hirst M, Farnham P, Kuhn RM, Zhu J, Smirnov I, et al. The Human Epigenome Browser at Washington University. Nat Methods. 2011; 8:989–90.
42. Lappalainen T, Sammeth M, Friedlander MR, t Hoen PA, Monlong J, Rivas MA, Gonzalez-Porta M, Kurbatova N, Griebel T, Ferreira PG, Barann M, Wieland T, Greger L, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013; 501:506–11.
43. Yin D, Xia X, Zhang J, Zhang S, Liao F, Zhang G, Zhang Y, Hou Q, Yang X, Wang H, Ma Z, Wang H, Zhu Y, et al. Impact of NUDT15 polymorphisms on thiopurines-induced myelotoxicity and thiopurines tolerance dose. Oncotarget. 2017; 8:13575–13585. doi: 10.18632/oncotarget.14594.
44. Bhojwani D, Kang H, Menezes RX, Yang W, Sather H, Moskowitz NP, Min DJ, Potter JW, Harvey R, Hunger SP, Seibel N, Raetz EA, Pieters R, et al. Gene expression signatures predictive of early response and outcome in high-risk childhood acute lymphoblastic leukemia: A Children's Oncology Group Study [corrected]. J Clin Oncol. 2008; 26:4376–84.
45. Kang H, Chen IM, Wilson CS, Bedrick EJ, Harvey RC, Atlas SR, Devidas M, Mullighan CG, Wang X, Murphy M, Ar K, Wharton W, Borowitz MJ, et al. Gene expression classifiers for relapse-free survival and minimal residual disease improve risk classification and outcome prediction in pediatric B-precursor acute lymphoblastic leukemia. Blood. 2010; 115:1394–405.
46. Bungaro S, Dell'Orto MC, Zangrando A, Basso D, Gorletta T, Lo Nigro L, Leszl A, Young BD, Basso G, Bicciato S, Biondi A, te Kronnie G, Cazzaniga G. Integration of genomic and gene expression data of childhood ALL without known aberrations identifies subgroups with specific genetic hallmarks. Genes Chromosomes Cancer. 2009; 48:22–38.
47. Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, Van Zutven LJ, Beverloo HB, Van der Spek PJ, Escherich G, Horstmann MA, Janka-Schaub GE, Kamps WA, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009; 10:125–34.
48. Holleman A, Cheok MH, den Boer ML, Yang W, Veerman AJ, Kazemier KM, Pei D, Cheng C, Pui CH, Relling MV, Janka-Schaub GE, Pieters R, Evans WE. Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N Engl J Med. 2004; 351:533–42.
49. Sorich MJ, Pottier N, Pei D, Yang W, Kager L, Stocco G, Cheng C, Panetta JC, Pui CH, Relling MV, Cheok MH, Evans WE. In vivo response to methotrexate forecasts outcome of acute lymphoblastic leukemia and has a distinct gene expression profile. PLoS Med. 2008; 5: e83.
50. Kirschner-Schwabe R, Lottaz C, Todling J, Rhein P, Karawajew L, Eckert C, von Stackelberg A, Ungethum U, Kostka D, Kulozik AE, Ludwig WD, Henze G, Spang R, et al. Expression of late cell cycle genes and an increased proliferative capacity characterize very early relapse of childhood acute lymphoblastic leukemia. Clin Cancer Res. 2006; 12:4553–61.
51. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen SC, Wei L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012; 481:157–63.
52. Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, De Vos J, Hernandez JM, Hofmann WK, Mills KI, Gilkes A, Chiaretti S, Shurtleff SA, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010; 28:2529–37.
53. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Graf S, Ha G, Haffari G, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012; 486:346–52.
54. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015; 43: D447-52.
55. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44–57.