
INTRODUCTION
As one of the most common malignancies worldwide, hepatocellular carcinoma (HCC) represents the second-leading cause of cancer deaths globally with 745,000 deaths per year [1]. The HCC were categorized into poorly-, moderately-, and well- differentiated types. Hepatic resection is currently the most optimal choice for HCC treatment. However, poorly differentiated HCC has a worse prognosis with high recurrence rate compared to other two types [2, 3]. In addition, it is hard to discriminate poorly differentiated HCC from other two types of HCC before treatment. The lack of good diagnostic markers and therapeutic targets has rendered HCC a major challenge.
Recently, high-throughput technologies like RNA sequencing make it possible and easy to illustrate the transcriptome characteristics of cancers including HCC [4]. Some potential biomarkers for HCC were identified using transcriptome sequencing e.g. SERPINA11 whose expression is correlated with pathology stages lack documented expression profiles in liver cancer [5]. Some signaling pathways like cell cycle were also involved via analysis of transcriptome sequencing data [6]. Those studies show a great promise of exploring the molecular basis of HCC.
In this study, we performed transcriptome sequencing for patients with poorly differentiated (PD), moderately differentiated (MD) and well differentiated (WD) HCC. 3 patients diagnosed as HCC of each grades were recruited, respectively. Differentially expressed genes (DEGs) were identified through analysis of transcriptome sequencing data. DEGs were then subjected to gene set enrichment analysis. Furthermore, RT-PCR was performed to validate potential biomarkers in 15 pairs of poorly differentiated tumor and adjacent non-tumorous samples. Our investigation may provide new clues on the molecular event responsible for the progression of HCC and potential biomarkers and therapeutic targets for diagnosis and treatment of HCC patients.
RESULTS
Overview of transcriptome sequencing statistics
Pair-end second-generation transcriptome sequencing was performed in 9 HCC patients. Sample characteristics were shown in Table 1. An average of 35,772,695 pair-end 125 bp clean reads was generated (Table 2). The average mapping rate was 93.17%, resulting an average coverage of depth of 32 × (Table 2). Expression levels of more than 25,200 genes were calculated using Tophat/Cufflinks (Supplementary Table 1). The heatmap of all expressed genes were drawn in Figure 1, which showed a big group of differentially expressed genes (DEGs) between tumor and adjacent non-tumorous samples of poorly differentiated HCC.
Table 1: Detailed characteristics of the patients
Patient |
Age |
Gender |
Hepatitis |
Serum AFP level (ng/mL) |
Metastasis |
Glisson capsule invasion |
Tumor size (mm) |
Differentiation Grade |
Multiple liver nodules |
|---|---|---|---|---|---|---|---|---|---|
P1 |
59 |
M |
HBV |
670.1 |
No |
− |
35 |
Poorly |
- |
P2 |
50 |
M |
HBV |
12483 |
No |
+ |
50 |
Poorly |
- |
P3 |
37 |
M |
HBV |
4.47 |
No |
+ |
41 |
Poorly |
- |
M1 |
59 |
M |
HBV |
3.73 |
No |
− |
6 |
moderately |
- |
M2 |
69 |
M |
HBV |
6.9 |
No |
+ |
160 |
moderately |
synchronous |
M3 |
61 |
M |
NBNC |
1.11 |
No |
+ |
80 |
moderately |
- |
W1 |
62 |
M |
HBV |
266.5 |
No |
+ |
32 |
Well |
- |
W2 |
76 |
M |
HCV |
5.84 |
No |
+ |
70 |
Well |
- |
W3 |
19 |
F |
NBNC |
2.85 |
No |
− |
29 |
Well |
- |
P4 |
48 |
M |
NBNC |
55.1 |
Yes |
+ |
180 |
Poorly |
- |
P5 |
41 |
M |
HBV |
4.7 |
Yes |
+ |
50 |
Poorly |
- |
P6 |
64 |
M |
HBV |
244.6 |
Yes |
+ |
65 |
Poorly |
synchronous |
P7 |
50 |
M |
HBV |
3.03 |
Yes |
+ |
55 |
Poorly |
synchronous |
P8 |
47 |
M |
HBV |
62.47 |
Yes |
+ |
105 |
Poorly |
- |
P9 |
64 |
M |
HBV |
36541 |
Yes |
+ |
140 |
Poorly |
- |
P10 |
52 |
M |
HBV |
2100 |
Yes |
− |
35 |
Poorly |
- |
P11 |
52 |
M |
NBNC |
5.3 |
No |
+ |
65 |
Poorly |
- |
P12 |
37 |
M |
HBV |
1810 |
No |
− |
55 |
Poorly |
- |
P13 |
48 |
M |
HBV |
2.07 |
No |
− |
35 |
Poorly |
- |
P14 |
52 |
F |
NBNC |
2.45 |
No |
+ |
22 |
Poorly |
- |
P15 |
58 |
M |
HBV |
6.52 |
Yes |
+ |
58 |
Poorly |
synchronous |
M4 |
63 |
M |
HBV |
2975 |
No |
+ |
70 |
moderately |
synchronous |
M5 |
47 |
M |
HBV |
5375 |
Yes |
+ |
130 |
moderately |
- |
M6 |
72 |
M |
HBV |
303.8 |
No |
+ |
40 |
moderately |
synchronous |
M7 |
66 |
M |
HBV |
60500 |
No |
+ |
50 |
moderately |
- |
M8 |
59 |
M |
NBNC |
54.84 |
No |
+ |
26 |
moderately |
- |
M9 |
67 |
M |
HBV |
321.1 |
No |
+ |
55 |
moderately |
synchronous |
M10 |
71 |
M |
HBV |
4606 |
Yes |
+ |
12 |
moderately |
- |
M11 |
18 |
F |
HBV |
37979 |
No |
+ |
70 |
moderately |
- |
M12 |
45 |
M |
HBV |
6528 |
No |
+ |
60 |
moderately |
synchronous |
M13 |
61 |
M |
HBV |
1.9 |
No |
+ |
30 |
moderately |
- |
M14 |
52 |
M |
HBV |
26.53 |
No |
+ |
20 |
moderately |
- |
M15 |
58 |
F |
HBV |
2.55 |
Yes |
+ |
55 |
moderately |
synchronous |
M16 |
42 |
M |
HBV |
3.38 |
No |
+ |
20 |
moderately |
synchronous |
M17 |
64 |
M |
HBV |
774.2 |
No |
+ |
10 |
moderately |
- |
M18 |
54 |
F |
NBNC |
23784 |
No |
+ |
22 |
moderately |
- |
W4 |
72 |
M |
HBV |
11.10 |
No |
+ |
20 |
Well |
- |
W5 |
55 |
M |
HBV |
1.82 |
No |
+ |
34 |
Well |
- |
W6 |
76 |
M |
NBNC |
2.1 |
No |
+ |
34 |
Well |
- |
W7 |
67 |
M |
NBNC |
3.30 |
No |
− |
18 |
Well |
- |
W8 |
50 |
M |
HBV |
260.9 |
No |
− |
25 |
Well |
- |
W9 |
71 |
M |
HBV |
11.65 |
No |
+ |
35 |
Well |
- |
W10 |
74 |
M |
NBNC |
5.9 |
No |
+ |
30 |
Well |
- |
W11 |
39 |
M |
HBV |
8.10 |
Yes |
+ |
22 |
Well |
synchronous |
W12 |
63 |
M |
HBV |
208 |
No |
+ |
40 |
Well |
- |
W13 |
48 |
M |
NBNC |
3.07 |
No |
− |
33 |
Well |
- |
W14 |
66 |
M |
HBV |
82.81 |
No |
+ |
34 |
Well |
- |
Note: The nine samples used for sequencing are labeled in bold and italic.
Table 2: Summary statistics of the transcriptome sequencing
Patient |
Differentiation grade |
Sample Type |
Total reads |
Mapped reads |
Total base (bp) |
Mapped base (bp) |
Mappping ratio |
Coverage (X) |
|---|---|---|---|---|---|---|---|---|
P1 |
Poorly |
T |
35,930,408 |
33,819,287 |
4,491,301,000 |
4,227,410,875 |
94.12% |
33 |
N |
34,902,228 |
32,863,973 |
4,362,778,500 |
4,107,996,625 |
94.16% |
32 |
||
P2 |
Poorly |
T |
35,315,378 |
33,125,921 |
4,414,422,250 |
4,140,740,125 |
93.80% |
32 |
N |
34,383,802 |
32,279,443 |
4,297,975,250 |
4,034,930,375 |
93.88% |
31 |
||
P3 |
Poorly |
T |
35,532,658 |
33,258,212 |
4,441,582,250 |
4,157,276,500 |
93.60% |
32 |
N |
34,537,914 |
32,152,860 |
4,317,239,250 |
4,019,107,500 |
93.09% |
31 |
||
M1 |
Moderately |
T |
34,566,526 |
32,670,216 |
4,320,815,750 |
4,083,777,000 |
94.51% |
31 |
N |
35,384,486 |
33,407,169 |
4,423,060,750 |
4,175,896,125 |
94.41% |
32 |
||
M2 |
Moderately |
T |
34,564,642 |
32,385,294 |
4,320,580,250 |
4,048,161,750 |
93.69% |
31 |
N |
34,771,048 |
32,792,475 |
4,346,381,000 |
4,099,059,375 |
94.31% |
32 |
||
M3 |
Moderately |
T |
35,371,920 |
32,571,695 |
4,421,490,000 |
4,071,461,875 |
92.08% |
31 |
N |
35,959,532 |
32,990,753 |
4,494,941,500 |
4,123,844,125 |
91.74% |
32 |
||
W1 |
Well |
T |
34,762,386 |
32,701,011 |
4,345,298,250 |
4,087,626,375 |
94.07% |
31 |
N |
35,314,918 |
33,021,170 |
4,414,364,750 |
4,127,646,250 |
93.50% |
32 |
||
W2 |
Well |
T |
35,610,444 |
32,509,118 |
4,451,305,500 |
4,063,639,750 |
91.29% |
31 |
N |
34,637,398 |
31,280,461 |
4,329,674,750 |
3,910,057,625 |
90.31% |
30 |
||
W3 |
Well |
T |
42,279,434 |
39,097,967 |
5,284,929,250 |
4,887,245,875 |
92.50% |
38 |
N |
40,083,384 |
36,852,292 |
5,010,423,000 |
4,606,536,500 |
91.90% |
35 |
||
Average |
35,772,695 |
33,321,073 |
4,471,586,847 |
4,165,134,146 |
93.17% |
32 |
Note: T and N represent tumor and adjacent non-tumor tissue, respectively.
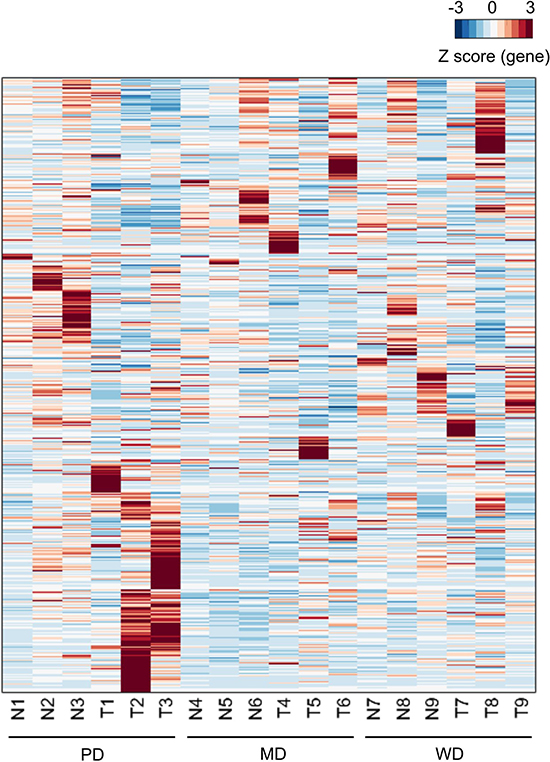
Figure 1: Heatmap of expressed genes. For the sample labels, N and T represented matched adjacent non-tumor tissue specimens and tumor tissue specimens respectively. Each row represented of Z scores of genes across different samples.
Identification of DEGs between tumor and adjacent non-tumor tissues
We next detected DEGs between tumor and adjacent non-tumorous samples of poorly differentiated, moderately differentiated and well differentiated HCC. Through paired-T test analysis (P value < 0.05, Fold change > 2), a total of 1020 DEGs were detected including 372 up-regulated genes (313, 47 and 12 for poorly-, moderately- and well-differentiated HCC, respectively) and 648 down-regulated genes (368, 249 and 31 for poorly-, moderately- and well-differentiated HCC, respectively) (Figure 2A; Supplementary Table 2). Only 1 down-regulated gene (CD44) was overlapped in all 3 grades. Top 20 up-regulated genes and Top 20 down-regulated genes of poorly differentiated HCC were shown in Table 3 and Table 4, respectively.
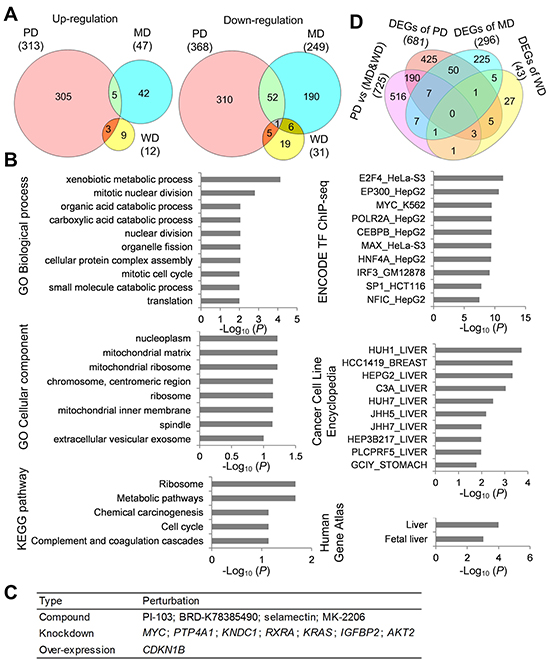
Figure 2: Functional characterization of DEGs. (A) Venn diagram of DEGs between poorly differentiated (PD), moderately differentiated (MD) and well differentiated (WD) HCC. (B) Functional enrichment analysis of DEGs of poorly differentiated HCC. (C) Top 12 perturbations with high negative connectivity scores with DEGs of poorly differentiated HCC. (D) Venn diagram of DEGs between PD HCC tumor tissues and MD&WD tumor tissues, DEGs of PD HCC, DEGs of MD HCC, and DEGs of WD HCC.
Table 3: Top 20 up-regulated genes of poorly differentiated HCC
Entrez_ID |
Gene Symbol |
N1 |
N2 |
N3 |
T1 |
T2 |
T3 |
Group mean _exp_N |
Group mean _exp_T |
Group _log2FC |
Group ttest _p value |
|---|---|---|---|---|---|---|---|---|---|---|---|
10130 |
PDIA6 |
116.76 |
123.98 |
126.57 |
255.14 |
260.71 |
263.04 |
122.44 |
259.63 |
1.08 |
1.90E-05 |
578 |
BAK1 |
6.41 |
5.00 |
9.99 |
20.24 |
18.51 |
23.98 |
7.14 |
20.91 |
1.43 |
1.03E-04 |
1650 |
DDOST |
53.21 |
72.67 |
59.72 |
115.80 |
139.67 |
121.44 |
61.87 |
125.64 |
1.01 |
6.57E-04 |
2786 |
GNG4 |
0.04 |
0.02 |
0.23 |
1.84 |
1.90 |
2.21 |
0.10 |
1.98 |
1.45 |
7.97E-04 |
84933 |
C8orf76 |
6.03 |
5.11 |
6.90 |
24.33 |
21.27 |
23.79 |
6.01 |
23.13 |
1.78 |
1.35E-03 |
22996 |
TTC39A |
0.04 |
0.29 |
0.46 |
2.38 |
2.35 |
2.73 |
0.26 |
2.49 |
1.46 |
1.46E-03 |
5691 |
PSMB3 |
119.36 |
132.67 |
145.10 |
301.66 |
342.81 |
347.47 |
132.37 |
330.64 |
1.31 |
1.74E-03 |
54522 |
ANKRD16 |
1.87 |
2.67 |
2.10 |
6.20 |
7.28 |
7.16 |
2.21 |
6.88 |
1.29 |
2.04E-03 |
28998 |
MRPL13 |
13.72 |
14.12 |
18.44 |
69.51 |
61.37 |
73.26 |
15.43 |
68.05 |
2.07 |
2.62E-03 |
50854 |
C6orf48 |
28.97 |
70.17 |
66.97 |
111.29 |
140.99 |
150.56 |
55.37 |
134.28 |
1.26 |
2.64E-03 |
3094 |
HINT1 |
270.52 |
305.61 |
289.43 |
605.26 |
590.80 |
625.23 |
288.52 |
607.10 |
1.07 |
2.74E-03 |
4738 |
NEDD8 |
131.06 |
138.11 |
152.30 |
291.78 |
329.80 |
338.89 |
140.49 |
320.16 |
1.18 |
2.84E-03 |
57538 |
ALPK3 |
0.47 |
1.81 |
1.41 |
3.16 |
4.41 |
3.61 |
1.23 |
3.73 |
1.08 |
3.65E-03 |
65244 |
SPATS2 |
2.33 |
3.32 |
3.91 |
6.11 |
7.99 |
8.07 |
3.18 |
7.39 |
1.00 |
3.73E-03 |
8624 |
PSMG1 |
15.75 |
16.67 |
25.69 |
37.42 |
39.74 |
44.17 |
19.37 |
40.44 |
1.02 |
4.12E-03 |
8836 |
GGH |
61.61 |
26.09 |
42.65 |
124.45 |
87.68 |
117.67 |
43.45 |
109.93 |
1.32 |
4.12E-03 |
23640 |
HSPBP1 |
7.35 |
11.32 |
9.61 |
19.05 |
25.08 |
24.27 |
9.42 |
22.80 |
1.19 |
4.27E-03 |
79075 |
DSCC1 |
0.34 |
0.51 |
1.13 |
4.16 |
4.12 |
4.17 |
0.66 |
4.15 |
1.63 |
4.36E-03 |
5591 |
PRKDC |
5.80 |
5.46 |
7.02 |
17.28 |
15.77 |
16.09 |
6.09 |
16.38 |
1.29 |
4.50E-03 |
84701 |
COX4I2 |
0.22 |
1.13 |
0.23 |
3.16 |
4.83 |
3.73 |
0.53 |
3.91 |
1.69 |
4.56E-03 |
Note: T1, T2, T3 represent tumor tissue of P1, P2, P3 with poorly differentiated HCC, respectively; N1, N2, N3 represent adjacent non-tumor tissue of P1, P2, P3 with poorly differentiated HCC, respectively.
Table 4: Top 20 down-regulated genes of poorly differentiated HCC
Note: T1, T2, T3 represent tumor tissue of P1, P2, P3 with poorly differentiated HCC, respectively; N1, N2, N3 represent adjacent non-tumor tissue of P1, P2, P3 with poorly differentiated HCC, respectively.
Significantly enriched functions for DEGs of poorly differentiated HCC
DEGs of poorly differentiated HCC were subjected to functional enrichment analyses to illustrate their biological function characteristics using Enrichr tool [7]. The top enriched terms for categories were shown in Figure 2B (P value < 0.1). From Human Gene Atlas and Cancer Cell Line Encyclopedia, those DEGs were enriched in liver tissue and various liver cancer cells. The enriched biological process and pathway included metabolism, cell cycle, translation and blood coagulation. The DEGs tended to lie in mitochondrial, chromosome and ribosome. From the ENCODE TF ChIP-seq data, 5 transcription factors in liver cancer cell HEPG2 were enriched to regulate the poorly differentiated DEGs.
Potential perturbations for reverse of abnormal gene expression change of poorly differentiated HCC
LINCS is a collection of signatures of gene expression for a broad range of conditions such as drug treatment, ligand treatment, gene knockdown, and gene over-expression in many different types of human cells [8]. We inputted the up-regulated genes and down-regulated genes into lincscloud to find perturbations that had opposite gene expression change with differentiated HCC. Top 12 perturbations with high negative connectivity scores (Figure 2C), which included 4 compounds, 7 knockdown genes and 1 over-expression gene.
Comparison of tumor tissues between PD HCC and MD&WD HCC
Besides DEGs between tumor and adjacent non-tumorous samples, we compared tumor tissues between PD HCC and MD&WD HCC. As a result, 725 differentially expressed genes were identified (Supplementary Table 2); among them, 209 were identified as DEGs when comparing tumor and adjacent non-tumorous samples (Figure 2D). Gene ontology enrichment analysis was also done based on this 725-gene list. Similarly, these genes were enriched in organelle fission, mitotic nuclear division, positive regulation of cell cycle phase transition and serine family amino acid catabolic process.
Validation of DEGs specific for poorly differentiated HCC by RT-PCR
4 DEGs specific for poorly differentiated HCC as well as related with cell cycle or proliferation were validated by RT-PCR. In the discovery phase, the expressions of NOVA1, NSMCE2 and KIAA0196 were significantly up-regulated, while expression of AQP9 was significantly down-regulated in poorly differentiated samples, as compared with that in adjacent non-tumorous samples, moderately differentiated samples and well differentiated samples (P < 0.05) (Table 5). RT-PCR results showed that all these 4 genes were successfully validated (Figure 3), and the dysregulation trend matched with those observed in the RNA-seq data.
Table 5: 4 DEGs specific for poorly differentiated HCC
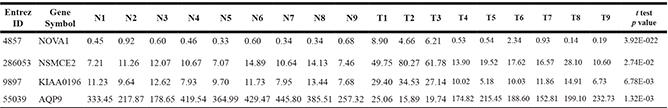
Note: N1, N2, N3 and T1, T2, T3 represent adjacent non-tumor tissue and tumor tissue of P1, P2, P3 with poorly differentiated HCC, respectively. N4, N5, N6 and T4, T5, T6 represent adjacent non-tumor tissue and tumor tissue of M1, M2, M3 with moderately differentiated HCC, respectively. N7, N8, N9 and T7, T8, T9 represent adjacent non-tumor tissue and tumor tissue of W1, W2, W3 with well differentiated HCC, respectively. p value refers to T1, T2, T3 vs N1, N2, N3, N4, N5, N6, N7, N8, N9, T4, T5, T6, T7, T8, T9 by t test.
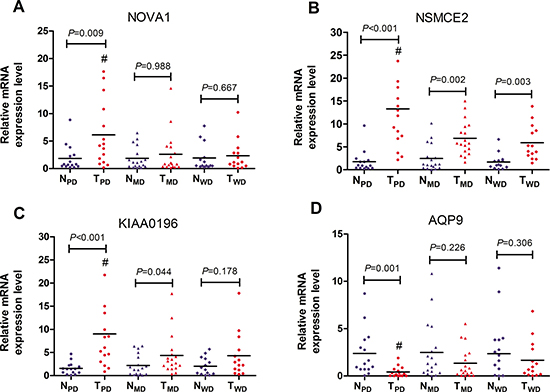
Figure 3: RT-PCR validation of 4 DEGs specific for poorly differentiated HCC. GAPDH mRNA was used as an internal control. Expression data were obtained as 2-ΔΔCT relative to adjacent non-tumor tissue values. NPD and TPD represented matched adjacent non-tumor tissue specimens (n = 15) and poorly differentiated HCC tissue specimens (n = 15) respectively. NMD and TMD represented matched adjacent non-tumor tissue specimens (n = 18) and moderately differentiated HCC tissue specimens (n = 18) respectively. NWD and TWD represented matched adjacent non-tumor tissue specimens (n = 14) and well differentiated HCC tissue specimens (n=14) respectively. (A) NOVA1; (B) NSMCE2; (C) KIAA0196; (D) AQP9. #P < 0.05 vs TMD and TWD.
DISCUSSION
We have applied the transcriptome sequencing approach to illustrate the gene expression characteristics of poorly differentiated HCC. The number of DEGs for poorly differentiated HCC is significantly bigger than that of other two stages. Those DEGs are enriched in liver tissue and cells, which is easily understood. Pathway analysis showed that two pathways, cell cycle and complement and coagulation cascades, were overrepresented with DEGs. Deregulation of the cell cycle pathway is expected since uncontrolled cell division is the major character of cancer cells. As for the complement and coagulation cascades pathway, consistent with our results, both gene expression [9] and proteomics [10] analysis have shown that this pathway is related to the pathogenesis of HCC. Some transcription factors were found to regulate the DEGs. For example, HNF4-α could represent a central regulator of gene transcription in hepatocytes, and a strong candidate to be involved in liver cancer cell development [11]. In addition, we also compared tumor tissues between PD HCC and MD&WD HCC. And similarly, the differentially expressed genes are enriched in the process of cell cycle and division.
After query of the poorly differentiated HCC signature in the LINCS server in our study, some compounds, knockdown genes and overexpression genes were found to have strong negative connections with the signature. PI-103 is a potent ATP-competitive dual inhibitor of phosphatidylinositol 3-kinase (PI3K) and mTOR (mammalian target of rapamycin). The combination of PI-103 and sorafenib was found to inhibit hepatocellular carcinoma cell proliferation by blocking Ras/Raf/MAPK and PI3K/AKT/mTOR pathways [12]. MK-2206 is an oral selective allosteric inhibitor of Akt that targets all three isoforms of human Akt (Akt-1, Akt-2 and Akt-3). MK2206 was found to inhibit hepatocellular carcinoma cellular proliferation via induction of apoptosis and cell cycle arrest [13]. Among 8 genetic perturbations, 5 genes are enriched in pathways in cancer or PI3K-Akt signaling pathway (p<0.001). Myc proto-oncogene protein (MYC) as a transcription factor can activate the transcription of growth-related genes. HCC is frequently associated with overexpression of MYC. In this study, MYC is up-regulated in the poorly differentiated HCC. Knockdown of oncogenic KRAS was found to suppress tumor growth in non-small cell lung cancers [14]. As mentioned, AKT2 is the target of MK2206, knockdown and inhibition of AKT2 may both help to reverse HCC. Overexpression of cyclin dependent kinase inhibitor 1B (CDKN1B) has the reverse effect in our study. CDKN1B binds to and prevents the activation of cyclin complexes, and thus controls the cell cycle progression at G1 [15]. All those perturbations may be the potential therapies or targets for treatment of HCC. In the validation cohort, further RT-PCR was performed to validate 4 DEGs specific for poorly differentiated HCC, of which NOVA1, NSMCE2 and KIAA0196 were significantly up-regulated, while AQP9 was significantly down-regulated in poorly differentiated samples, as compared with that in 9 adjacent non-tumor samples, 3 moderately differentiated samples and 3 well differentiated samples. The RT-PCR results showed consistency with RNA-seq. Interestingly, these DEGs were closely related with cell cycle or proliferation. High expression of NOVA1 was found correlated with poor survival rate and increased recurrence rate in HCC [16]. NSMCE2 was required for G1-S transition in breast cancer cells and manipulation of NSMCE2-mediated sumoylation may alter the growth rates of breast cancer cells [17]. KIAA0196, involving in meiosis-related spindle assembly [18], has been showed increased expression in clinical prostate carcinomas and also amplified in 30-40% of xenografts and hormone-refractory tumors [19]. However, AQP9 has been found to be down-regulated in hepatocellular carcinoma and its over-expression suppresses hepatoma cell invasion through inhibiting epithelial-to-mesenchymal transition [20]. In conclusion, this study explored the molecular mechanism of hepatocarcinogenesis through assessment of RNA seq data of HCC and validation of 4 DEGs specific for poorly differentiated HCC in an independent cohort. It provides useful information on the transcriptomic landscape as well as a mechanistic overview of HCC. Our findings offer novel insights and useful support in biomarker development and suggest new potential targets in poorly differentiated HCC characterization.
MATERIALS AND METHODS
Ethics statement
Our study design was approved by the Ethics Committee of the Fujian Provincial Hospital. Written informed consent was obtained from all subjects.
Subjects
47 subjects were diagnosed as primary HCC in the Fujian Provincial Hospital (Table 1), of which 33 subjects were present with cirrhosis on the non-neoplastic background, including 29 subjects with hepatitis B virus (HBV), 1 subjects with hepatitis C virus (HCV) and 3 subjects with NBNC. HBV related tumors were defined according to the presence of HB surface antigen (HBsAg) in serum, and HCV related tumors were according to the presence of antibody to HCV (HCVAb) in serum. NBNC tumor was defined according to the absence of both HBsAg and HCVAb in serum. Primary tumor and adjacent non-tumorous samples were obtained from all patients who underwent surgical tumor resection. All samples were frozen immediately at –80°C until RNA extraction. Total RNA was isolated by using RecoverAll™ Total Nucleic Acid Isolation Kit (Life Technologies, Carlsbad, CA, USA). Integrity of RNA was assessed by Agilent 2100 bioanalyzer (Agilent, Santa Clara, CA, USA). RNA from nine samples was subjected to sequencing and all samples were used in the validation experiments.
Transcriptome sequencing
Sequencing libraries were prepared by using TruSeq RNA Sample Prep Kit (Illumina, San Diego, CA, USA) according to standard protocols. Briefly, total RNA was firstly randomly fragmented and poly-A-selected. Secondly, the RNA fragments were reverse transcribed to cDNA, end-repaired and ligated with adapters. The libraries then underwent size selection, PCR and purification. The quality of libraries was assessed by using Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA). Sequencing was then performed on an Illumina HiSeq2500 sequencer with 125 bp pair-end reads.
Reads processing
Raw sequencing reads were firstly filtered for adapters and ribosomal RNA. Reads containing five or more low quality (quality score < 20) bases were also removed. The remained high-quality reads were then aligned to human genome (hg19) by using Tophat [21]. The mapped reads were then subjected to alignment against the human transcriptome (Ensembl, GRCh37.73). Gene expression level measured by FPKM (fragments per kilobase per million) was calculated by Cufflinks [22].
Differentially expressed genes (DEGs) analysis
For each differentiated HCC group, DEGs between the tumor and matched non-tumorous tissues were identified with pair-wise t test and the significant threshold was set as adjusted p-value of less than 0.05 and |log2(fold change, FC)| > = 1. Enrichr was used to do functional enrichment analysis like Gene Ontology (GO) and KEGG pathway [7]. The significant threshold for enrichment was set as p < 0.1. In addition, the list of up-regulated genes and down-regulated was uploaded into lincscloud [8] to find perturbations which can reverse the cancer condition. Using similar methods (t test instead of paired t test), DEGs were identified and analyzed between tumor tissues of PD HCC and those of MD&WD HCC.
DEGs specific for poorly differentiated HCC validated by RT-PCR
DEGs specific for poorly differentiated HCC (Compared with 3 matched adjacent non-tumor tissue of poorly differentiated HCC, P<0.05, |log2(fold change, FC)| > = 1; Compared with all adjacent non-tumor tissue, 3 moderately differentiated tissue and 3 well differentiated tissue, P<0.05, |log2(fold change, FC)| > = 1 ) were subjected to validation using RT-PCR. The validation cohort included 15 poorly differentiated HCC, 18 moderately differentiated HCC and 14 well differentiated HCC (Table 1). For the RT-PCR reactions, total RNA was converted to cDNA with random hexamer primers using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR was performed with SYBR Green I (Applied Biosystems, United States) on ABI 7300 (Applied Biosystems). The primers used were described in Table 6.
Table 6: Primers used in this study
Gene |
Primers (5′–3′) |
Length (bp) |
|---|---|---|
NOVA1 |
GACCAATACGGGCGAAGACG |
295 |
CTGGGGTTGTAGAATGCTGACTG |
||
NSMCE2 |
AGACCAACTTCACCTGCCCC |
131 |
CTTTTTCTTCCGCTTTTGCCTG |
||
KIAA0196 |
GAGGGAGGGGTGGAAACTGG |
208 |
ATTGTGAGGCGGACCGACTAC |
||
AQP9 |
TGGAGGGGTCATCACTATCAAT |
226 |
CATAAGTCCATCATAGTAAATGCCAAA |
Statistical analysis
T-test was used to compare continuous measurement data with a normal distribution between two groups. Mann-Whitney U test was used to evaluate continuous data with a non-normal distribution. Statistical analyses were performed with the SPSS13.0 software (SPSS, United States). P < 0.05 was considered statistically significant.
Abbreviations
HCC, hepatocellular carcinoma; PD, poorly differentiated; MD, moderately differentiated; WD, well differentiated; DEGs, differentially expressed genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PI3K, phosphatidylinositol 3-kinase; Akt, protein kinase B; mTOR, mammalian target of rapamycin; MAPK, mitogen activated kinase-like protein; MYC, Myc proto-oncogene protein; CDKN1B, cyclin dependent kinase inhibitor 1B; FPKM, fragments per kilobase per million; HBV, Hepatitis B virus; HBsAg, HB surface antigen; HCV, Hepatitis C virus; NBNC tumor, non-B non-C tumor; NOVA1, NOVA alternative splicing regulator 1; NSMCE2, NSE2/MMS21 homolog, SMC5-SMC6 complex SUMO ligase; AQP9, aquaporin 9.
Authors’ contributions
YH: guarantor of integrity of the entire study, study concepts, study design, definition of intellectual content, literature research, experimental studies, data acquisition, data analysis, statistical analysis, manuscript preparation, manuscript editing, manuscript review; JP: guarantor of integrity of the entire study, study concepts, study design, definition of intellectual content, literature research, experimental studies, data acquisition, data analysis, statistical analysis, manuscript preparation; DC: guarantor of integrity of the entire study, study concepts, study design, definition of intellectual content, literature research, experimental studies, data acquisition, data analysis, statistical analysis; JZ: experimental studies, data acquisition, data analysis, statistical analysis; FQ: experimental studies, data acquisition, data analysis; FL: experimental studies, data acquisition, data analysis; YW: data analysis, statistical analysis, manuscript preparation; Wenbing Wu experimental studies, data acquisition; XH: data analysis, statistical analysis; JQ: guarantor of integrity of the entire study, study concepts, study design, definition of intellectual content, literature research, manuscript review. YH and JP contributed equally to this study. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
We thank AIC biotechnology for their support in transcriptome sequencing and analysis.
CONFLICTS OF INTEREST
All authors declare that they have no any conflicts of interests.
FUNDING
This study was supported by the Middle-Young Age Backbone Talent Cultivation Program of Fujian Health System, No. 2013-ZQN-JC-2; and Key Projects of Science and Technology Plan of Fujian Province, No. 2014Y0009.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65:5–29.
2. Oishi K, Itamoto T, Amano H, Fukuda S, Ohdan H, Tashiro H, Shimamoto F, Asahara T. Clinicopathologic features of poorly differentiated hepatocellular carcinoma. J Surg Oncol. 2007; 95:311–316.
3. Arii S, Yamaoka Y, Futagawa S, Inoue K, Kobayashi K, Kojiro M, Makuuchi M, Nakamura Y, Okita K, Yamada R. Results of surgical and nonsurgical treatment for small-sized hepatocellular carcinomas: a retrospective and nationwide survey in Japan. The Liver Cancer Study Group of Japan. Hepatology. 2000; 32:1224–1229.
4. Simon EP, Freije CA, Farber BA, Lalazar G, Darcy DG, Honeyman JN, Chiaroni-Clarke R, Dill BD, Molina H, Bhanot UK, La Quaglia MP, Rosenberg BR, Simon SM. Transcriptomic characterization of fibrolamellar hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2015; 112: E5916-5925.
5. Lin KT, Shann YJ, Chau GY, Hsu CN, Huang CY. Identification of latent biomarkers in hepatocellular carcinoma by ultra-deep whole-transcriptome sequencing. Oncogene. 2014; 33:4786–4794.
6. Ho DW, Kai AK, Ng IO. TCGA whole-transcriptome sequencing data reveals significantly dysregulated genes and signaling pathways in hepatocellular carcinoma. Front Med. 2015; 9:322–330.
7. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016; 44: W90-97.
8. Duan Q, Flynn C, Niepel M, Hafner M, Muhlich JL, Fernandez NF, Rouillard AD, Tan CM, Chen EY, Golub TR, Sorger PK, Subramanian A, Ma’ayan A. LINCS Canvas Browser: interactive web app to query, browse and interrogate LINCS L1000 gene expression signatures. Nucleic Acids Res. 2014; 42: W449-460.
9. Zhang L, Guo Y, Li B, Qu J, Zang C, Li F, Wang Y, Pang H, Li S, Liu Q. Identification of biomarkers for hepatocellular carcinoma using network-based bioinformatics methods. Eur J Med Res. 2013; 18: 35.
10. Tsai TH, Song E, Zhu R, Di Poto C, Wang M, Luo Y, Varghese RS, Tadesse MG, Ziada DH, Desai CS, Shetty K, Mechref Y, Ressom HW. LC-MS/MS-based serum proteomics for identification of candidate biomarkers for hepatocellular carcinoma. Proteomics. 2015; 15:2369–2381.
11. Babeu JP, Boudreau F. Hepatocyte nuclear factor 4-alpha involvement in liver and intestinal inflammatory networks. World J Gastroenterol. 2014; 20:22–30.
12. Gedaly R, Angulo P, Hundley J, Daily MF, Chen C, Koch A, Evers BM. PI-103 and sorafenib inhibit hepatocellular carcinoma cell proliferation by blocking Ras/Raf/MAPK, PI3K/AKT/mTOR pathways. Anticancer Res. 2010; 30:4951–4958.
13. Wilson JM, Kunnimalaiyaan S, Gamblin TC, Kunnimalaiyaan M. MK2206 inhibits hepatocellular carcinoma cellular proliferation via induction of apoptosis and cell cycle arrest. J Surg Res. 2014; 191:280–285.
14. Sunaga N, Shames DS, Girard L, Peyton M, Larsen JE, Imai H, Soh J, Sato M, Yanagitani N, Kaira K, Xie Y, Gazdar AF, Mori M, et al. Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol Cancer Ther. 2011; 10:336–346.
15. Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013; 140:3079–3093.
16. Zhang YA, Zhu JM, Yin J, Tang WQ, Guo YM, Shen XZ, Liu TT. High expression of neuro-oncological ventral antigen 1 correlates with poor prognosis in hepatocellular carcinoma. PLoS One. 2014; 9: e90955.
17. Ni HJ, Chang YN, Kao PH, Chai SP, Hsieh YH, Wang DH, Fong JC. Depletion of SUMO ligase hMMS21 impairs G1 to S transition in MCF-7 breast cancer cells. Biochim Biophys Acta. 2012; 1820:1893–1900.
18. Wang F, Zhang L, Zhang GL, Wang ZB, Cui XS, Kim NH, Sun SC. WASH complex regulates Arp2/3 complex for actin-based polar body extrusion in mouse oocytes. Sci Rep. 2014; 4: 5596.
19. Porkka KP, Tammela TL, Vessella RL, Visakorpi T. RAD21 and KIAA0196 at 8q24 are amplified and overexpressed in prostate cancer. Genes Chromosomes Cancer. 2004; 39:1–10.
20. Zhang WG, Li CF, Liu M, Chen XF, Shuai K, Kong X, Lv L, Mei ZC. Aquaporin 9 is down-regulated in hepatocellular carcinoma and its over-expression suppresses hepatoma cell invasion through inhibiting epithelial-to-mesenchymal transition. Cancer Lett. 2016; 378:111–119.
21. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111.
22. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010; 28:511–515.