
INTRODUCTION
In vertebrates, postnatal hematopoiesis mainly occurs in the bone marrow (BM). Through a complex but precisely regulated differentiation process the hematopoietic stem cells (HSC) give rise to mature blood cells of all lineages. HSC niche-derived signals, originating from cells as diverse as the osteoblasts, endothelial cells, Cxcl12-abundant reticular (CAR) cells, Nestin+ mesenchymal stem cells etc., and a host of cell-intrinsic factors, such as Notch1, Runx1, GATA2, and SCL etc. play a key role in the maintenance of HSCs in the BM [1-10]. HSCs possess the unique traits of quiescence, and the ability to self-renew, which are absolutely critical for the general well-being and longevity of any organism as HSCs are the sole source for replenishment of all types of blood cells in case of demands that arise under various physiological and pathological conditions [11, 12].
Phenotypically HSCs are characterized as the lineage marker-negative (Lin-), stem cell antigen-1 (Sca1) and the stem cell factor receptor (SCFR or c-Kit) positive (Lin-Sca1+c-Kit+: LSK) cells in the BM. Various in vivo and in vitro analyses of LSK cells identified two distinct populations, the long- and the short-term repopulating stem cells (LT-HSC and ST-HSC). The LT-HSCs, which do not express the fetal liver kinase-2 (Flk2) on their surface, retain the ability for life-long self-renew and at the same time give rise to the ST-HSCs, whereas the Flk2 expressing ST-HSCs are capable of limited self-renewal and give rise to the multipotent progenitor cells for all blood lineages [13]. Maintenance of HSC quiescence is vital to prevent exhaustion and thereby for the integrity of the hematopoietic system [14, 15]. Cytokines and growth factors form an important class of regulatory molecules, which are indispensable for the maintenance of HSC quiescence [16-21]. Though, recent studies have identified many such cytokines, chemokines and other hematopoietic growth factors, a role for the primarily lymphocyte-specific growth factor interleukin-2 (IL-2) has not been identified.
In T cells, IL-2 binds to a heterotrimeric receptor consisting of the IL-2 receptor α (IL-2Rα, CD25), IL-2Rβ (CD122) and the common γ (CD132, γc) chain [22, 23]. Upon ligation, IL-2 activates the receptor bound tyrosine kinases Jak1 and Jak3, which in turn phosphorylate the signal transducer and activator of transcription 5 (STAT5) transcription factors (TF) [24-26]. Activated STAT5 subsequently regulate the expression of various target genes that executes IL-2 signals [27-29]. IL-2 is mainly produced by activated CD4+ and CD8+ T cells, and plays a critical role in the survival and proliferation of lymphocytes [22, 23, 30-33]. Recent studies have established the indispensable role of IL-2 in the maintenance of the CD4+CD25+Foxp3+ regulatory T (Treg) cells, which are vital to keep immune homeostasis in place [23, 34, 35]. Lack of IL-2 signaling results in the death of Treg cells leading to rapid development of various autoimmune disorders, such as lymphoproliferation, colitis etc., [36-39]. A similar phenotype has also been reported for mice deficient in Foxp3 or any component of IL-2 signaling pathway [30, 40]. In addition, humans having an inactivating mutation in the Foxp3 gene suffer from a severe autoimmune pathology called IPEX (Immunodysregulation Polyendocrinopathy Enteropathy X-linked) syndrome [41]. Thus, the role of IL-2 in T cell function and in maintaining immune homeostasis is well established.
We have recently reported that IL-2 plays a critical role in maintaining erythropoiesis by modulating Treg cell activity in the BM [42]. However, the influence of IL-2 on HSC generation or maintenance in the BM has not yet been investigated. In this study we have analysed whether lack of IL-2 signaling has any influence on hematopoiesis and report that the IL-2-Treg-Teff cell axis plays an indispensable role in maintaining steady state HSC population in the BM. Deficiency in IL-2 signaling results in an IFN-γ-mediated disruption of equilibrium in HSC physiology leading to impaired hematopoiesis, which might be a major contributing factor towards the severe phenotype observed in all IL-2 signaling deficient mice, and most likely also in humans.
RESULTS
Impaired HSC maintenance in Il2-/- mice
To investigate whether IL-2 deficiency affects hematopoiesis, we first of all analysed the distribution of HSCs in Il2-/- mice. Evaluation of BM cells revealed a strong increase in LSK population in Il2-/- mice compared to WT mice (Figure 1A). The increased LSK population in Il2-/- mice was gender-independent and was observed both in proportion (Figure 1B) and in absolute numbers (Figure 1C). Interestingly, though the Il2-/- LSK cells were slightly bigger in size (Figure 1D, 1F), they showed a markedly higher granularity compared to WT LSK cells (Figure 1E, 1F). We also observed a consistently strong increase in Lin-Sca1+c-Kit- (Sca1+) cells in Il2-/- mice, whereas the distribution of Lin-Sca1-c-Kit+ (c-Kit+) cells was similar compared to WT mice (Figure 1A). However, the increase in LSK cells in Il2-/- mice was independent of the impairment in Sca1+ population in these mice. Compared to LSK cells, Il2-/- Sca1+ and c-Kit+ cells showed distinct patterns in their cell size and granularity, comparable with that of WT mice (Figure 1G, 1H and Supplementary Figure 1A, B). In addition, HSC analysis in BM using CD48 and CD150 as markers also showed an increase in HSC (Lin-c-Kit+CD48-CD150+) population in the Il2-/- mice compared to WT mice (Figure 1I). This further ruled out any possible contamination from cells expressing Sca1 in the perturbed HSC (LSK) population in Il2-/- mice. Similar to BM, impaired HSC distribution was also observed in the spleen of Il2-/- mice suggesting a systemic defect in HSC maintenance in the absence of IL-2 signaling (Supplementary Figure 1C, D).
Analysis of mature hematopoietic cell populations in WT and Il2-/- mice revealed a drastically altered distribution of lymphoid (CD3+, B220+ and NK1.1+), myeloid (CD11b+, CD11c+, Gr1+ and F4/80+) and erythroid (Ter119+ and CD41+) lineage cells in Il2-/- mice BM (Supplementary Figure 1E). In general, in Il2-/- mice myelopoiesis was strongly increased over the impaired lympho- and erythropoiesis, indicating a myeloid bias of the Il2-/- HSC compared to WT controls (Supplementary Figure 1E). This myeloid-biased hematopoiesis was supported by an increased expression of myeloid-specific genes (Cebpa, Csf3r, Csf2ra, Csf1r and Il3ra) in the Il2-/- mice (Figure 1J). To dissect this severely perturbed hematopoiesis, we next investigated whether the distribution of the LT- and the ST-HSCs are altered in the context of IL-2 deficiency. Intriguingly, analysis of Flk2 expression revealed a significant increase in Flk2- LT-HSC and a corresponding decrease in the Flk2+ ST-HSC in Il2-/- mice compared to that in WT mice (Figure 1K, 1L). Moreover, the defective hematopoiesis in absence of IL-2 signaling was readily evident from the forward (FSC) and side scatter (SSC) distribution of the BM cells as a clear loss of FSCloSSClo and an increase in FSChiSSChi cells was observed in Il2-/- mice compared to the WT mice (Figure 1M, 1N). The impairment in Il2-/- HSC was further confirmed as we observed a suppressed Flk2 and an enhanced Slamf1 (Cd150) expression in the LSK cells from Il2-/- mice, which was exactly opposite in the WT LSK cells (Figure 1O). Previous studies have implicated the criticality of Notch signaling and the Runx TFs in the generation and proliferation of HSC [7]. Interestingly, a strong increase in Notch (Notch1 and Notch2) and Runx1 expression was evident in the Il2-/- LSK cells suggesting that enhanced Notch-Runx1 activity could be the factor promoting dysregulated HSC maintenance in Il2-/- mice (Figure 1P). Additionally, in Il2-/- mice the expression of Scl, Cxcr4 and cMpl, the key signalling molecules regulating HSC maintenance were also altered [9, 10, 18, 43-46] (Figure 1P). Further, analysis of TFs involved in HSC quiescence revealed a dose-dependent increase in their expression in Il2-/- HSCs [47-56] compared to WT cells (Figure 1Q). Interestingly, the Il2-/- LSK cells expressed higher levels of Bcl2 and Bcl2l1, indicating that the dysregulated hematopoiesis in Il2-/- mice was not due to the death of HSCs (Figure 1R). Also, in line with the increased number of LSK cells, expression of the cell cycle inhibitor p27kip1 (Cdkn1b) was strongly downregulated and genes regulating HSC expansion were enhanced in the Il2-/- mice (Figure 1R).
Next, we investigated whether the HSC defects observed in Il2-/- mice are replicated in all mice that lack IL-2 signaling. Analysis of Il2ra-/- or Jak3-/- mice showed a similar increase in LSK cells, and a loss of FSCloSSClo BM cells with corresponding increase in FSChiSSChi cells as was observed in Il2-/- mice suggesting a specific influence of lack of IL-2 signaling on HSC maintenance (Supplementary Figure 1F-K).
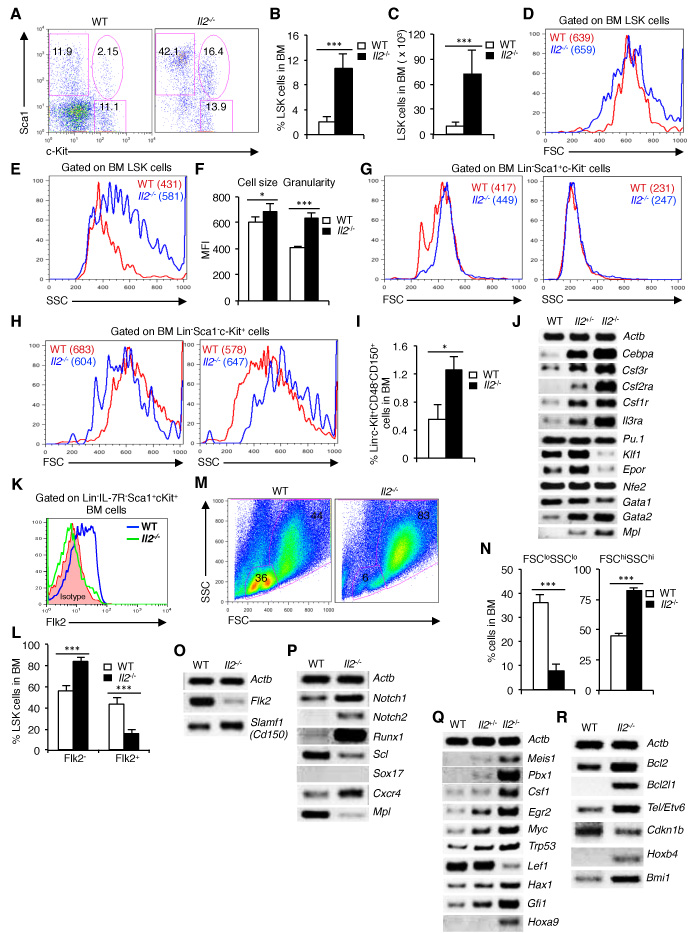
Figure 1: Impaired maintenance of HSCs in Il2-/- mice BM. A. Flow cytometry analysis of BM cells for LSK population in WT and Il2-/- mice. B. Proportion of LSK cells in the BM of WT and Il2-/- mice (n = 20 per group). C. LSK cells distribution in absolute numbers in WT and Il2-/- mice (n = 20 per group). D. Cell size of WT and Il2-/- HSCs as reflected by their FSC distribution pattern. E. SSC distribution pattern of WT and Il2-/- HSCs. F. Mean cell size and granularity of WT and Il2-/- HSCs. G. FSC and SSC profiles of BM Lin-Sca1+c-Kit- cells from WT and Il2-/- mice. H. Profiles of WT and Il2-/- BM Lin-Sca1-c-Kit+ cells according to their FSC and SSC distribution pattern. I. Proportion of Lin-c-Kit+CD48-CD150+ cell population in the BM from WT and Il2-/- mice (n = 5 per group). J. Myeloid, lymphoid and erythroid lineage-specific genes expression in WT, Il2+/- and Il2-/- mice. K. Flow cytometry revealing the distribution of LT-HSCs and ST-HSCs among LSK cells in indicated mice (n = 5 for WT and 4 for Il2-/- mice). L. Population distribution of LT- and ST-HSC in the BM of WT and Il2-/- mice based on Flk2 expression (n = 7 per group). M. Flow cytometry profiles of BM cells in WT and Il2-/- mice according to their FSC and SSC distribution pattern (n = 20 per group). N. Quantification of FSCloSSClo and FSChiSSChi cells in WT and Il2-/- mice. O. RT-PCR analysis of LT- and ST-HSC-specific genes expression in sorted LSK cells from WT and Il2-/- mice. P. Semi-quantitative RT-PCR on WT and Il2-/- LSK cells for critical genes involved in HSC maintenance. Q. Gene expression analysis of TFs and signaling molecules in LSK cells from WT, Il2+/- and Il2-/- mice. R. Cell survival and HSC proliferation-specific genes expression in LSK cells from WT, Il2+/- and Il2-/- mice. Numbers inside each dot plot represent percent respective population, and in histograms represent the mean fluorescence intensity (MFI). Data are representative of 2-5 independent experiments and shown as mean ± s.d., in (B, C & N) ***P < 0.0001, in (F) *P = 0.0389, ***P = 0.0001, in (I) *P = 0.0348, and in (L) ***P = 0.0002, unpaired t-test.
IL-2 signaling influences hematopoiesis
To explore if IL-2 signals influence HSC maintenance, we adoptively transferred CD45.1+ WT BM cells to lethally irradiated CD45.2+ WT or Il2-/- mice and analysed donor-derived cells-specific hematopoiesis in the recipient mice. Analysis of LSK cells in the BM 5 weeks post-transfer revealed an increase in donor-derived LSK cells in the Il2-/- recipients compared to the WT recipients replicating a phenotype observed in Il2-/- mice (Figure 2A). This pattern persisted even at 11 weeks after BM cell transfer (Figure 2A), suggesting a disturbed HSC maintenance in an IL-2-deficient environment. In addition, similar to Il2-/- mice, Il2-/- recipients showed increased Sca1+ cells in the BM both at 5 and 11 weeks after cell transfer (Supplementary Figure 2A). Further, the thymus, spleen and BM cellularity in the Il2-/- recipients was severely reduced compared to WT recipients indicating an inefficient hematopoietic reconstitution in the Il2-/- recipients despite having higher number of HSCs (Figure 2B). At 5 weeks, the Il2-/- recipients were consistently underweight (Figure 2C), and their spleens were smaller in size (Figure 2D) compared to WT recipients. Analysis of lineage-positive cells in lymphoid organs demonstrated a marked defect in lymphopoiesis and erythropoiesis, and replicated the myeloid bias of HSCs in an IL-2-deficient environment similar to that observed in the Il2-/- mice (Figure 2E). Though, the thymic, splenic and BM cellularity improved at 11 weeks after transfer, the Il2-/- recipients still remained underweight (Supplementary Figure 2B, C). However, the increase in cellularity was not due to a normal hematopoiesis rather it was the outcome of the dysregulated hematopoiesis as shown in Figure 2E, with myeloid cells being the major population in these organs. Also, due to impaired hematopoiesis splenomegaly was observed at 11 weeks after cell transfer in all Il2-/- recipients (Supplementary Figure 2D).
To further clarify the role of IL-2 in HSC maintenance and lineage differentiation, we transferred CD45.2+ WT or Il2-/- mice BM cells to lethally irradiated CD45.1+ WT hosts. Analysis at 4 weeks post-transfer again revealed an increased LSK cell population in the Il2-/- BM transferred WT recipients compared to the WT BM transferred mice (Figure 2F). At this time point an increased Sca1+ population, characteristic of the Il2-/- mice was also observed in the Il2-/- BM transferred WT recipients (Supplementary Figure 2E). Analysis of cellularity in the lymphoid organs revealed that Il2-/- BM transferred WT recipients had strongly reduced thymocyte numbers, whereas spleen and BM cell numbers were comparable with that in WT BM transferred WT recipients (Figure 2G). However, again, the comparable cellularity in spleen and BM was due to impaired hematopoiesis, as all Il2-/- BM transferred WT recipients showed significantly enlarged spleen (Figure 2H), and in the BM, myelopoiesis was enhanced over lympho- and erythropoiesis (Figure 2I). This was significant as in an IL-2-sufficient environment even with increased number of LSK cells, Il2-/- BM cells failed to efficiently reconstitute the hematopoietic system.
Interestingly, 13 weeks after BM transfer the HSC population in the Il2-/- BM transferred WT recipients were similar to that in the WT BM transferred WT recipients (Figure 2F), which in turn gave rise to efficient hematopoietic reconstitution resulting in comparable cellularity, normal body weight as well as spleen size (Supplementary Figure 2F-H). Also, the myeloid-biased hematopoiesis in IL-2-deficient background was largely reversed (Supplementary Figure 2I). The contrasting observations at an early (4 weeks) compared to a late time point (13 weeks) after BM cell transfer suggests that the reconstitution capacity of the Il2-/- HSCs is significantly impaired relative to the WT HSCs and require prolonged sensitization in an IL-2-sufficient environment in order to promote normal hematopoiesis. Altogether, both sets of experiments underline the critical requirement of IL-2 signaling for HSC maintenance and normal hematopoiesis.
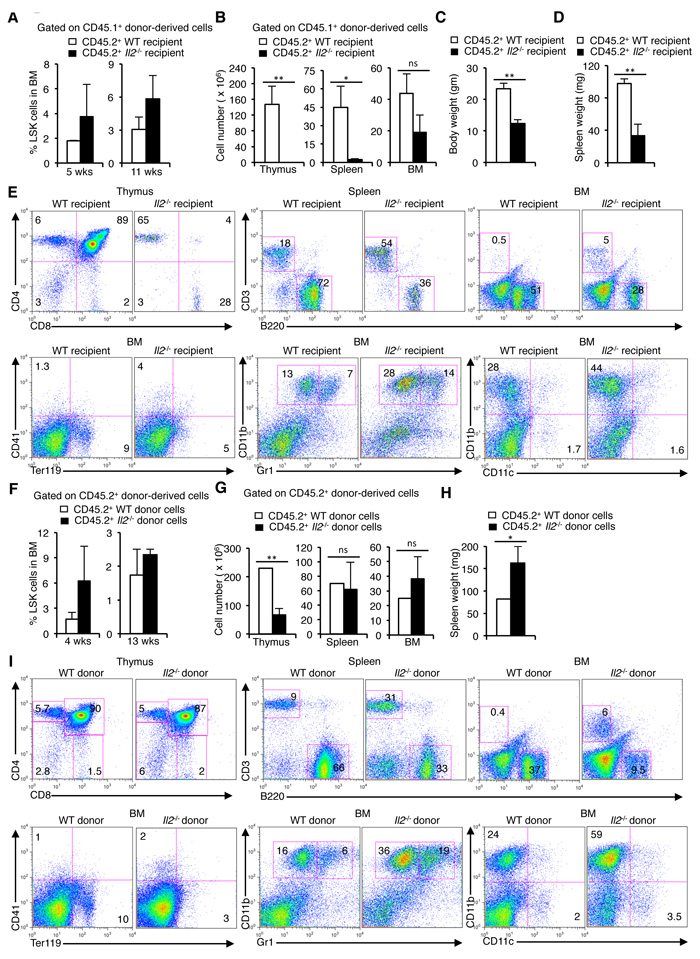
Figure 2: HSC maintenance in BM depends on IL-2 signaling. A. Enumeration of CD45.1+ donor-derived LSK cells distribution 5 and 11 weeks post-transfer in the BM of irradiated CD45.2+ WT or Il2-/- recipient mice. B. Number of CD45.1+ donor-derived cells in the thymus, spleen and BM of irradiated CD45.2+ WT or Il2-/- recipient mice 5 weeks after transfer. C. Evaluation of body weight, and D. spleen weight of the recipient mice 5 weeks after cell transfer. E. Flow cytometry profiles of the distribution of various donor-derived mature hematopoietic cell populations in the thymus, spleen and BM of recipient mice. Data are representative of two independent experiments (n = 4 per group/experiment). F. Distribution of CD45.2+ WT or Il2-/- donor-derived LSK cells distribution 4 and 13 weeks post-transfer in the BM of irradiated CD45.1+ WT recipient mice. G. Number of CD45.2+ WT or Il2-/- donor-derived cells in the Thymus, spleen and BM of irradiated CD45.1+ WT recipient mice 4 weeks after transfer. H. Spleen weight of the recipient mice 4 weeks after cell transfer. I. Flow cytometry profiles of the distribution of various CD45.2+ WT or Il2-/- donor-derived mature hematopoietic cell populations in the thymus, spleen and BM of CD45.1+ WT recipient mice. Data are representative of two independent experiments (n = 4 per group/experiment). Numbers inside each FACS plot represent percent respective population. Data are shown as mean ± s.d., in (B) **P = 0.0090, *P = 0.0176, (C) **P = 0.0034, (D) **P = 0.0096, (G) **P = 0.0028, (H) *P = 0.0198, and ns = not significant, unpaired t-test.
Hematopoietic defects in Il2-/- mice are T cell-mediated
To show that IL-2 signaling directly influences HSC maintenance, we treated Il2-/- mice with exogenous IL-2. Surprisingly, IL-2 treatment significantly reversed the altered pattern in HSC distribution in Il2-/- mice, which was now comparable to that in WT mice (Figure 3A, 3B). IL-2 treatment restored the missing FSCloSSClo population in the BM (Figure 3C), and eventually led to resolving the defective hematopoiesis in Il2-/- mice (Supplementary Figure 3A). Also, IL-2 treatment reversed the distribution of HSC subpopulations, as it enhanced Flk2 and reduced Slamf1 expression in Il2-/- LSK cells (Figure 3D). These data show that IL-2 signals exert a beneficial influence on HSC maintenance that is essential for normal hematopoiesis.
Next, to investigate the basis of this strong effect of IL-2 signaling on HSC maintenance, we hypothesized that the massive T-lymphocyte proliferation reported in Il2-/- mice could in some way be responsible for the hematopoietic defects in these mice [30, 57]. To verify this issue, we analysed Il2-/-Il7ra-/- mice in which lymphoproliferation was effectively prevented [42]. However, analysis of LSK cells in these mice showed, the persistence of HSC defects both in the BM and in spleen as was in the Il2-/- mice (Figure 3E, 3F and Supplementary Figure 3B, C). To resolve this intriguing observation we hypothesized that most likely the residual T-lymphocytes do not function normally in the absence of IL-2 signaling and thereby induced the HSC defects in the Il2-/-Il7ra-/- mice. To investigate this possibility, we bred Il2-/- mice with Rag1-/- mice in order to abolish all T cells. Surprisingly, the Il2-/-Rag1-/- mice demonstrated a normal LSK population both in the BM (Figure 3G, 3H) and in the spleen (Supplementary Figure 3D, E), as well as a normal hematopoiesis in contrast to the Il2-/- mice. These observations suggest that the defective HSC maintenance and dysregulated hematopoiesis in the Il2-/- mice are to a large part mediated by T cells.
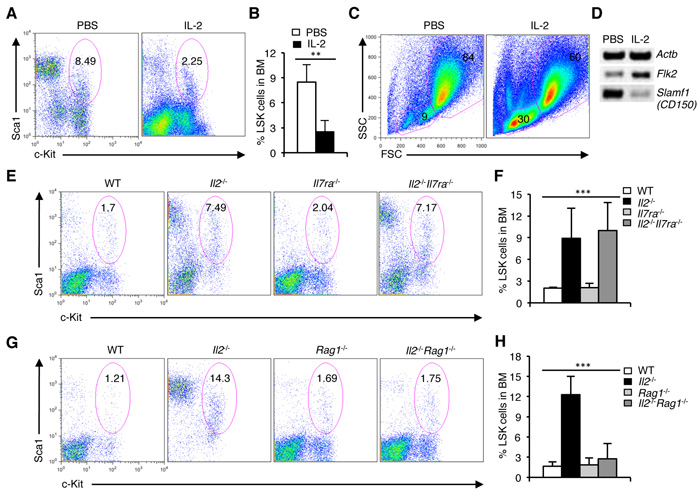
Figure 3: Defective HSC maintenance in Il2-/- mice is T cell-mediated. A. Distribution of LSK cells in the BM of PBS or IL-2 treated Il2-/- mice. B. Proportion of LSK cells in the BM of Il2-/- mice treated either with PBS or IL-2. C. FSC/SSC distribution of BM cells from PBS or IL-2 treated Il2-/- mice. D. RT-PCR analysis of Flk2 and Slamf1 expression in sorted BM LSK cells from Il2-/- mice treated either with PBS or IL-2. E. Flow cytometry analysis of LSK cells distribution in the BM of WT, Il2-/-, Il7ra-/- and Il2-/-Il7ra-/- mice. F. Quantification of the distribution of BM LSK population in indicated mice. G. Distribution of LSK cells in the BM of WT, Il2-/-, Rag1-/- and Il2-/-Rag1-/- mice. H. Proportion of LSK cells in BM cells from WT, Il2-/-, Rag1-/- and Il2-/-Rag1-/- mice. Numbers inside each FACS plot represent percent respective population. Data are representative of 3 independent experiments, (n = 4 per group) and shown as mean ± s.d., in (F) ***P = 0.0004 and (H) ***P < 0.0001, one-way ANOVA.
Treg cell activity is indispensable for HSC integrity
The persistence of HSC defects in the Il2-/-Il7ra-/- but not in Il2-/-Rag1-/- mice raised the question of how T cell activity could influence hematopoiesis in the BM. IL-2 signaling has been reported to be indispensable for Treg cell survival and accordingly, Il2-/- mice show a strongly reduced Treg cell population (Supplementary Figure 4A). We have shown previously that in the Il2-/-Il7ra-/- mice Treg cell population is also drastically reduced [42]. To prove that the lack of adequate number of Treg cells in the Il2-/-Il7ra-/- and Il2-/- mice influences HSC maintenance and hematopoiesis, we adopted an inducible Treg cell depletion model [58]. Treatment of DEREG mice with diphtheria toxin (DT) rapidly depleted Treg cells in these mice creating a situation similar to the Il2-/- mice (Supplementary Figure 4B). Analysis of LSK cells in the DT-treated DEREG mice revealed a strong increase compared to the control PBS-treated mice (Figure 4A, 4B). Due to defective hematopoiesis in DT-treated DEREG mice cellularity in BM and peripheral lymphoid organs were enhanced compared to the control mice (Figure 4C). Similar to the Il2-/- mice, DT-treated mice, consistently lost body weight (Figure 4D), and showed enlarged spleen (Figure 4E) very quickly after DT treatment indicating overall defects in hematopoiesis following depletion of Treg cells. These defects were readily evident in the BM as a strong reduction in FSCloSSClo cells and an increase in FSChiSSChi cells was observed in DT-treated animals compared to the control animals (Figure 4F, 4G). Ablation of Treg cell activity also enhanced the LSK population in the spleen further resembling the hematopoietic defects observed in the Il2-/- mice (Supplementary Figure 4C, D). Analysis of lineage-positive cells in the BM further supported the critical role of Treg cells in hematopoiesis as lack of Treg cell activity in the DT-treated DEREG mice promoted myelopoiesis (Supplementary Figure 4E-H), an outcome similar to that in the Il2-/- mice. These observations suggest that in the absence of Treg cells most likely the activated effector T cells (Teff) activity was responsible for the hematopoietic defects observed in the IL-2 signaling-deficient mice.
To further consolidate our observation regarding the essentiality of Treg activity in HSC maintenance, we adoptively transferred Treg-depleted CD4+ Teff cells to Rag1-/- mice. In contrast to control mice, Teff cells transferred Rag1-/- mice rapidly developed a hematopoietic phenotype similar to that of the Il2-/- mice. A strong increase in the BM LSK population (Figure 4H, 4I), concomitant with a decrease in FSCloSSClo BM cells (Figure 4J) reflected the severity of defects in HSC maintenance and hematopoiesis when Treg cell activity was lacking.
Next we investigated whether restoration of Treg cell activity can reverse the defects in HSC maintenance and facilitate normal hematopoiesis in the Il2-/- mice. Analysis of Il2-/-Bim-/- mice in which Bim deficiency completely restored Treg population [42], showed normal hematopoiesis even if IL-2 signaling was still absent. In contrast to the Il2-/- mice, LSK population in BM (Figure 4K, 4L), and spleen (Supplementary Figure 5A, B) of the Il2-/-Bim-/- mice was comparable to that of the control mice. As an immediate sign of normal hematopoiesis, the FSC/SSC distribution of BM cells in Il2-/-Bim-/- mice showed normal pattern compared to the drastically reduced FSCloSSClo BM cells in the Il2-/- mice (Supplementary Figure 5C). Also, the spleen size (Figure 4M), and the distribution of lineage-positive cells in the Il2-/-Bim-/- mice were comparable with that of control mice (Supplementary Figure 5D). Thus, our analysis shows that the lack of Treg cell activity is detrimental for HSC maintenance and a Treg-Teff homeostasis is essential for normal hematopoiesis.
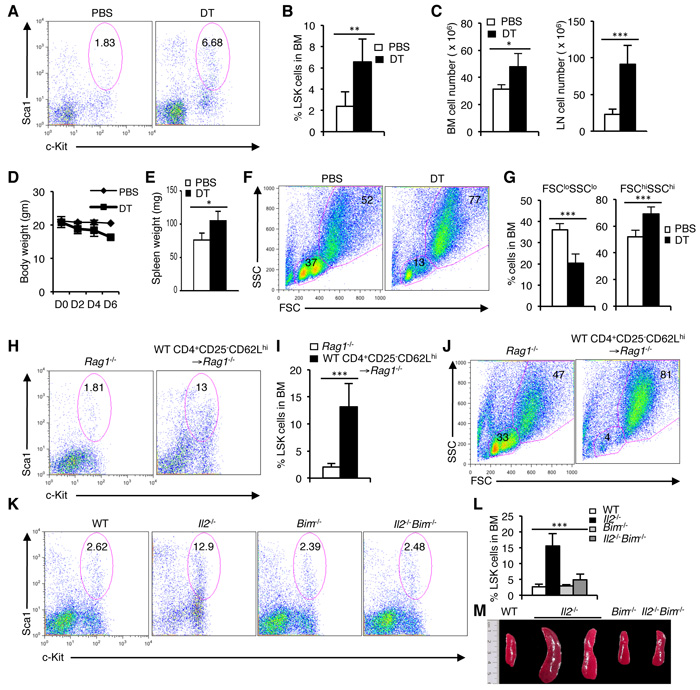
Figure 4: Treg cell activity is critical for steady state hematopoiesis. A. LSK cells distribution in the BM of DEREG mice treated with PBS or diphtheria toxin (DT). B. Quantification of the LSK population distribution in DEREG mice BM after PBS or DT treatment. C. Cellularity in the BM and LNs of DEREG mice following PBS or DT treatment. D. Evaluation of body weight following each PBS or DT injection to the DEREG mice. E. Spleen weight of DEREG mice treated with PBS or DT. F. FSC/SSC distribution of BM cells from PBS or DT treated DEREG mice. G. Evaluation of FSCloSSClo and FSChiSSChi BM cells in PBS or DT treated DEREG mice. H. Distribution of LSK population in the BM of Rag1-/- mice, 3 weeks after transfer of WT CD4+CD25-CD62Lhi cells, compared to normal WT or Rag1-/- mice. I. Quantification of BM LSK cells in Rag1-/- mice, 3 weeks post-transfer of WT CD4+CD25-CD62Lhi cells in comparison to control Rag1-/- mice. J. FSC/SSC distribution of BM cells from WT CD4+CD25-CD62Lhi cells transferred Rag1-/- mice compared to control Rag1-/- mice. K. Flow cytometry profiles of LSK cells in BM of WT, Il2-/-, Bim-/- and Il2-/-Bim-/- mice. L. Quantification of the distribution of BM LSK population in indicated mice. M. Photograph of spleens from WT, Il2-/-, Bim-/- and Il2-/-Bim-/- mice. Numbers inside each FACS plot represent percent respective population. Data are shown as mean ± s.d., in (B) **P = 0.0043, (C) *P = 0.0113 and ***P = 0.0005, (E) *P = 0.01734, (G) ***P = 0.0003, ***P = 0.0008, (I) ***P = 0.0006, and in (L) ***P < 0.0001 unpaired t-test. Data are representative of 3 independent experiments, (n = 4 per group).
Enhanced Teff cell activity impairs HSC maintenance
As in steady state Treg cells keep Teff cell activity under control and avoid unnecessary T cell activation, we next investigated the phenotypes of T cells in the Il2-/- mice. Similar to an earlier report [42], the majority of CD4+ T cells in the BM of Il2-/- mice were of CD62L-CD44- activated effector phenotype in contrast to the prevalence of CD62L-CD44+ effector memory cells in WT mice (Figure 5A). These CD4+CD62L-CD44- T cells in the BM of Il2-/- mice were smaller in size (Figure 5B) and much less granular (Figure 5C) compared to the WT cells.
To prove our assumption that activated CD4+ T cells can dysregulate HSC maintenance in the BM, we co-cultured WT BM cells with unstimulated, or PMA plus ionomycin stimulated WT CD4+ T cells (Supplementary Figure 6A), in presence or absence of IL-2 on WT BM stromal cell layer (Figure 5D). Interestingly, co-culture of BM cells with activated CD4+ T cells showed an increase in the proportion of LSK cells, which was further enhanced in the presence of IL-2 (Figure 5E, 5F) compared to the medium controls. Even IL-2 treatment alone, or in combination with unstimulated CD4+ T cells showed a significant increase in LSK cells (Figure 5E, 5F). Additionally, co-culture with activated CD4+ T cells also showed a strong increase in the Sca1+ cells similar to the phenotype in Il2-/- mice, which was further increased in presence of IL-2 (Figure 5E, 5G). However, under each culture condition these anomalies in LSK and Sca1+ cells hardly affected the distribution of c-Kit+ cells (Figure 5E, 5H) suggesting that the increase in LSK cells is not due to a mere redistribution of Lin-c-Kit+ and Lin-Sca1+ cells.
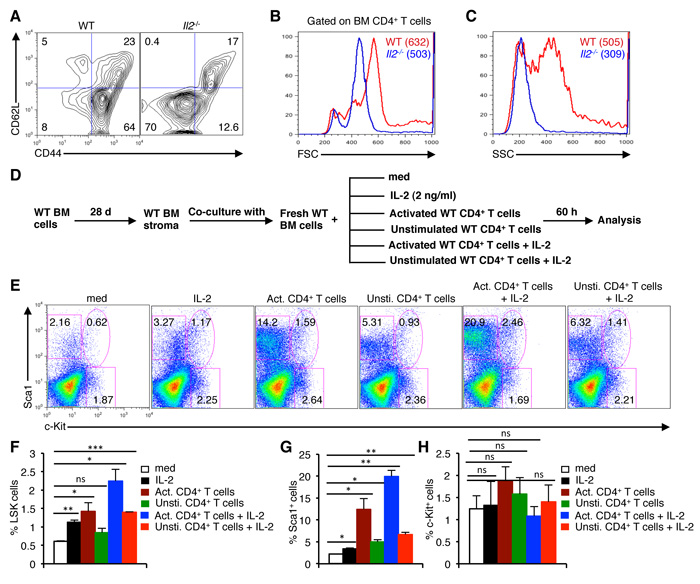
Figure 5: Activated T cells induce HSC defects in the BM. A. Distribution of CD4+ T cells in the BM of Il2-/- mice based on CD62L and CD44 expression compared to WT mice. B. Profiles of FSC distribution, and C. SSC distribution of the BM CD4+ T cells in WT and Il2-/- mice. D. Experimental plan to analyze the involvement of activated CD4+ T cells in BM HSC maintenance. E. Flow cytometry profiles showing the influence of IL-2, of naïve or activated CD4+ T cells on the maintenance of HSCs in the BM cells-stromal cells co-culture assays. F. Quantification of LSK cells in the respective BM cells-stromal cells co-culture assays as indicated. G. Distribution of BM Lin-Sca1+, and H. Lin-c-Kit+ cells as evaluated from the BM cells-stromal cells co-culture assays in respective conditions as indicated. Numbers inside each dot plot represent percent respective population. Data are representative of 3 independent experiments, (n = 3 per group) and shown as mean ± s.d., in (F) **P = 0.0061, *P = 0.0185 or 0.0185 and ***P = 0.0002, (G) *P = 0.0135 or 0.0279 or 0.0143 and **P = 0.0029 or 0.0067, ns = not significant, one-way ANOVA.
IL-10-independent defects in HSC maintenance in Il2-/- mice
IL-10, produced by the Treg cells is a key player in the Treg-mediated immune suppression [59-61]. Deficiency of IL-10 has been shown to induce anemia and other hematopoietic anomalies [62]. Therefore, we hypothesized that IL-10 deficiency in the Il2-/- mice might contribute to the HSC defects as they have a strongly reduced Treg population (Supplementary Figure 4A). To check if IL-10 influences HSC maintenance, we analysed Cd4-CreIl10fl/fl mice in which IL-10 production was ablated not only in Treg cells, but also in all T cells. Surprisingly, LSK cells distribution was unaffected in presence, or in T cell-specific loss of IL-10 activity (Figure 6A, 6B). A slightly decreased distribution of FSCloSSClo BM cells (Figure 6C), and an unaffected Lin-c-Kit+ or Sca1+ cells was observed in the Cd4-CreIl10fl/fl mice (Figure 6D). This was contrary to a recent report implicating IL-10 in maintaining HSC homeostasis [63]. To disprove the notion that IL-10 produced by other hematopoietic cells most likely compensated for the loss of IL-10 production by T cells in the Cd4-CreIl10fl/fl mice, we ablated IL-10 production in all hematopoietic cells (Vav-CreIl10fl/fl mice). Analysis of Vav-CreIl10fl/fl mice showed a slight increase in BM LSK cells (Figure 6E, 6F), and an insignificant change in the distribution of BM FSCloSSClo and FSChiSSChi cells (Figure 6G). Also, the distribution of BM Lin-c-Kit+ or Sca1+ cells was unaffected in Vav-CreIl10fl/fl mice compared to the WT controls (Figure 6H). The non-involvement of IL-10 in HSC maintenance was also clear as we observed a strong increase in IL-10 production in the Il2-/- T cells (Figure 6I). Thus, despite having increased level of IL-10, HSC maintenance was drastically altered in the Il2-/- mice.
To investigate what other factors produced by the activated T cells in the Il2-/- mice could be involved in the hematopoietic abnormality, we analysed the expression pattern of several cytokines in these T cells. A strong increase in Ifng expression in addition to slightly increased expression of Il4, Il12b, Il23a and Tnfa was clearly evident in Il2-/- mice compared to WT controls (Figure 6I). Compared to WT mice, upon stimulation, a huge increase in IFN-γ producing cells was observed in the Il2-/- mice (Figure 6J). These observations suggest that an enhanced IFN-γ activity might be involved in the dysregulated hematopoiesis in Il2-/- mice.
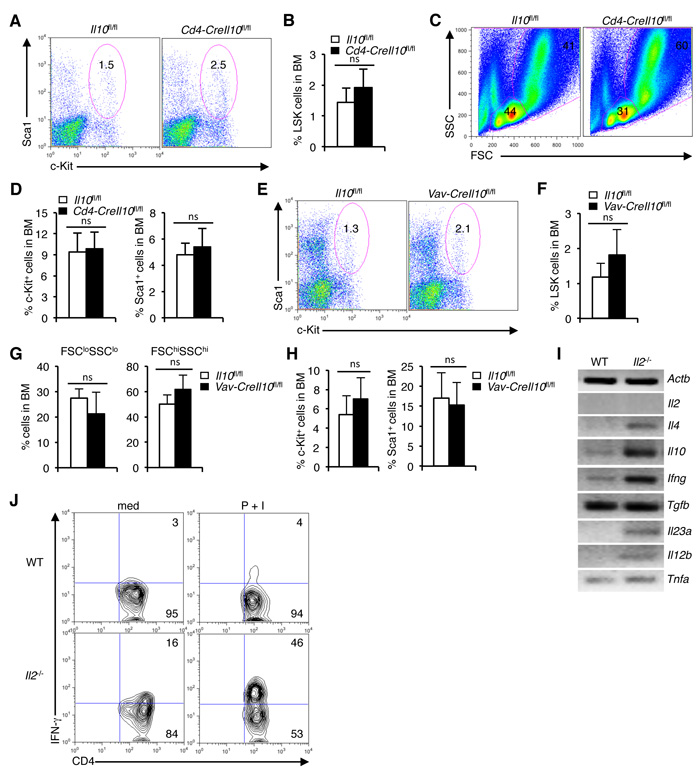
Figure 6: Dysregulated hematopoiesis in Il2-/- mice is IL-10-independent. A. Flow cytometry revealing the distribution of LSK cells in Cd4-CreIl10fl/fl mice compared to control mice. B. Proportion of LSK cells in the BM of Cd4-CreIl10fl/fl and Il10fl/fl mice. C. Profiles of BM cells in Cd4-CreIl10fl/fl mice compared to control mice according to their FSC/SSC distribution pattern. D. Evaluation of c-Kit+ and Sca1+ cells distribution among lineage-negative BM cells in Cd4-CreIl10fl/fl and Il10fl/fl mice. E. BM LSK cells distribution in Vav-CreIl10fl/fl mice compared to littermate control mice. F. Analysis of the proportion of BM LSK cells in Vav-CreIl10fl/fl and Il10fl/fl mice. G. Evaluation of FSCloSSClo and FSChiSSChi cells in the BM of Vav-CreIl10fl/fl mice compared to Il10fl/fl mice. H. Proportion of c-Kit+ and Sca1+ cells distribution among lineage-negative BM cells in Vav-CreIl10fl/fl and Il10fl/fl mice. I. Semi-quantitative RT-PCR analysis of cytokine gene expression in Il2-/- T cells compared to WT cells. J. Flow cytometry analysis of IFN-γ production by unstimulated or P+I stimulated CD4+ T cells in WT and Il2-/- mice. Numbers inside each dot plot represent percent respective population and in histograms represent the MFI. Data are representative of 3 independent experiments, (n = 3 per group) and shown as mean ± s.d., ns = not significant, one-way ANOVA.
Loss of IFN-γ activity rescues HSC defects in Il2-/- mice
To explore if IFN-γ is indeed involved in the dysregulated hematopoiesis, and whether by abolishing IFN-γ activity HSC maintenance could be restored in the Il2-/- mice, we analysed the Il2-/-Ifng-/- mice. In stark contrast to the Il2-/- mice, LSK cells distribution in the Il2-/-Ifng-/- mice was comparable with that of the littermate controls (Figure 7A, 7B). As a result, HSC maintenance was normal in the Il2-/-Ifng-/- mice and the BM regained its usual distribution of FSCloSSClo and FSChiSSChi cells (Figure 7C). IFN-γ deficiency in the Il2-/-Ifng-/- mice also reversed the high Sca1+ population observed in the Il2-/- mice to WT levels without significantly affecting the c-Kit+ population (Figure 7D).
Abolition of IFN-γ activity not only restored the distribution of the LSK cells, but it also reversed the highly granular nature of Il2-/- HSCs to that of WT levels in Il2-/-Ifng-/- mice (Figure 7E). IFN-γ activity seems to be specifically influencing the granularity of HSCs, as the cell size was relatively unaffected in presence or absence of it (Figure 7E). In contrast, IFN-γ deficiency did neither influence cell size nor the granularity of Lin-Sca1+ or c-Kit+ cells (Supplementary Figure 6B, C). The beneficial effects of IFN-γ deficiency on HSCs in the Il2-/- mice were comparable to that observed in case of Il2-/-Bim-/- mice. Similar to the Il2-/-Ifng-/- mice, LSK cells in Il2-/-Bim-/- mice reverted back to the normal granularity when Treg activity was restored (Figure 7F). However, that was not the case in case of Il2-/-Il7ra mice, where the HSCs maintained higher granularity as in Il2-/- mice (Supplementary Figure 6D), and the defects in hematopoiesis remained (Figure 3E, 3F and Supplementary Figure 3B, C). As a result of normal HSC activity, Il2-/-Ifng-/- mice regained body weight (Figure 7G), and also these mice no more developed splenomegaly (Figure 7H, 7I). Significantly, IFN-γ-deficiency dramatically increased the life span of Il2-/- mice, which otherwise died very early in their life (Figure 7J).
The hyperactive T cells in Il2-/- mice BM were smaller and less granular compared to the littermate control mice, where majority of CD4+ T cells were larger and more granular (Figure 7K). Abolition of IFN-γ activity reversed the CD4+ T cell phenotype of Il2-/- mice to that of WT mice (Figure 7K) suggesting this as an underlying mechanism, which facilitates HSC maintenance. In agreement with this, in Il2-/-Bim-/- mice also the BM CD4+ T cells regained the phenotype as in case of littermate control mice (Figure 7L). However, in case of Il2-/-Il7ra-/- mice, where HSC defects were still intact, the BM CD4+ T cells retained their smaller size and low granularity (Figure 7M). Thus, our study shows that abolition of IFN-γ activity in the absence of Treg cells (Il2-/-Ifng-/-), or restoring full Treg activity (Il2-/-Bim-/-) can reverse the abnormalities not only in HSCs but also in the CD4+ T cells in the BM, which were the prime cause of the hematopoietic abnormalities in Il2-/- mice.
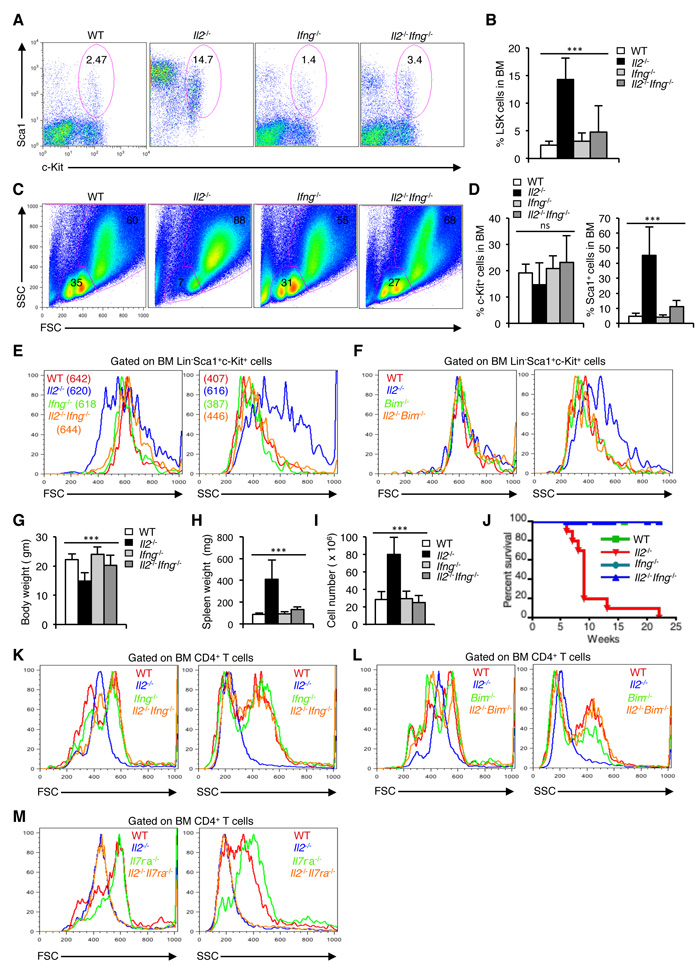
Figure 7: Enhanced IFN-γ activity leads to dysregulated hematopoiesis in Il2-/- mice. A. Distribution of LSK cells in BM from WT, Il2-/-, Ifng-/- and Il2-/-Ifng-/- mice. B. Quantification of BM LSK cells in Il2-/-Ifng-/- mice compared to WT, Il2-/-and Ifng-/- mice. C. Distribution of BM cells in indicated mice according to their FSC and SSC pattern. D. Evaluation of c-Kit+ and Sca1+ cells distribution among lineage-negative BM cells in Il2-/-Ifng-/- mice compared to littermate control mice. E. FSC and SSC patterns of Il2-/-Ifng-/- LSK cells compared to that of WT, Il2-/- and Ifng-/- cells. F. FSC and SSC patterns of Il2-/-Bim-/- LSK cells compared to that of WT, Il2-/- and Bim-/- cells. G. Body weight, H. spleen weight, and I. splenocytes number in Il2-/-Ifng-/- mice compared to WT, Il2-/-and Ifng-/- mice. J. Survival curve for Il2-/-Ifng-/- mice compared to WT, Il2-/-and Ifng-/- mice. K. Profile of Il2-/-Ifng-/- BM CD4+ T cells according to their FSC and SSC distribution pattern compared to that of littermate control mice. L. FSC and SSC distribution of Il2-/-Bim-/- BM CD4+ T cells compared to that of littermate control mice. M. Profile of Il2-/-Il7ra-/- BM CD4+ T cells according to their FSC and SSC distribution pattern compared to that of WT, Il2-/-and Il7ra-/- mice. Numbers inside each dot plot represent percent respective population and in histograms represent the MFI. Data are representative of 3 independent experiments, (n = 3 per group) and shown as mean ± s.d., in B, D, G, H & I ***P < 0.0001, ns = not significant, one-way ANOVA.
DISCUSSION
We have previously reported a severely defective erythropoiesis in Il2-/- mice due to the lack of KLF1 activity in erythroid precursor cells [42]. Also, earlier studies have reported severe autoimmunity, colitis and other pathological conditions in Il2-/- mice [57, 64, 65]. However, defects at the level of HSC resulting in a highly dysregulated hematopoiesis threatening the survival of these animals have so far not been investigated. Maintenance of HSC quiescence and subsequent multiple lineage differentiation are governed by an array of signalling pathways and TFs. A role for IL-2 in HSC maintenance has not been elucidated though altered HSC distribution in these mice has been reported [66]. Here using multiple gene-manipulated mice we show that IL-2 signaling is an essential component of the complex regulatory mechanisms that need to be in place to maintain HSC integrity and steady state hematopoiesis.
Our findings of dysregulated Notch-Runx and other signalling molecules in HSCs lacking IL-2 signals is a critical attribute in explaining how the HSCs in Il2-/- mice lose their quiescence and give rise to a myeloid-biased hematopoiesis. The HSCs in Il2-/- mice are phenotypically distinct from that of WT mice, as they are highly granular (Figure 1D). This was a specific effect of IL-2 deficiency on HSCs only, as the Sca1+ or c-Kit+ cells were similar to that of WT cells, even though the Sca1+ population was strongly increased in Il2-/- mice (Figure 1A). The observations regarding altered Flk2 and Slamf1 expression in Il2-/- HSCs (Figure 1O), further vindicate an abnormal HSC maintenance in Il2-/- mice. Besides, from our gene expression analysis of TFs and various signalling molecules involved in maintaining HSC quiescence (Figure 1P, 1Q), it is clear that the defects in Il2-/- HSCs are specific defects arising out of IL-2 deficiency. The loss of FSCloSSClo cells and the accumulation of FSChiSSChi cells in the BM was a characteristic phenotype and could be used as a diagnostic trait for defective hematopoiesis in absence of IL-2 signals (Figure 1M, 1N). The fact that all these defects could be reversed with the treatment of IL-2 to the Il2-/- mice (Figure 3A-3D and Supplementary Figure 3A) suggests that IL-2 signaling has an important contribution in maintaining HSCs in the BM. The hematopoietic phenotype observed in both our settings (Figure 2 and Supplementary Figure 2) at an early and late time points after adoptive transfer of BM cells, indicate that IL-2 signaling is critical in maintaining normal BM hematopoiesis.
Contrary to Il2-/-Rag1-/- mice (Figure 3G, 3H and Supplementary Figure 3D, E) the persistence of the HSC defects in Il2-/-Il7ra-/- mice (Figure 3E, 3F and Supplementary Figure 3B, C) underlines the important role T cells play in the maintenance of HSCs. The phenotype of T cells in the BM of Il2-/- mice is very distinct compared to that in WT mice (Figure 5A), and we have previously reported that these activated T cells in the Il2-/- mice cause severe defects in erythrocyte development [42]. Here also, we have unravelled a link between intact Treg activity establishing control over Teff cell activities, which is absolutely necessary for maintaining HSC integrity and normal hematopoiesis. Lack of optimal Treg population in the Il2-/- mice initiates the cascading effect of activated Teff cells producing excess amount of IFN-γ, which in turn destabilizes the HSC physiology leading to defective hematopoiesis. Involvement of the Treg cells in maintaining HSC integrity and in hematopoiesis was clearly evident in our inducible Treg depletion model (Figure 4A-4G). Further, our analysis of the Il2-/-Bim-/- mice, where restoration of Treg activity prevented the loss of HSC quiescence and maintained hematopoiesis (Figure 4K, 4L and Supplementary Figure 5), suggests that the Treg cells could actually fix the hematopoietic defects in Il2-/- mice.
How do the Treg cells exert such a profound influence on HSC integrity? Treg cells are a prime source of IL-10, and a recent study has implicated IL-10 signaling to be critical for maintaining HSC quiescence [60, 61, 63]. However, in addition to the normal HSC phenotype and hematopoiesis observed in the Cd4-CreIl10fl/fl and Vav-CreIl10fl/fl mice (Figure 6A-6H), our finding of an increased IL-10 production in the Il2-/- mice (Figure 6I) suggests that the HSC defects in these mice are IL-10-independent. On the other hand, the reversal in HSC defects in Il2-/-Ifng-/- mice (Figure 7) demonstrated that increased IFN-γ activity was the main reason behind the dysregulated hematopoiesis in Il2-/- mice. Our study unravelled the contrasting activities of IL-2 and IFN-γ, produced by the lymphocytes on HSC physiology and on hematopoiesis. IL-2 being the guarantor of Treg survival and activity keeps the T cell activation in the BM under check resulting in minimal IFN-γ in the HSC niche. This in turn ensures optimal Notch-Runx signalling maintaining the HSC quiescence and normal hematopoiesis (Supplementary Figure 7). Any defects in IL-2 signaling will rapidly lead to Treg cell death and breaking the delicate balance between Treg and Teff cell activity resulting in an excess IFN-γ having detrimental effect on hematopoiesis (Supplementary Figure 7). IFN-γ signalling has been reported to play a role in HSC maintenance and our study further supports this activity [67-69].
Our finding that an optimal number of Treg cells are critical for HSC integrity will have wide impact in the clinical contexts of HSC reconstitution. Hematopoietic dysfunction is common following radio- or chemotherapy of patients suffering from various forms of cancer. Further, the observation regarding abolition of IFN-γ activity restoring hematopoiesis even in the absence of Treg cells will foster the use of anti-IFN-γ in facilitating hematopoietic reconstitution in varied settings. In addition, while deciding on clinical remedies in the context of hematopoietic reconstitution the contrasting activities of both IL-2 and IFN-γ need to be carefully evaluated keeping in mind their important role in T cell function.
MATERIALS AND METHODS
Mice
C57BL/6 wild-type, Il2-/-, Il2ra-/-, Il7ra-/-, Jak3-/-, Rag1-/-, Bim-/-, DEREG, Il2-/-Il7ra-/-, Il2-/-Rag1-/-, Il2-/-Bim-/-, Il10fl/fl, Cd4-CreIl10fl/fl, Vav-CreIl10fl/fl, Ifng-/- and Il2-/-Ifng-/- mice of 6-8 weeks age unless mentioned otherwise were used in this study. All mice used in the study were on C57BL/6 background. Animals were housed either in the central animal facility (ZEMM) or in the animal facility of the Institute of Virology and Immunobiology, University of Würzburg, according to standard animal care protocols. All animal experiments were performed with extreme care, and were according to established guidelines (approved by the Regierung von Unterfranken, Wuerzburg, Permit Number 55.2-2531.01-53/10B).
Flow cytometry
All antibodies for flow cytometry and cell isolation were purchased either from BD Pharmingen or eBioscience. Anti-Ly-6A/E (Sca1; D7), anti-c-Kit (2B8), Biotin mouse lineage panel (# 559971), anti-CD45R/B220 (RA3-6B2), anti-CD3ε (145-2C11), anti-CD4 (GK1.5), anti-CD8α (53-6.7), anti-CD11b (M1/70), anti-CD11c (N418), anti-CD25 (PC61), anti-CD41 (MWReg30), anti-CD44 (IM7), anti-CD45.1 (A20), anti-CD45.2 (104), anti-CD48 (HM48-1), anti-CD62L (MEL-14), anti-Ly-6G (Gr1; RB6-8C5), anti-F4/80 (BM8), anti-NK1.1 (PK136), anti-Ter119 (TER-119), anti-CD127 (A7R34), anti-CD135 (Flk2; A2F10), anti-CD150 (9D1), anti-Foxp3 (FJK-16s), anti-IFN-γ (XMG1.2), anti-IgM (R6-60.2) and anti-IgD (11-26c), antibodies either directly conjugated with fluorochromes or with biotin were used throughout this study. Biotinylated antibodies were revealed with secondary streptavidin-allophycocyanin or phycoerythrin-Cy5 (PE-Cy5) antibodies. HSC (LSK) population in BM was analyzed by gating either on lineage-negative cells not expressing IL-7Rα and high levels of both Sca1 and c-Kit molecules, or on LSK cells negative for CD48 and positive for high levels of CD150 expression. LT- and ST-HSCs were analyzed by further gating on LSK cells for Flk2 (CD135) expression. Flow cytometry and data analysis were performed following standard procedure using FACSCalibur and CellQuest or FlowJo software.
Cell sorting
For cell sorting, femur and tibia from both hind limbs were collected and the bone marrow was flushed out by pumping BSS containing 0.1% BSA with the help of a syringe and needle. Single cell suspension was prepared and cells were washed once in BSS/BSA and then with PBS/0.1% BSA. Afterwards, total BM cells were incubated with biotinylated antibodies against lineage markers (anti-CD3, anti-B220, anti-CD11b/CD11c, anti-Gr1, and anti-Ter119) followed by incubation with anti-biotin microbeads. Lineage-positive and lineage-negative cells were isolated by magnetic separation. Lineage-negative fraction was further incubated with anti-IL-7Rα (CD127), anti-Sca1, anti-c-Kit and anti-Flk2 antibodies for sorting of LSK cells, or the LT- and ST-HSCs. Cells were sorted by using a FACSAria (BD Biosciences) flow cytometer.
Intracellular staining
For intracellular IFN-γ staining, 5 x 106 total BM cells from WT and Il2-/- mice were either cultured in complete-RPMI (10% FCS) medium or stimulated with PMA plus Ionomycin (100 ng each) for 5 h in presence of GolgiPlug (1:1000 dilution) and Monensin (1:1500 dilution). For intracellular Foxp3 staining, freshly isolated 5 x 106 splenocytes from WT and Il2-/- mice, or BM, spleen and LN cells from PBS or DT treated DEREG mice were used. Cells were first surface stained for CD4, CD8 and CD25 followed by intracellular IFN-γ or Foxp3 staining according to eBioscience Foxp3 staining protocol. IFN-γ expression in the BM was analyzed by gating on CD4+ T cells. Similarly, Foxp3 expression in each case was analyzed by gating on CD4+CD25+ T cells (WT and Il2-/- mice) or by evaluating GFP levels in DEREG mice. Flow cytometry was performed using a FACSCalibur (BD Biosciences) and data were analyzed using FlowJo software.
Photographs
Photograph of spleens were taken using a Nikon Coolpix 4500 digital camera, and the photographs were processed using Adobe Photoshop software.
Adoptive cell transfer
Single cell suspensions of BM cells were prepared and RBCs were lysed before transfer. For adoptive transfer, in the first set of experiments 4 x 106 BM cells from CD45.2+ WT or Il2-/- donors were transferred to lethally (9 Gy) irradiated CD45.1+ congenic WT recipients. In the reverse set of experiments BM cells from CD45.1+ WT donors were transferred to lethally irradiated CD45.2+ WT or Il2-/- recipients by retro-orbital injection of the venous sinus. Post-cell transfer, recipient mice were maintained with antibiotic-supplemented drinking water and were treated with extra care. BM hematopoiesis was analyzed at 5 and 11 weeks (CD45.2+ WT or Il2-/- donor cells), or at 4 and 13 weeks (CD45.1+ WT donor cells) after transfer by gating on donor-derived cells in each set of experiments. For the transfer of naïve CD4+ Teff cells, 2.0 x 106 CD4+CD25-CD62Lhi cells from LN and spleen were transferred intraperitoneally to Rag1-/- mice, and, three weeks post-transfer, frequency of LSK cells in the BM was analyzed.
In vivo injections
6-8 weeks old Il2-/- mice were injected every alternate day with 1 µg recombinant murine IL-2 (rmIL-2, Peprotech) or an equal volume of PBS intraperitoneally for two weeks. Mice were analyzed to investigate the effects of rmIL-2 on BM hematopoiesis four days after the last injection. DEREG mice of 6 weeks of age were either injected with 1 µg (100 µl) diphtheria toxin (DT) or an equal volume of PBS intraperitoneally for four consecutive days. Subsequently, three days after last injection mice were analyzed for the effect of lack of Treg cell activity on BM hematopoiesis.
In vitro co-culture assay
Establishment of BM stromal cell layer: 20 x 106 total BM cells from WT mice were plated into each well of a 24-well plate in 2 ml complete-RPMI (10% FCS) medium. Cells were incubated at 37° C and after 3 days all floating cells were removed and the adherent stromal cells were washed with complete-RPMI (10% FCS) medium. Cells were washed every 4 days and fresh medium was added to allow the stromal cells to grow up to confluency. At day 28, 5 x 106 freshly isolated WT BM cells were added to the stromal cell layer and were co-cultured with 4 x 105 unstimulated or 6 h PMA plus ionomycin stimulated WT CD4+ T cells in presence or absence of IL-2 (2 ng/ml). After 60 h, the cultures were analyzed to evaluate the influence of T cell activity on LSK cells in respective culture condition.
Semiquantitative RT-PCR
Sorted LSK cells were used to synthesize cDNA using Miltenyi Biotec cDNA synthesis kit and protocol. Semiquantitative RT-PCR was performed to analyze the expression of indicated genes. Primer sequences are available in the supplementary information online.
Statistics
Results are presented as mean ± s.d. Statistical significance was assessed using Student’s t-test for comparison between two groups and ANOVA for the differences between groups.
Author contributions
SG and GW helped with mice breeding, isolated cells and performed experiments. ES contributed to the study design, data evaluation and manuscript writing. AP conceived the project, designed and performed majority experiments, analysed the data and wrote the manuscript.
ACKNOWLEDGMENTS
The authors thank C. Linden (University of Würzburg) for excellent cell sorting; D. J. Murphy (University of Glasgow, UK), T. Sparwasser (Institute of Infection Immunology, TWINCORE, Center for Experimental and Clinical Infection Research, Hannover, Germany), and A. Roers (Institute of Immunology, Technical University of Dresden, Dresden, Germany) for the Bim-/-, DEREG, and Il10fl/fl mice respectively; M. Chopra and A. Beilhack (University of Würzburg) for assistance with adoptive cell transfer experiments; D. Langenhorst (University of Würzburg) with DEREG mice experiments; and E. Schmitt (University of Würzburg) for the excellent photographs of mice spleens.
CONFLICTs OF INTEREST
The authors declare that they have no financial or commercial conflict of interest.
FUNDING
This work was supported by a Deutsche Forschungsgemeinschaft (DFG) grant TRR52 (ES), by the Wilhelm Sander-Stiftung (ES), ‘PostDoc Plus Funding’ grant from the Graduate School of Life Sciences (GSLS), University of Wuerzburg, Germany (AP), the Deutsche José Carreras Leukämie-Stiftung e.V. grant (DJCLS R 15/12) (AP), and by a fellowship from the Vandervell Foundation (AP).
REFERENCES
1. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014; 505:327-334.
2. Anthony BA, Link DC. Regulation of hematopoietic stem cells by bone marrow stromal cells. Trends in immunology. 2014; 35:32-37.
3. Coskun S, Chao H, Vasavada H, Heydari K, Gonzales N, Zhou X, de Crombrugghe B, Hirschi KK. Development of the fetal bone marrow niche and regulation of HSC quiescence and homing ability by emerging osteolineage cells. Cell reports. 2014; 9:581-590.
4. Kumano K, Chiba S, Kunisato A, Sata M, Saito T, Nakagami-Yamaguchi E, Yamaguchi T, Masuda S, Shimizu K, Takahashi T, Ogawa S, Hamada Y, Hirai H. Notch1 but not Notch2 is essential for generating hematopoietic stem cells from endothelial cells. Immunity. 2003; 18:699-711.
5. Varnum-Finney B, Halasz LM, Sun M, Gridley T, Radtke F, Bernstein ID. Notch2 governs the rate of generation of mouse long- and short-term repopulating stem cells. The Journal of clinical investigation. 2011; 121:1207-1216.
6. Cai Z, de Bruijn M, Ma X, Dortland B, Luteijn T, Downing RJ, Dzierzak E. Haploinsufficiency of AML1 affects the temporal and spatial generation of hematopoietic stem cells in the mouse embryo. Immunity. 2000; 13:423-431.
7. Burns CE, Traver D, Mayhall E, Shepard JL, Zon LI. Hematopoietic stem cell fate is established by the Notch-Runx pathway. Genes & development. 2005; 19:2331-2342.
8. de Pater E, Kaimakis P, Vink CS, Yokomizo T, Yamada-Inagawa T, van der Linden R, Kartalaei PS, Camper SA, Speck N, Dzierzak E. Gata2 is required for HSC generation and survival. The Journal of experimental medicine. 2013; 210:2843-2850.
9. Souroullas GP, Salmon JM, Sablitzky F, Curtis DJ, Goodell MA. Adult hematopoietic stem and progenitor cells require either Lyl1 or Scl for survival. Cell stem cell. 2009; 4:180-186.
10. Lacombe J, Herblot S, Rojas-Sutterlin S, Haman A, Barakat S, Iscove NN, Sauvageau G, Hoang T. Scl regulates the quiescence and the long-term competence of hematopoietic stem cells. Blood. 2010; 115:792-803.
11. Pietras EM, Warr MR, Passegue E. Cell cycle regulation in hematopoietic stem cells. The Journal of cell biology. 2011; 195:709-720.
12. Jagannathan-Bogdan M, Zon LI. Hematopoiesis. Development. 2013; 140:2463-2467.
13. Christensen JL, Weissman IL. Flk-2 is a marker in hematopoietic stem cell differentiation: a simple method to isolate long-term stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:14541-14546.
14. Nakamura-Ishizu A, Takizawa H, Suda T. The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development. 2014; 141:4656-4666.
15. Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends in cell biology. 2005; 15:494-501.
16. Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, Link DC. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013; 495:227-230.
17. Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, Nakamura Y, Gomei Y, Iwasaki H, Matsuoka S, Miyamoto K, Miyazaki H, Takahashi T, Suda T. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell stem cell. 2007; 1:685-697.
18. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006; 25:977-988.
19. Qian H, Buza-Vidas N, Hyland CD, Jensen CT, Antonchuk J, Mansson R, Thoren LA, Ekblom M, Alexander WS, Jacobsen SE. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell stem cell. 2007; 1:671-684.
20. Mayani H, Guilbert LJ, Janowska-Wieczorek A. Biology of the hemopoietic microenvironment. European journal of haematology. 1992; 49:225-233.
21. Metcalf D. Hematopoietic cytokines. Blood. 2008; 111:485-491.
22. Kim HP, Imbert J, Leonard WJ. Both integrated and differential regulation of components of the IL-2/IL-2 receptor system. Cytokine & growth factor reviews. 2006; 17:349-366.
23. Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity. 2010; 33:153-165.
24. Boussiotis VA, Barber DL, Nakarai T, Freeman GJ, Gribben JG, Bernstein GM, D’Andrea AD, Ritz J, Nadler LM. Prevention of T cell anergy by signaling through the gamma c chain of the IL-2 receptor. Science. 1994; 266:1039-1042.
25. Miyazaki T, Kawahara A, Fuji H, Nakagawa Y, Minami Y, Liu ZJ, Oishi I, Silvennoinen O, Witthuhn BA, Ihle JN, Taniguchi T. Functional activation of Jak1 and Jak3 by selective association with IL-2 receptor subunits. Science. 1994; 266:1045-1047.
26. Russell SM, Johnston JA, Noguchi M, Kawamura M, Bacon CM, Friedmann M, Berg M, McVicar DW, Witthuhn BA, Silvennoinen O, Goldman AS, Schmalstieg FC, Ihle JN, et al. Interaction of IL-2R beta and gamma c chains with Jak1 and Jak3: implications for XSCID and XCID. Science. 1994; 266:1042-1045.
27. Friedmann MC, Migone TS, Russell SM, Leonard WJ. Different interleukin 2 receptor beta-chain tyrosines couple to at least two signaling pathways and synergistically mediate interleukin 2-induced proliferation. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93:2077-2082.
28. Lin JX, Leonard WJ. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene. 2000; 19:2566-2576.
29. Lin JX, Li P, Liu D, Jin HT, He J, Ata Ur Rasheed M, Rochman Y, Wang L, Cui K, Liu C, Kelsall BL, Ahmed R, Leonard WJ. Critical Role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity. 2012; 36:586-599.
30. Leonard WJ. Cytokines and immunodeficiency diseases. Nature reviews Immunology. 2001; 1:200-208.
31. Paliard X, de Waal Malefijt R, Yssel H, Blanchard D, Chretien I, Abrams J, de Vries J, Spits H. Simultaneous production of IL-2, IL-4, and IFN-gamma by activated human CD4+ and CD8+ T cell clones. Journal of immunology. 1988; 141:849-855.
32. Siegel JP, Sharon M, Smith PL, Leonard WJ. The IL-2 receptor beta chain (p70): role in mediating signals for LAK, NK, and proliferative activities. Science. 1987; 238:75-78.
33. Mingari MC, Gerosa F, Carra G, Accolla RS, Moretta A, Zubler RH, Waldmann TA, Moretta L. Human interleukin-2 promotes proliferation of activated B cells via surface receptors similar to those of activated T cells. Nature. 1984; 312:641-643.
34. Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. 2010; 140:845-858.
35. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008; 133:775-787.
36. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nature immunology. 2005; 6:1142-1151.
37. Barron L, Dooms H, Hoyer KK, Kuswanto W, Hofmann J, O’Gorman WE, Abbas AK. Cutting edge: mechanisms of IL-2-dependent maintenance of functional regulatory T cells. Journal of immunology. 2010; 185:6426-6430.
38. Chougnet CA, Tripathi P, Lages CS, Raynor J, Sholl A, Fink P, Plas DR, Hildeman DA. A major role for Bim in regulatory T cell homeostasis. Journal of immunology. 2011; 186:156-163.
39. Tai X, Erman B, Alag A, Mu J, Kimura M, Katz G, Guinter T, McCaughtry T, Etzensperger R, Feigenbaum L, Singer DS, Singer A. Foxp3 transcription factor is proapoptotic and lethal to developing regulatory T cells unless counterbalanced by cytokine survival signals. Immunity. 2013; 38:1116-1128.
40. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nature genetics. 2001; 27:68-73.
41. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nature genetics. 2001; 27:20-21.
42. Chopra M, Langenhorst D, Beilhack A, Serfling E, Patra AK. Interleukin-2 critically regulates bone marrow erythropoiesis and prevents anemia development. European journal of immunology. 2015; 45:3362-3374.
43. Schlaeger TM, Mikkola HK, Gekas C, Helgadottir HB, Orkin SH. Tie2Cre-mediated gene ablation defines the stem-cell leukemia gene (SCL/tal1)-dependent window during hematopoietic stem-cell development. Blood. 2005; 105:3871-3874.
44. Nie Y, Han YC, Zou YR. CXCR4 is required for the quiescence of primitive hematopoietic cells. The Journal of experimental medicine. 2008; 205:777-783.
45. de Graaf CA, Metcalf D. Thrombopoietin and hematopoietic stem cells. Cell cycle. 2011; 10:1582-1589.
46. Chou FS, Mulloy JC. The thrombopoietin/MPL pathway in hematopoiesis and leukemogenesis. Journal of cellular biochemistry. 2011; 112:1491-1498.
47. Ariki R, Morikawa S, Mabuchi Y, Suzuki S, Nakatake M, Yoshioka K, Hidano S, Nakauchi H, Matsuzaki Y, Nakamura T,Goitsuka R. Homeodomain transcription factor Meis1 is a critical regulator of adult bone marrow hematopoiesis. PloS one. 2014; 9:e87646.
48. Ficara F, Murphy MJ, Lin M, Cleary ML. Pbx1 regulates self-renewal of long-term hematopoietic stem cells by maintaining their quiescence. Cell stem cell. 2008; 2:484-496.
49. Hoffman B, Amanullah A, Shafarenko M, Liebermann DA. The proto-oncogene c-myc in hematopoietic development and leukemogenesis. Oncogene. 2002; 21:3414-3421.
50. Baena E, Ortiz M, Martinez AC, de Alboran IM. c-Myc is essential for hematopoietic stem cell differentiation and regulates Lin(-)Sca-1(+)c-Kit(-) cell generation through p21. Experimental hematology. 2007; 35:1333-1343.
51. Laurenti E, Varnum-Finney B, Wilson A, Ferrero I, Blanco-Bose WE, Ehninger A, Knoepfler PS, Cheng PF, MacDonald HR, Eisenman RN, Bernstein ID, Trumpp A. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell stem cell. 2008; 3:611-624.
52. Yamashita M, Nitta E, Suda T. Regulation of hematopoietic stem cell integrity through p53 and its related factors. Annals of the New York Academy of Sciences. 2016; 1370:45-54.
53. Hock H, Hamblen MJ, Rooke HM, Schindler JW, Saleque S, Fujiwara Y, Orkin SH. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature. 2004; 431:1002-1007.
54. Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, Antipin J, Reva B, Koff A, et al. p53 regulates hematopoietic stem cell quiescence. Cell stem cell. 2009; 4:37-48.
55. Zeng H, Yucel R, Kosan C, Klein-Hitpass L, Moroy T. Transcription factor Gfi1 regulates self-renewal and engraftment of hematopoietic stem cells. The EMBO journal. 2004; 23:4116-4125.
56. Abramovich C, Pineault N, Ohta H, Humphries RK. Hox genes: from leukemia to hematopoietic stem cell expansion. Annals of the New York Academy of Sciences. 2005; 1044:109-116.
57. Sadlack B, Lohler J, Schorle H, Klebb G, Haber H, Sickel E, Noelle RJ, Horak I. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. European journal of immunology. 1995; 25:3053-3059.
58. Lahl K, Sparwasser T. In vivo depletion of FoxP3+ Tregs using the DEREG mouse model. Methods in molecular biology. 2011; 707:157-172.
59. Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. The Journal of experimental medicine. 1999; 190:995-1004.
60. Annacker O, Pimenta-Araujo R, Burlen-Defranoux O, Barbosa TC, Cumano A, Bandeira A. CD25+ CD4+ T cells regulate the expansion of peripheral CD4 T cells through the production of IL-10. Journal of immunology. 2001; 166:3008-3018.
61. Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR Jr, Muller W, Rudensky AY. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008; 28:546-558.
62. Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993; 75:263-274.
63. Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, Gao W, Saito TI, Lo Celso C, Tsuyuzaki H, Sato T, Cote D, Sykes M, et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature. 2011; 474:216-219.
64. Horak I, Lohler J, Ma A, Smith KA. Interleukin-2 deficient mice: a new model to study autoimmunity and self-tolerance. Immunological reviews. 1995; 148:35-44.
65. Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993; 75:253-261.
66. Chen J, Astle CM, Harrison DE. Hematopoietic stem cell functional failure in interleukin-2-deficient mice. Journal of hematotherapy & stem cell research. 2002; 11:905-912.
67. Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010; 465:793-797.
68. de Bruin AM, Voermans C, Nolte MA. Impact of interferon-gamma on hematopoiesis. Blood. 2014; 124:2479-2486.
69. Sawamiphak S, Kontarakis Z, Stainier DY. Interferon gamma signaling positively regulates hematopoietic stem cell emergence. Developmental cell. 2014; 31:640-653.