
INTRODUCTION
NSCLC remains an emerging worldwide health problem, accounting for 85% of the reported cases of lung cancer. Pathological features classify it in three main categories: adenocarcinoma, squamous cell carcinoma and large-cell carcinoma [1-3]. The outcome of patients with NSCLC is poor for nearly half of them with metastatic disease when first diagnosis [4]. Nowadays, patients with advanced NSCLC harboring sensitizing mutations can achieve survival rates at least double by treated with epidermal growth factor receptor tyrosine kinase inhibitors (EGFR TKIs). EGFR TIKs show dramatic tumor response and favorable clinical outcomes via competitively inhibiting the tyrosine kinase domain of EGFR [5]. Furthermore, all guidelines recommended that chemotherapy is the mainstay of treatment in NSCLC and adjuvant radiotherapy is commonly useful for patients with limited disease [6]. However, primary and acquired resistance to these therapies remains a major clinical problem.
NON-CODING RNAs OPEN NEW AVENUES INTO THE BIOLOGY OF NSCLC
The advent of advanced techniques has uncovered that protein-coding genes only represent < 2% of the total human genome sequence. Importantly, these technologies also have revealed the widespread expression of ncRNAs [7]. Currently, this novel class of RNAs are well recognized as versatile and effective regulatory molecules in a wide range of biological progresses [8]. Based on size, ncRNAs can be divided into two classes: small ncRNAs less than 200 bps and long ncRNAs (lncRNAs) greater than 200 bps.
MicroRNAs (miRNAs), with size from 19 to 25 nucleotides, are the best characterized family of small ncRNAs [9]. Increasing evidence has shown that miRNAs play important roles in various biological processes through regulating the expression of genes. About 60% of messenger RNAs (mRNAs) contain target sites of miRNAs, and following polymerase II-mediated transcription, miRNAs bind to mRNAs to mediate gene expression at the post-transcriptional level [10]. MiRNAs are suggested to be implicated with cancer initiation, progression and metastasis due to their frequent deregulation in various types of human cancers, including NSCLC [11]. For example, Let-7 is the first identified miRNA in NSCLC [12]. The let-7 family was later shown to inhibit cancer progression via downregulating the tumor promoting protein, such as RAS, MYC, and HMGA2. To date, a number of miRNAs are reported to have a similar tumor-suppressing action, including miR-138, miR-136 and miR-221. In contrast, other miRNAs (miR-21, miR-137 and miR-182-3p, etc) are reported to participate in promoting the development and progression of NSCLC [9]. Taking this into account, the vital field of miRNAs provides a reservoir of new biomarkers for NSCLC. Recently, most studies on the roles of miRNAs have focused on their involvement in chemo- or radioresistance of tumor cells [13]. For example, it was reported that let-7c not only has a suppressor activity, but also reverses chemo- or radioresistance of lung adenocarcinoma cells [14].
On the other hand, lncRNA is the least characterized class, but emerged as a versatile regulator of pathophysiological key pathways. To carry on its functions, lncRNA regulates gene expression through acting on the epigenetic, transcriptional, and posttranscriptional levels [15]. Similar to miRNAs, lncRNAs are gaining increasing attention in the field of NSCLC research. Although dozens of lncRNAs are identified to contribute the development and progression cancer, only few validated lncRNAs have been reported to change expression in NSCLC today [16]. High expression level of metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) was observed to be correlated with poor prognostic in patients suffering from NSCLC. Schmidt et al. has first demonstrated that MALAT1 can mediate metastasis development [17]. Other well-characterized lncRNAs contain HOTAIR, ANRIL and H19, whose expression level also increased in NSCLC, whereas GAS5 and MEG3 are associated with tumor-suppressive function [18-22].
As one of the epithelial tumors, NSCLC recapitulates multiple development programs that promote the occurrence of metastases, including EMT [23]. Recent studies have demonstrated that post-transcriptional regulatory networks, such as miRNAs and lncRNAs, mediate the progresses of EMT.
MOLECULAR BASIS OF EMT IN NSCLC
During the past 20 years, it has gained more and more attention that the activation of tumor invasion is induced by a phenotype change that recapitulates the EMT event, which not only plays a crucial role in the development of embryo [24]. Increasing evidence has pointed to the role of EMT by which tumor cells would weaken E-cadherin-dependent cell-cell junctions and enhance motility. Recently, loss of epithelial markers such as E-cadherin has been reported to be associated with poor survival in several carcinomas including NSCLC.
EMT is known as a series of events where epithelial cells convert into mesenchymal cells. Invasion causes cancer metastasis, and extracellular matrix (ECM) is a key player in the activities that cause cell translocation into the stroma [25]. ECM, surrounding mesenchymal cells in interstitial spaces, is a source of growth factors, including fibroblast growth factors (FGF), epidermal growth factor (EGF) and hepatocyte growth factor (HGF). In most studied systems, growth factors and their cell surface receptors seem to be implicated as signals for triggering EMT, and ECM-related molecules generate cascades of intracellular signals that converge on the downregulation of E-cadherin. Therefore, the cadherin/catenin/cytoskeleton complex is perturbed, leading to the loss of cell-cell adhesion and reorganisation of the actin cytoskeleton [26]. Finally, the basal surface emanates a cytoplasmic protrusion, allowing cell body detachment from its neighbouring cells. The tumor cell will take on the typical properties of mesenchymal cell, which finally migrate in the adjacent tissue [27].
E-cadherin is thought to be a crucial component of the cellular signaling network , and the loss of its expression is a fundamental event in EMT [28] (Figure 1). Inaction of E-cadherin play an important role in tumor progression, and action of inappropriate tumor stroma components can also induce EMT in lung cancer. According to the effects on E-cadherin promoter, the transcription factors can be classified into two groups. Factors, such as Snail, Zeb, E47 and KLF8 repress the activity of E-cadherin by binding to them, and another group is consisted of Twist, Goosecoid, E2.2 and FoxC2, which repress E-cadherin expression directly [29]. In stage I NSCLC, abnormal expression of Twist, Slug and Foxc2 is a significant predictor of overall survival [30]. Knockdown of Snail or Twist, zinc finger transcription factors and key E-cadherin repressor molecules can lead to the restoration of chemosensitivity during EMT [31, 32]. Central in E-cadherin repression is the transcription factor Snail, that is responsible for the disruption of E-cadherin mediated cell-cell contacts and invasion [33]. Snail family is the zinc finger protein, containing SNAI1 and SNAI2 (also known as Slug), that represses E-cadherin transcription through binding the E-box E-pal element in developmental EMT. Different signaling pathways triggering EMT have been found to be linked with the induction of Snail family members [34]. Cigarette smoke extract (CSE) can decrease the expression of E-cadherin through upregulation of Slug and LEF-1 in lung cancer cell lines, demonstrating that cigarette smoking is significantly associated with inducing EMT [35, 36]. Meanwhile, some of these oncogenic pathways regulate Snail by modulating GSK-3β activity. Thus, Snail cooperates with GSK-3β to mediate many signaling pathways that lead to EMT [37].
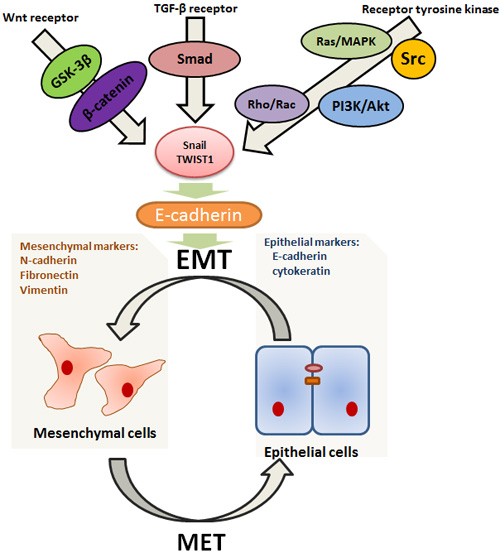
Figure 1: Highly simplified diagram illustrating some better elaborated transduction pathways related to EMT/MET. EMT is a highly complex process under the strict control of growth factors and downstream transcription factors. It seems to occur in a context where main roles are played by tyrpsine kinase receptors, Wnt and TGF-β pathways. These pathways are involved in the upregulation of Snail and Twist family via signaling through downstream transcriptional factors (such as Ras, PI3K, Rac, Src, Smad and β-catenin) , which then represses E-cadherin. What’s more, the inhibition of E-cadherin can induce EMT and, on the other hand, its re-activation can stimulate the reverse process, suggesting that E-cadherin takes center stage in mediating the switches between EMT and MET.
In most model systems, EMT takes place in a context where growth factors and receptor tyrosine kinases play a main positive role. Upon growth factors binding, receptor tyrosine kinases initiate a series of cytoplasmic transduction events such as activation of Ras/MAPK, PI3K/Akt, Rho/Rac and Src pathways [26]. Ras is a member of small GTP-binding protein that mediates its effects by activating the serine-threonine kinase Raf, which then activate mitogen-activated protein kinase (MAPK) leading directly to Snail family upregulation [38]. In addition to activating the MAPK cascade, Ras can also induce EMT by triggering other effectors such as the phosphatidylinositol-3-kinase (PI3K) and Rho/Rac signaling [39, 40]. PI3K is a lipid kinase which generates phosphatidylinositol-3, 4, 5 triphosphate (PIP3), and in turn, targets the serine-threonine kinase Akt, causing its translocation at the inner cell membrane. In addition to up-regulating Snail, PI3K-Akt signaling is required for TGF-β-mediated EMT [40]. Growth factor-mediated stimulation of receptor tyrosine kinases and their downstream mediators Ras and PI3K, all able can lead to activation of Rho family GTPases. The small GTPase Rho and Ras are involved in cell motility, and the balance between Rho and Rac activities seems to be one of major switch of EMT [27]. Src behaves as a potent trigger of EMT by activating focal adhesion kinase (FAK) directly, which results in activation of the MEK-MAPK signaling [41]. In addition, Src activation induces β-catenin phosphorylation at tyrosine residues, and leads to its binding to E-cadherin, favouring cell dissociation and EMT [42]. Repeated exposure to gefitinib or erlotinib can drive tumor cells into mesenchymal cells. Then cellular dependence on EGFR signaling is suppressed, suggesting that acquired clinical resistance to EGFR-TIKs is associated with EMT [43]. Lung adenocarcinomas harboring EGFR mutations have been shown to exhibit losing expression of IL-6, which is a major activator of the JAK/STAT3 and PI3K/AKT pathways. Based on these results, Li et al. has proved that suppress the IL-6/STAT3 pathway can cause EMT reversal and re-sensitize EGFR-TKI-resistant human lung cancer cells to erlotinib or gefitinib [44]. The NSCLC tumors insensitive to EGFR TKIs include increased protein expression of vimentin, combined with the loss of E-cadherin, claudin 4, and claudin 7 by immunoblotting. Two zinc-finger E-box-binding homeobox factors, ZEB1 and ZEB2, contain a conserved central homeobox region and two zinc-finger domains, and function as inhibitors of E-cadherin during EMT. Indeed, an inverse relationship between E-cadherin and ZEB1 expression has been observed in gefitinib-resistant NSCLC cell lines [45]. Cigarette smoking is also demonstrated to stimulate the EGFR-TKI resistance via inhibiting Src activation in NSCLC. And the Src family functions as a regulator of ERK and AKT pathway, which are important signal pathways mediating EMT [41, 46, 47].
Wnts are interesting regulators in EMT and the central component of their signaling pathway is β-catenin. Paradoxically, loss of β-catenin is associated with worsened prognosis in NSCLC, possibly because of its interactions with E-cadherin [48]. This signaling is initiated when Wnt ligand binds to Frizzled proteins, and therefore GSK-3β is inhibited, finally leading to cytoplasmic accumulation of β-catenin. Then, β-catenin functions as a transcription cofactor with TCF/LEF to activate target gene in EMT. Meanwhile, a variety of growth factors or receptors, including E-cadherin, ILK, PI3K/Akt and Ras/MAPK pathways, are intersect with Wnt/β-catenin signaling, and this can enhance their influence on EMT induction [49].
Meanwhile, TGF-β is another potent inducer of EMT. The TGF-β signalling pathway acts through TGF-β type I and II transmembrane serine-threonine kinase receptors to phosphorylate the cytoplasmic Smad2 and Smad3, thus resulting in the activation of the Snail family [50]. Moreover, synergism between IL-6/JAK/STAT3 and TGF-β/Smad signaling, as well as TGF-β and Raf/MAPK, is required to induce EMT in lung carcinomas, suggesting that EMT is orchestrated by several signaling pathways [51]. In addition, it shows that collagen type I can stimulate TGF-β to promote the development of EMT through PI3K-extracellular signal-regulated kinase pathway [52].
Furthermore, lung cancer cells with TGF-β1-induced EMT can acquire stem cell properties, and then exhibit therapeutic resistance [53]. Cancer stem cell (CSC) is a rare subset with the capacity for self-renewal and differentiation with tumors. Emerging evidence supported that lung CSCs is involved in metastasis, as well as resistance to radiation and chemotherapy [54]. TGF-β1-induced EMT in lung cancer cells can drive mobile CSCs from stationary CSCs through the acquisition of a mesenchymal profile which is related to increased levels of stem cell markers [55]. Meanwhile, several signaling pathways, including Wnt, Notch and Hedgehog that mediate EMT, also drive CSCs self-renewal and maintenance [56]. The special properties of CSCs is significantly involved in drug-resistance, such as their high capacity for DNA repair, and high expression of various drug resistance membrane transporters [57].
Interestingly, EMT is not irreversible and the reverse process is mesenchymal-epithelial transition (MET). At the site of metastases, the mesenchymal tumor cells must undergo the reverse transition after the occurrences of EMT, which is critical for distant colonization [23]. Indeed, the alternation between EMT and MET is ongoing between tumor progression [58]. Similar to EMT, MET is also mediated by various effectors. For example, the re-activation of E-cadherin can result in MET [23].
MICRORNAS-RELATED EMT IN NSCLC
MicroRNAs function as suppressors of EMT
MiRNAs are associated with almost all basic signaling pathways, which make them to be one of most relevant determination of cancer biology. As the list of these miRNAs keeps increasing, the molecular knowledge of miRNAs related to EMT is now considered to be an important focus for NSCLC research [56]. Increasing evidence indicates that specific miRNAs function as powerful suppressors of EMT, such as miR-124, miR-135a, miR-148a and miR-193a-3p/5p, which are essential in tumor cell invasion. Their expression is often downregulated in lung cancer [59-62] (Figure 2).
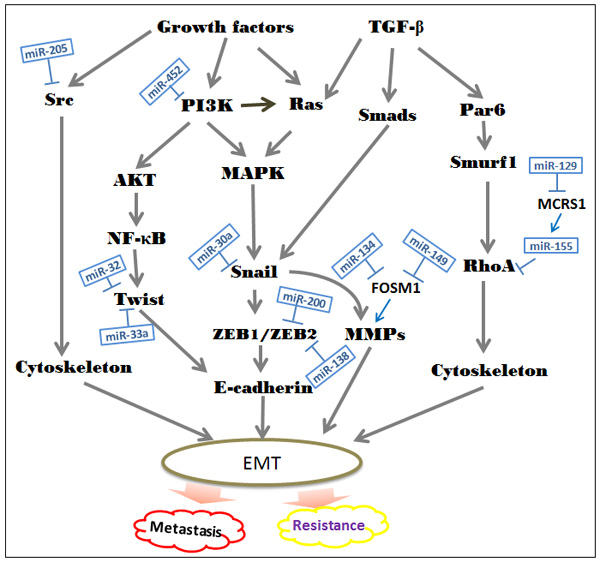
Figure 2: Suppressive roles of special miRNAs in the EMT-associated regulatory networks. The diagram shows the major signal transduction pathways leading to EMT in NSCLC. Currently, dozens of miRNAs are identified to function as vital inhibitors in the development of EMT. In NSCLC, loss of these specific miRNAs leads to expression of target oncogenes, which in turn contributes to cancer cell invasion and resistance to chemotherapy, radiotherapy and EGFR-TKIs.
The miR-200 family, represent by miR-200a, -200b, -200c, -141 and -429, is involved in the regulation of EMT/MET [63]. The EMT event allows the tumor cell to migrate away from the original tissue, and MET enables it to colonize and produce metastases in distant organs. Namely, both downregulation and upregulation of miR-200 family members have been reported to correlate with worse prognosis of patients. Early studies have reported that low levels of miR-200a, miR-200b and miR-429 are correlated with short overall survival in NSCLC patients. In contrast, high miR-141 and miR-200c function as poor prognostic makers [64]. In general, the expression of miR-200 is lost in mesenchymal-like tumor cells and negatively correlated with NCSLC metastasis. Restoring its expression inhibits ZEB1 and ZEB2 leading to the re-expression of E-cadherin [65]. Then, re-activation of E-cadherin will make cancerous cell to loss aggressive, invasive phenotype [66]. Re-expression of miR-200 also drives the significant downregulation of ECM proteins, including collagens and matrix stiffening proteins. These ECM products may function as an effector of TGF-βto induce EMT [67]. Although these miRNAs belong to the same family, they differ in their targets. Hereditary hemochromatosis (HFE) is a predicate target of miR-200a, miR-200b and miR-200c. Inhibition of either HFE or its upstream beta-2-microglobulin (B2M) can reverse EMT [66]. Moreover, recent findings have suggested that miR-200c affects the invasion suppression at least in part through the induction of USP25, which is a member of ubiquitinating-specific proteases family. EMT is intimately interwoven with the ubiquitin proteasome system to regulate the process related to carcinogenesis [68]. ZEB2 is one of the targets of miR-200c and miR-132 that suppress EMT via upregulating E-cadherin, downregulating N-cadherin and vimentin expression [69]. In addition to the miR-200 family, miR-489 and miR-638 also have been reported to induce EMT via decreasing epithelial makers level and increasing mesenchymal makers levels [70, 71]. FOXM1 is a member of forkhead box (FOX) transcription factors, and participates in early steps of metastasis via inducing MMP-2, MMP-9 and VEGF. Through binding to FOXM1 directly, both miR-149 and miR-134 can activate the TGF-β-induced EMT in NSCLS cells [72].
Microspherule protein 1 (MCRS1) is identified as a vital regulator in gene transcription, and observed to promote EMT and metastasis in NSCLC cells. It is directly and negatively mediated by miR-129. MiR-155 may be a functional downstream effector of MCRS1 and regulates TGF-βinduced EMT by targeting RhoA. The miR-129/MCRS1/miR-155 axis seems to have a potential role in mediating EMT-induced metastasis [73]. It is well recognized that the PI3K/AKT pathway contributes to the regulation of EMT. MiR-452 inhibits NSCLC proliferation and metastasis through blocking the activation of PI3K/AKT pathway [74]. TWIST is a key promoter of E-cadherin in NSCLC. Both miR-32 and miR-33a are reported to target TWIST1 directly [75, 76]. Snail, another E-cadherin promoter, was also proved to function as a direct target for specific miRNAs. MiRNA-30a inhibits EMT via regulating Snail in NSCLC cells [77]. Additionally, suppression of Let-7 is consistent with the increased expression of the HMGA2, upregulating the TGF-β co-receptor TGFBR3 and thereby promoting EMT traits [78].
In the context, we show the emerging roles of EMT in inducing resistance of lung cancer cells to conventional chemotherapy and radiotherapy, and recent studies also have shown that miRNAs is significantly associated with the regulation of resistance. For example, miR-138 has been demonstrated to sensitize NSCLC cells to ADM via directly target ZEB2 [79]. Besides, miR-200/ZEB axis is also responsible for sensitivity to nintedanib in NSCLC cells [80]. The activation of ZEB1 and Src related to induction of EMT is in part mediated by Human CRIPTO1, which is also known as teratocarcinoma-derived growth factor 1 (TDGF1). Overexpression of miR-205 inhibits CRIPTO1-dependent ZEB1, as well as the activation of Src, leading to the restoration of erlotinib sensitivity [81].
The hedgehog (Hh) signaling pathway is a vital mediator of embryogenesis especially in the lung tissue. Activation of the Hh pathway exists in many cancers including NSCLC, which also establishes a mechanistic role in EMT-associated drug resistance phenotype. Re-expression of miR-200 and let-7 family, especially miR-220b and let-7c, causes a direct inhibition of Hh signaling, and thereby diminishes the erlotinib and cisplatin resistance of A549M cells [82]. Meanwhile, miR-200c reversals EMT and then radiosensitizes A549 cells via targeting VEGF-VEGFR2 pathway partly [83]. Through cross-talking with several transcriptional factors, Notch signaling pathway promotes EMT, and then affects the sensitivity of cancer cells to treatment in NSCLC. Rhamnetin and cirsiliol can act as novel radiosensitizers via increasing the expression of tumor-suppressive microRNA, miR-34a, which leads to the inhibition of Notch-1 expression [84]. Besides, Over-expression of miR-17 can decrease cisplatin-resistant via inhibition of TGF-β signal pathway partly. It directly targets and represses TGF-beta receptor 2 (TGF-βR2), and the latter is an important component of TGF-β signal pathway [85].
MicroRNAs function as promoters of EMT
Breakdown of tight junctions is one of the defining functional changes occurring in the EMT progress, and it is associated with the loss of epithelial markers and acquisition of mesenchymal makers [27]. Overexpression of miR-30c and miR-544a has been reported to downregulate E-cadherin and upregulate vimentin that thereby stimulate EMT [86, 87]. Meanwhile, anti-miR-1246 and anti-miR-1290 can decrease the expression of CSCs markers and EMT markers in NSCLC [88]. MiR-221 and miR-222 exert their oncogenic abilities in part via upregulating the EMT-inducing gene ZEB2 through TRPS1 [89]. It suggests that inhibition of these miRNAs suppress the invasiveness of NSCLC cells.
Furthermore, a plenty of miRNAs have been shown to reduce sensitivity and acquired resistance to EGFR-TKI in lung cancer cells, including miR-23a. It is regulated by the TGF-β/Smad pathway and affects the EMT event through target E-cadherin. Because epithelial phenotype is less resistance to EGFR-TKIs, restoration of E-cadherin may increase the sensitivity of lung cancer cells to EGFR-TKIs. This study highlights that miR-23a may be useful as a new therapeutic target to overcome the EGFR-TKI resistance in NSCLC [90]. In addition to miR-23a, overexpression of miR-134 and miR-487b has reported to promote the EMT phenomenon and induce the drug resistance to gefitinib. Similarly, knockdown of these miRNAs can inhibit EMT and then reverse TGF-β1-induced resistance to gefitinib in NSCLC. Membrane-associated guanylate kinase, WW, and PDZ domain-containing protein 2 (MAGI2) is a direct target of miR-134 and miR-487b. Its suppression causes the loss of PTEN stability and PI3K/AKT activation. Kitamura et al. has reported that absence of PTEN-MAGI2 is related to acquire EGFR-TKI resistance in vitro [91].
LNCRNAS-RELATED EMT IN NSCLC
LncRNAs suppress tumor metastasis by reversing EMT
To date, a growing number of publications emphasize the role of lncRNAs play in tumor initiation, progression, and metastasis. This novel class of ncRNAs gives hope for a more profound understanding of lung cancer. Recent research has linked the dysregulation of lncRNAs with cell invasion in NSCLC [15]. BRAF-activated non-coding RNA (BANCR) is on chromosome 9 with the size of 693 bp. It has been found that the expression of BANCR was significantly downregulated at the later stages of tumor development, which is consist with lower overall survival time of patients with NSCLC. Most investigations agree on a major role for BANCR in enforcing EMT. Indeed, sun and colleagues reported that alteration of BANCR expression promoted cell migration and invasion in vitro [92]. MMP2 is a member of Matrix metalloproteases (MMPs). Upregulation of BANCR leads to a decrease of its protein level in NSCLC cells, which finally stimulating E-cadherin expression and reducing Vimentin expression. Taking this into account, BANCR possibly suppress the metastasis ability of NSLC cell partly through regulation of EMT, shedding new light on therapeutic approach [92].
LncRNAs promote tumor metastasis by inducing EMT
Currently, lncRNAs is possible the key in the development of novel cancer treatment because targeting oncogenic lncRNAs is more direct to induce anticancer effect [16]. MALAT1 (also known as NEAT2), as the first identified lncRNA in lung cancer, is well studied in recent decades. This highly conserved lncRNA locates on 11q13, whose length is more than 8000 nt [93]. High level of MALAT1 has been reported to associate with lung cancer metastasis, which is crucially related to poor prognosis. In this study, alteration of E-cadherin and Vimentin also has been observed in the highly invasive subline of brain metastasis lung cancer cells. In conclusion, MALAT1 can promote brain metastasis via inducing EMT in NSCLC [94]. Besides, increasing evidence shows that downregulation of MALAT1 will alter the expression of EMT-associated genes, finally decreasing ZEB1, ZEB2 and Slug levels and increasing E-cadherin level [16].
HOTAIR belongs to lncRNA chain with 2158 nucleotide long, which is located on chromosome 12q13.13. Recent studies have reported that the level of HOTAIR expression is distinctly upregulated in tumor tissues, including breast cancer, liver cancer, ovarian cancer, gastric cancer, and NSCLC [95]. Alves et al. first showed that HOTAIR is required to tumor metastasis for its key role in different signaling mechanisms related to EMT [96]. HOTAIR selectively binds to polycomb repressive complex 2(PRC2) to regulate the chromatin methylation state. PRC2 is required for H3K27 trimethylation, which contains H3K27 histone methyl transferase EZH2, SUZ12 and EED [97]. Both EZH2 and SUZ12 have been identified to mediate the repression of gene E-cadherin by H3K27 trimethylation [98, 99]. These findings suggest that the activation of EMT is due, in part, to the interaction of HOTAIR with the PRC2 in lung cancer cells.
Although cigarette smoke is recognized as a strong risk factor for the development of lung cancer, the related molecular mechanisms remain unclear. In CSE-exposed HBE cells, MALAT1 affects EMT via binding to EZH2, and is negatively regulated by miR-217 [100]. Moreover, CSE has been demonstrated to induce secretion of IL-6, and then act on STAT3, which bind to HOTAIR promoter directly. Thus, the activity of HOTAIR finally contributes to EMT and CSCs [101]. These results provide a better understanding of the processes associated with NSCLC metastasis caused by cigarette smoke.
CONCLUSIONS AND PERSPECTIVES
Despite great progress in early detection of NSCLC, almost 50% patients combine with developing distant metastases when they are first diagnosed [1]. Worse still, resistance to conventional therapeutics or advanced therapies presents an emerging challenge in the treatment of patients with NSCLC [6]. New biomarkers and druggable targets must be considered.
Currently, evidence is accumulating that shows a crucial role of EMT in tumor metastasis [26]. Some investigators have reported that losing expression of EMT markers such as E-cadherin and cytokeratin, and gaining expression of mesenchymal markers such as N-cadherin and vitmentin is significantly associated with advanced NSCLC stage as well as poor prognosis [102]. Furthermore, EMT-derived NSCLC cells acquire stem cell properties and exhibit resistance to chemotherapy, radiotherapy and target agents [53]. Molecules in the regulatory networks linking EMT are starting to emerge as potential molecular biomarkers and therapeutic targets for improving current cancer therapies and overcoming resistance. Non-coding RNAs has gained more attention in recent studies for its dramatic role in regulating EMT progress. Although without the ability of encoding proteins, they are emerging as central players in various cellular pathways, including EMT, through the targeting of a plenty of mRNAs [103].
NcRNAs are emerging as central players in the development of NSCLC via regulating the EMT progress (Table 1). MiRNAs is the small ncRNA chain, and gaining better understanding in the last decades. The functional properties make miRNAs appealing biomarkers and potential therapeutic targets in clinic. It is well known that targeting one miRNAs may affect the expression of various vital molecules simultaneously. For example, miR-200c can suppress the induction of EMT in part through targeting HFE, ZEB2 and USP25 respectively [66, 80]. However, in different lung cell lines, the same miRNAs seem to play an opposite role. Tumor-suppressive effect of miR-221 and miR-222 has been observed in H3255and HCC4006. In the contrary, both of these miRNAs promote invasiveness in H460 [89]. Furthermore, miR-134 seems to have a dual role in the development of lung cancer progression. It has been reported to promote the EMT phenomenon via targeting MAGI2 in A549 cells, whereas it also shows oncogenic ability in the same cell lines [91, 104]. The trait of these miRNAs may be a useful tool to predict whether they function as a tumor suppressor or not when efficient methods is developed.
Table 1: MicroRNAs related to EMT in NSCLC.
MicroRNA |
Role of microRNA |
Target gene |
Reference |
|
miR-124 |
inhibit EMT |
CDH2 |
[56] |
|
miR-138 |
sensitize NSCLC cells to ADM |
ZEB2 |
[77] |
|
miR-32 |
inhibit EMT |
TWIST1 |
[73] |
|
miR-200 family |
||||
miR-200c |
inhibit EMT, radiosensitize A549 cells |
ZEB2, SNAIL, N-cadherin, VEGF-VEGFR2 pathway |
||
miR-200b |
inhibit EMT and diminish the erlotinib resistance of A549M cells |
DLC1, HNRNPA3 |
||
miR-452 |
inhibit EMT |
PI3K/AKT pathway |
[72] |
|
miR-33a |
inhibit EMT |
TWIST1 |
[74] |
|
miR-135a |
inhibit EMT |
KLF8 |
[57] |
|
miR-205 |
inhibit EMT |
ZEB1,Src |
||
miR-489 |
inhibit EMT |
SUZ12 |
[67] |
|
miR-129 |
inhibit EMT |
MCRS1 |
[71] |
|
let 7 |
inhibit EMT |
HMGA2 |
[76] |
|
let 7c |
inhibit EMT and diminished the erlotinib resistance |
unclear |
[80] |
|
miR-638 |
inhibit EMT |
SOX2 |
[68] |
|
miR-17 |
Inhibit EMT, diminish cisplatin-resistance |
TGFβR2 |
[83] |
|
miR-132 |
inhibit EMT |
ZEB1 |
[66] |
|
miR-193a-3p |
inhibit EMT |
ERBB4,S6K2 |
[59] |
|
miR-193a-5p |
inhibit EMT |
PIK3R3,mTOR |
[59] |
|
miR-34a |
inhibition of EMT, radiosensitize NSCLC cells |
Notch-1 |
[82] |
|
miR-149 |
inhibit EMT |
FOXM1 |
[69] |
|
miR-30a |
inhibit EMT |
Snial1,Vimentin |
[75] |
|
miR-148a |
inhibit EMT |
ROCK1 |
[58] |
|
miR-1246&miR-1290 |
induce EMT |
unclear |
[86] |
|
miR-221&miR-222 |
induce EMT |
PTEN |
[87] |
|
miR-30c |
induce EMT |
E-cadherin, snail and vimentin |
[84] |
|
miR-134/487b/655 |
induce EMT, affect the drug resistance to gefitinib |
MAGI2 |
[89] |
|
miR-23a |
induce EMT, promote resistant to gefitinib |
E-cadherin |
[88] |
|
miR-544a |
induce EMT |
CDH2,vimentin |
[85] |
|
LncRNAs, another important component of ncRNAs, open new opportunities in understanding the role of EMT in metastases (Table 2). Recently, the competing endogenous RNA (ceRNA) hypothesis has described the lncRNA-miRNA-mRNA ceRNA network, where lncRNAs harbor miRNA response elements (MREs). Special lncRNAs could regulate the accumulation of miRNAs and in turn influencing the stability of protein complex [105]. For example, lncRNA unigene56159 inhibits the expression of Slug via directly binding to miR-140-5p, thereby promoting EMT in hepatocellular carcinoma (HCC) [106]. Similarly, lncRNA HULC/miR-200a-3p/ZEB1 axis facilitates HCC cells migration and invasion [107]. Also, in NSCLC, NEAT1 functions as a ceRNA for miR-377-3P. MiR-377-3p suppresses cancer progression through the activation of E2F3 pathway [108]. According to recent evidence, it is promising that lncRNAs regulate EMT via acting as ceRNAs in NSCLC cell lines. More effort is necessary to find the underlying function of lncRNAs.
In the prospective studies of EMT, circular RNA (circRNA), a potential class of ncRNAs, may capture the interest of researchers. Lately, abundant circRNAs have been demonstrated to exist in animal cells with regulatory potency. Contrary to miRNAs, circRNAs may partially act as post-transcriptional regulators [109]. Hundreds of cicrRNAs are regulated by specific factors during EMT [110]. Hence, it is with great anticipation that EMT-related circRNAs will been found in diseases, including NSCLC.
In summary, the inhibition of EMT could be a useful approach for inhibition cell invasiveness in NSCLC. And as such, the reversal of EMT, particularly through manipulating relevant ncRNAs, could also be useful for resensitization of NSCLC cells to conventional therapeutics, which would likely contribute significant improvements in treatment response.
Table 2: LncRNAs function as regulators of EMT during NSCLC metastasis.
LncRNA |
Roles of lncRNA |
Target gene |
Reference |
BANCR |
Inhibit EMT |
MMP2 |
[90] |
MALAT1 |
Promote EMT |
E-cadherin, Vimentin, EZH2 |
|
HOTAIR |
Induce EMT |
PRC2 |
[99] |
Author contributions
Chen Y and Wang R reviewed the literature and wrote the manuscript. All authors approved the final version of the manuscript.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (No. 81172335 and 81472266) and the Excellent Youth Foundation of Jiangsu Province, China (BK20140032).
CONFLICTs OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Chheang S, Brown K. Lung cancer staging: clinical and radiologic perspectives. Semin Intervent Radiol. 2013; 30:99-113.
2. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136: E359-86.
3. Hoffman PC, Mauer AM, Vokes EE. Lung cancer. The Lancet. 2000; 355: 479-85.
4. Patel AN, Simone CB, Jabbour SK. Risk factors and management of oligometastatic non-small cell lung cancer. Ther Adv Respir Dis. 2016; 10:338-348.
5. Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010; 10:760-74.
6. Goldstraw P, Ball D, Jett JR, Le Chevalier T, Lim E, Nicholson AG, Shepherd FA. Non-small-cell lung cancer. The Lancet. 2011; 378:1727-40.
7. Clark MB, Choudhary A, Smith MA, Taft RJ, Mattick JS. The dark matter rises: the expanding world of regulatory RNAs. Essays Biochem. 2013; 54:1-16.
8. Brosnan CA, Voinnet O. The long and the short of noncoding RNAs. Curr Opin Cell Biol. 2009; 21:416-25.
9. Del Vescovo V, Denti MA. microRNA and Lung Cancer. Adv Exp Med Biol. 2015; 889:153-177.
10. Booton R, Lindsay MA. Emerging role of MicroRNAs and long noncoding RNAs in respiratory disease. Chest. 2014; 146:193-204.
11. Gao W, Xu J, Shu YQ. miRNA expression and its clinical implications for the prevention and diagnosis of non-small-cell lung cancer. Expert Rev Respir Med. 2011; 5:699-709.
12. Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, Mitsudomi T, Takahashi T. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004; 64:3753-3756.
13. MacDonagh L, Gray SG, Finn SP, Cuffe S, O’Byrne KJ, Barr MP. The emerging role of microRNAs in resistance to lung cancer treatments. Cancer Treat Rev. 2015; 41:160-169.
14. Cui SY, Huang JY, Chen YT, Song HZ, Feng B, Huang GC, Wang R, Chen LB, De W. Let-7c governs the acquisition of chemo- or radioresistance and epithelial-to-mesenchymal transition phenotypes in docetaxel-resistant lung adenocarcinoma. Mol Cancer Res. 2013; 11:699-713.
15. Roth A, Diederichs S. Long Noncoding RNAs in Lung Cancer. Curr Top Microbiol Immunol. 2016; 394:57-110.
16. Li CH, Chen Y. Targeting long non-coding RNAs in cancers: progress and prospects. Int J Biochem Cell Biol. 2013; 45:1895-1910.
17. Schmidt LH, Spieker T, Koschmieder S, Schaffers S, Humberg J, Jungen D, Bulk E, Hascher A, Wittmer D, Marra A, Hillejan L, Wiebe K, Berdel WE, et al. The long noncoding MALAT-1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J Thorac Oncol. 2011; 6:1984-1992.
18. Nakagawa T, Endo H, Yokoyama M, Abe J, Tamai K, Tanaka N, Sato I, Takahashi S, Kondo T, Satoh K. Large noncoding RNA HOTAIR enhances aggressive biological behavior and is associated with short disease-free survival in human non-small cell lung cancer. Biochemical and Biophysical Research Communications. 2013; 436:319-324.
19. Nie FQ, Sun M, Yang JS, Xie M, Xu TP, Xia R, Liu YW, Liu XH, Zhang EB, Lu KH, Shu YQ. Long noncoding RNA ANRIL promotes non-small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol Cancer Ther. 2015; 14:268-277.
20. Zhang E, Li W, Yin D, De W, Zhu L, Sun S, Han L. c-Myc-regulated long non-coding RNA H19 indicates a poor prognosis and affects cell proliferation in non-small-cell lung cancer. Tumour Biol. 2016; 37: 4007-15.
21. Shi X, Sun M, Liu H, Yao Y, Kong R, Chen F, Song Y. A critical role for the long non-coding RNA GAS5 in proliferation and apoptosis in non-small-cell lung cancer. Mol Carcinog. 2015; 54:E1-E12.
22. Lu KH, Li W, Liu XH, Sun M, Zhang ML, Wu WQ, Xie WP, Hou YY. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer. 2013; 13:461.
23. Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW. Epithelial--mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J Cell Physiol. 2007; 213:374-383.
24. Savagner P. The epithelial-mesenchymal transition (EMT) phenomenon. Ann Oncol. 2010; 21:vii89-92.
25. Mareel M, Leroy A. Clinical, cellular, and molecular aspects of cancer invasion. Physiol Rev. 2003; 83:337-376.
26. Guarino M, Rubino B, Ballabio G. The role of epithelial-mesenchymal transition in cancer pathology. Pathology. 2007; 39: 305-318.
27. Guarino M. Epithelial-mesenchymal transition and tumour invasion. Int J Biochem Cell Biol. 2007; 39:2153-2160.
28. Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998; 392:190-1903.
29. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009; 139: 871-90.
30. Jiang W, Pang XG, Wang Q, Shen YX, Chen XK, Xi JJ. Prognostic role of Twist, Slug, and Foxc2 expression in stage I non-small-cell lung cancer after curative resection. Clin Lung Cancer. 2012; 13:280-287.
31. Zhuo W, Wang Y, Zhuo X, Zhang Y, Ao X, Chen Z. Knockdown of Snail, a novel zinc finger transcription factor, via RNA interference increases A549 cell sensitivity to cisplatin via JNK/mitochondrial pathway. Lung Cancer. 2008; 62:8-14.
32. Zhuo WL, Wang Y, Zhuo XL, Zhang YS, Chen ZT. Short interfering RNA directed against TWIST, a novel zinc finger transcription factor, increases A549 cell sensitivity to cisplatin via MAPK/mitochondrial pathway. Biochem Biophys Res Commun. 2008; 369:1098-1102.
33. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007; 7:415-428.
34. Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002; 3:155-166.
35. Wu Y, Liu HB, Ding M, Liu JN, Zhan P, Fu XS, Lu G. The impact of E-cadherin expression on non-small cell lung cancer survival: a meta-analysis. Mol Biol Rep. 2012; 39:9621-9628.
36. Nagathihalli NS, Massion PP, Gonzalez AL, Lu P, Datta PK. Smoking induces epithelial-to-mesenchymal transition in non-small cell lung cancer through HDAC-mediated downregulation of E-cadherin. Mol Cancer Ther. 2012; 11:2362-2372.
37. Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004; 6:931-940.
38. Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006; 66:8319-8326.
39. Edme N, Downward J, Thiery JP, Boyer B. Ras induces NBT-II epithelial cell scattering through the coordinate activities of Rac and MAPK pathways. J Cell Sci. 2002; 115:2591-2601.
40. Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000; 275:36803-36810.
41. Avizienyte E, Frame MC. Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Curr Opin Cell Biol. 2005; 17:542-547.
42. Lilien J, Balsamo J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr Opin Cell Biol. 2005; 17:459-465.
43. Nurwidya F, Takahashi F, Murakami A, Kobayashi I, Kato M, Shukuya T, Tajima K, Shimada N, Takahashi K. Acquired resistance of non-small cell lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Respir Investig. 2014; 52:82-91.
44. Li L, Han R, Xiao H, Lin C, Wang Y, Liu H, Li K, Chen H, Sun F, Yang Z, Jiang J, He Y. Metformin sensitizes EGFR-TKI-resistant human lung cancer cells in vitro and in vivo through inhibition of IL-6 signaling and EMT reversal. Clin Cancer Res. 2014; 20:2714-2726.
45. Frederick BA, Helfrich BA, Coldren CD, Zheng D, Chan D, Bunn PA, Raben D. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Molecular Cancer Therapeutics. 2007; 6:1683-1691.
46. Li D, Zhang L, Zhou J, Chen H. Cigarette smoke extract exposure induces EGFR-TKI resistance in EGFR-mutated NSCLC via mediating Src activation and EMT. Lung Cancer. 2016; 93:35-42.
47. Liu M, Zhou C, Zheng J. Cigarette smoking impairs the response of EGFR-TKIs therapy in lung adenocarcinoma patients by promoting EGFR signaling and epithelial-mesenchymal transition. Am J Transl Res. 2015; 7:2026-2035.
48. Stewart DJ. Wnt Signaling Pathway in Non-Small Cell Lung Cancer. J Natl Cancer Inst. 2013; 106:djt356.
49. Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004; 303:1483-1487.
50. Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell. 2005; 16:1987-2002.
51. Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, Beug H, Grunert S. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol. 2002; 156:299-313.
52. Shintani Y, Maeda M, Chaika N, Johnson KR, Wheelock MJ. Collagen I promotes epithelial-to-mesenchymal transition in lung cancer cells via transforming growth factor-beta signaling. Am J Respir Cell Mol Biol. 2008; 38:95-104.
53. Mittal V. Epithelial Mesenchymal Transition in Aggressive Lung Cancers. Adv Exp Med Biol. 2016; 890:37-56.
54. Suresh R, Ali S, Ahmad A, Philip PA, Sarkar FH. The Role of Cancer Stem Cells in Recurrent and Drug-Resistant Lung Cancer. Adv Exp Med Biol. 2016; 890:57-74.
55. Tirino V, Camerlingo R, Bifulco K, Irollo E, Montella R, Paino F, Sessa G, Carriero MV, Normanno N, Rocco G, Pirozzi G. TGF-beta1 exposure induces epithelial to mesenchymal transition both in CSCs and non-CSCs of the A549 cell line, leading to an increase of migration ability in the CD133+ A549 cell fraction. Cell Death Dis. 2013; 4:e620.
56. Wang Z, Li Y, Ahmad A, Azmi AS, Kong D, Banerjee S, Sarkar FH. Targeting miRNAs involved in cancer stem cell and EMT regulation: An emerging concept in overcoming drug resistance. Drug Resist Updat. 2010; 13:109-18.
57. Trumpp A, Wiestler OD. Mechanisms of Disease: cancer stem cells--targeting the evil twin. Nat Clin Pract Oncol. 2008; 5: 337-47.
58. Brabletz T, Hlubek F, Spaderna S, Schmalhofer O, Hiendlmeyer E, Jung A, Kirchner T. Invasion and metastasis in colorectal cancer: epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin. Cells Tissues Organs. 2005; 179:56-65.
59. Ma T, Zhao Y, Wei K, Yao G, Pan C, Liu B, Xia Y, He Z, Qi X, Li Z, Wang J, Shao Y. MicroRNA-124 Functions as a Tumor Suppressor by Regulating CDH2 and Epithelial-Mesenchymal Transition in Non-Small Cell Lung Cancer. Cell Physiol Biochem. 2016; 38:1563-1574.
60. Shi H, Ji Y, Zhang D, Liu Y, Fang P. MiR-135a inhibits migration and invasion and regulates EMT-related marker genes by targeting KLF8 in lung cancer cells. Biochem Biophys Res Commun. 2015; 465:125-130.
61. Li J, Song Y, Wang Y, Luo J, Yu W. MicroRNA-148a suppresses epithelial-to-mesenchymal transition by targeting ROCK1 in non-small cell lung cancer cells. Mol Cell Biochem. 2013; 380:277-282.
62. Yu T, Li J, Yan M, Liu L, Lin H, Zhao F, Sun L, Zhang Y, Cui Y, Zhang F, Li J, He X, Yao M. MicroRNA-193a-3p and -5p suppress the metastasis of human non-small-cell lung cancer by downregulating the ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Oncogene. 2015; 34:413-423.
63. Cufi S, Bonavia R, Vazquez-Martin A, Oliveras-Ferraros C, Corominas-Faja B, Cuyas E, Martin-Castillo B, Barrajon-Catalan E, Visa J, Segura-Carretero A, Joven J, Bosch-Barrera J, Micol V, et al. Silibinin suppresses EMT-driven erlotinib resistance by reversing the high miR-21/low miR-200c signature in vivo. Sci Rep. 2013; 3:2459.
64. Tejero R, Navarro A, Campayo M, Vinolas N, Marrades RM, Cordeiro A, Ruiz-Martinez M, Santasusagna S, Molins L, Ramirez J, Monzo M. miR-141 and miR-200c as markers of overall survival in early stage non-small cell lung cancer adenocarcinoma. PLoS One. 2014; 9:e101899.
65. Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, Diao L, Wang J, Roybal JD, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014; 5:5241.
66. Pacurari M, Addison JB, Bondalapati N, Wan YW, Luo D, Qian Y, Castranova V, Ivanov AV, Guo NL. The microRNA-200 family targets multiple non-small cell lung cancer prognostic markers in H1299 cells and BEAS-2B cells. Int J Oncol. 2013; 43:548-560.
67. Schliekelman MJ, Gibbons DL, Faca VM, Creighton CJ, Rizvi ZH, Zhang Q, Wong CH, Wang H, Ungewiss C, Ahn YH, Shin DH, Kurie JM, Hanash SM. Targets of the tumor suppressor miR-200 in regulation of the epithelial-mesenchymal transition in cancer. Cancer Res. 2011; 71:7670-7682.
68. Li J, Tan Q, Yan M, Liu L, Lin H, Zhao F, Bao G, Kong H, Ge C, Zhang F, Yu T, Li J, He X, et al. miRNA-200c inhibits invasion and metastasis of human non-small cell lung cancer by directly targeting ubiquitin specific peptidase 25. Mol Cancer. 2014; 13:166.
69. You J, Li Y, Fang N, Liu B, Zu L, Chang R, Li X, Zhou Q. MiR-132 suppresses the migration and invasion of lung cancer cells via targeting the EMT regulator ZEB2. PLoS One. 2014; 9:e91827.
70. Xie Z, Cai L, Li R, Zheng J, Wu H, Yang X, Li H, Wang Z. Down-regulation of miR-489 contributes into NSCLC cell invasion through targeting SUZ12. Tumour Biol. 2015; 36:6497-6505.
71. Xia Y, Wu Y, Liu B, Wang P, Chen Y. Downregulation of miR-638 promotes invasion and proliferation by regulating SOX2 and induces EMT in NSCLC. FEBS Lett. 2014; 588: 2238-45.
72. Ke Y, Zhao W, Xiong J, Cao R. miR-149 Inhibits Non-Small-Cell Lung Cancer Cells EMT by Targeting FOXM1. Biochem Res Int. 2013; 2013:506731.
73. Liu MX, Zhou KC, Cao Y. MCRS1 overexpression, which is specifically inhibited by miR-129*, promotes the epithelial-mesenchymal transition and metastasis in non-small cell lung cancer. Mol Cancer. 2014; 13:245.
74. Zhang Y, Han L, Pang J, Wang Y, Feng F, Jiang Q. Expression of microRNA-452 via adenoviral vector inhibits non-small cell lung cancer cells proliferation and metastasis. Tumour Biol. 2016; 37:8259-8270.
75. Li L, Wu D. miR-32 inhibits proliferation, epithelial-mesenchymal transition, and metastasis by targeting TWIST1 in non-small-cell lung cancer cells. Onco Targets Ther. 2016; 9:1489-1498.
76. Yang L, Yang J, Li J, Shen X, Le Y, Zhou C, Wang S, Zhang S, Xu D, Gong Z. MircoRNA-33a inhibits epithelial-to-mesenchymal transition and metastasis and could be a prognostic marker in non-small cell lung cancer. Sci Rep. 2015; 5:13677.
77. Kumarswamy R, Mudduluru G, Ceppi P, Muppala S, Kozlowski M, Niklinski J, Papotti M, Allgayer H. MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snai1 and is downregulated in non-small cell lung cancer. Int J Cancer. 2012; 130:2044-2053.
78. Alam M, Ahmad R, Rajabi H, Kufe D. MUC1-C Induces the LIN28B-->LET-7-->HMGA2 Axis to Regulate Self-Renewal in NSCLC. Mol Cancer Res. 2015; 13:449-460.
79. Jin Z GL, Song Y, Xiang GM, Chen SX, Gao B. MicroRNA-138 regulates chemoresistance in human non-small cell lung cancer via epithelial mesenchymal transition. Eur Rev Med Pharmacol Sci. 2016; 20:1080-1086.
80. Nishijima N, Seike M, Soeno C, Chiba M, Miyanaga A, Noro R, Sugano T, Matsumoto M, Kubota K, Gemma A. miR-200/ZEB axis regulates sensitivity to nintedanib in non-small cell lung cancer cells. Int J Oncol. 2016; 48:937-944.
81. Park KS, Raffeld M, Moon YW, Xi L, Bianco C, Pham T, Lee LC, Mitsudomi T, Yatabe Y, Okamoto I, Subramaniam D, Mok T, Rosell R, et al. CRIPTO1 expression in EGFR-mutant NSCLC elicits intrinsic EGFR-inhibitor resistance. J Clin Invest. 2014; 124:3003-3015.
82. Ahmad A, Maitah MY, Ginnebaugh KR, Li Y, Bao B, Gadgeel SM, Sarkar FH. Inhibition of Hedgehog signaling sensitizes NSCLC cells to standard therapies through modulation of EMT-regulating miRNAs. J Hematol Oncol. 2013; 6:77.
83. Shi L, Zhang S, Wu H, Zhang L, Dai X, Hu J, Xue J, Liu T, Liang Y, Wu G. MiR-200c increases the radiosensitivity of non-small-cell lung cancer cell line A549 by targeting VEGF-VEGFR2 pathway. PLoS One. 2013; 8:e78344.
84. Kang J, Kim E, Kim W, Seong KM, Youn H, Kim JW, Kim J, Youn B. Rhamnetin and cirsiliol induce radiosensitization and inhibition of epithelial-mesenchymal transition (EMT) by miR-34a-mediated suppression of Notch-1 expression in non-small cell lung cancer cell lines. J Biol Chem. 2013; 288:27343-27357.
85. Jiang Z, Yin J, Fu W, Mo Y, Pan Y, Dai L, Huang H, Li S, Zhao J. MiRNA 17 family regulates cisplatin-resistant and metastasis by targeting TGFbetaR2 in NSCLC. PLoS One. 2014; 9:e94639.
86. Zhong Z, Xia Y, Wang P, Liu B, Chen Y. Low expression of microRNA-30c promotes invasion by inducing epithelial mesenchymal transition in non-small cell lung cancer. Mol Med Rep. 2014; 10:2575-2579.
87. Mo X, Zhang F, Liang H, Liu M, Li H, Xia H. miR-544a promotes the invasion of lung cancer cells by targeting cadherina 1 in vitro. Onco Targets Ther. 2014; 7:895-900.
88. Kim G, An HJ, Lee MJ, Song JY, Jeong JY, Lee JH, Jeong HC. Hsa-miR-1246 and hsa-miR-1290 are associated with stemness and invasiveness of non-small cell lung cancer. Lung Cancer. 2016; 91:15-22.
89. Yamashita R, Sato M, Kakumu T, Hase T, Yogo N, Maruyama E, Sekido Y, Kondo M, Hasegawa Y. Growth inhibitory effects of miR-221 and miR-222 in non-small cell lung cancer cells. Cancer Med. 2015; 4: 551-564.
90. Cao M, Seike M, Soeno C, Mizutani H, Kitamura K, Minegishi Y, Noro R, Yoshimura A, Cai L, Gemma A. MiR-23a regulates TGF-beta-induced epithelial-mesenchymal transition by targeting E-cadherin in lung cancer cells. Int J Oncol. 2012; 41:869-875.
91. Kitamura K, Seike M, Okano T, Matsuda K, Miyanaga A, Mizutani H, Noro R, Minegishi Y, Kubota K, Gemma A. MiR-134/487b/655 cluster regulates TGF-beta-induced epithelial-mesenchymal transition and drug resistance to gefitinib by targeting MAGI2 in lung adenocarcinoma cells. Mol Cancer Ther. 2014; 13:444-453.
92. Sun M, Liu XH, Wang KM, Nie FQ, Kong R, Yang JS, Xia R, Xu TP, Jin FY, Liu ZJ, Chen JF, Zhang EB, De W, et al. Downregulation of BRAF activated non-coding RNA is associated with poor prognosis for non-small cell lung cancer and promotes metastasis by affecting epithelial-mesenchymal transition. Mol Cancer. 2014; 13:68.
93. Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, Thomas M, Berdel WE, Serve H, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003; 22:8031-8041.
94. Shen L, Chen L, Wang Y, Jiang X, Xia H, Zhuang Z. Long noncoding RNA MALAT1 promotes brain metastasis by inducing epithelial-mesenchymal transition in lung cancer. J Neurooncol. 2015; 121:101-108.
95. Zhou X, Chen J, Tang W. The molecular mechanism of HOTAIR in tumorigenesis, metastasis, and drug resistance. Acta Biochim Biophys Sin (Shanghai). 2014; 46:1011-1015.
96. Padua Alves C, Fonseca AS, Muys BR, de Barros ELBR, Burger MC, de Souza JE, Valente V, Zago MA, Silva WA Jr. Brief report: The lincRNA Hotair is required for epithelial-to-mesenchymal transition and stemness maintenance of cancer cell lines. Stem Cells. 2013; 31:2827-2832.
97. Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007; 129:1311-1323.
98. Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, Mehra R, Laxman B, Cao X, Yu J, Kleer CG, Varambally S, Chinnaiyan AM. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene. 2008; 27:7274-7284.
99. Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. 2010; 39:761-772.
100. Lu L, Luo F, Liu Y, Liu X, Shi L, Lu X, Liu Q. Posttranscriptional silencing of the lncRNA MALAT1 by miR-217 inhibits the epithelial-mesenchymal transition via enhancer of zeste homolog 2 in the malignant transformation of HBE cells induced by cigarette smoke extract. Toxicol Appl Pharmacol. 2015; 289:276-285.
101. Liu Y, Luo F, Xu Y, Wang B, Zhao Y, Xu W, Shi L, Lu X, Liu Q. Epithelial-mesenchymal transition and cancer stem cells, mediated by a long non-coding RNA, HOTAIR, are involved in cell malignant transformation induced by cigarette smoke extract. Toxicol Appl Pharmacol. 2015; 282:9-19.
102. Shintani Y, Okimura A, Sato K, Nakagiri T, Kadota Y, Inoue M, Sawabata N, Minami M, Ikeda N, Kawahara K, Matsumoto T, Matsuura N, Ohta M, et al. Epithelial to mesenchymal transition is a determinant of sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann Thorac Surg. 2011; 92:1794-1804.
103. Cano A, Nieto MA. Non-coding RNAs take centre stage in epithelial-to-mesenchymal transition. Trends Cell Biol. 2008; 18:357-359.
104. Li J, Wang Y, Luo J, Fu Z, Ying J, Yu Y, Yu W. miR-134 inhibits epithelial to mesenchymal transition by targeting FOXM1 in non-small cell lung cancer cells. FEBS Lett. 2012; 586:3761-3765.
105. Sarkar D, Leung EY, Baguley BC, Finlay GJ, Askarian-Amiri ME. Epigenetic regulation in human melanoma: past and future. Epigenetics. 2015; 10:103-121.
106. Lv J, Fan HX, Zhao XP, Lv P, Fan JY, Zhang Y, Liu M, Tang H. Long non-coding RNA Unigene56159 promotes epithelial-mesenchymal transition by acting as a ceRNA of miR-140-5p in hepatocellular carcinoma cells. Cancer Lett. 2016; 382:166-175.
107. Li SP, Xu HX, Yu Y, He JD, Wang Z, Xu YJ, Wang CY, Zhang HM, Zhang RX, Zhang JJ, Yao Z, Shen ZY. LncRNA HULC enhances epithelial-mesenchymal transition to promote tumorigenesis and metastasis of hepatocellular carcinoma via the miR-200a-3p/ZEB1 signaling pathway. Oncotarget. 2016; 7:42431-42446. doi: 10.18632/oncotarget.9883.
108. Sun CC, Li SJ, Zhang F, Xi YY, Wang L, Bi YY, Li DJ. Long non-coding RNA NEAT1 promotes non-small cell lung cancer progression through regulation of miR-377-3p-E2F3 pathway. Oncotarget. 2016; 7:51784-51814. doi: 10.18632/oncotarget.10108.
109. Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, Loewer A, Ziebold U, Landthaler M, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013; 495:333-338.
110. Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015; 160:1125-1134.