
INTRODUCTION
Acute lymphoblastic leukemia (ALL), a malignancy characterized by clonal proliferation of arrested immature blood cell precursors (blasts), progresses rapidly and often appears without an obvious prodrome [1]. It can be classified according to either immunophenotype or by molecular identification of a growing number of somatic genetic alterations characterized during the last decades [2, 3]. In pediatric B-cell acute lymphoblastic leukemia (ALL), the most frequent chromosomal lesion is t(12;21)(p13;q22), which results in its molecular genetic counterpart, the ETV6/RUNX1 (also known as TEL/AML1) fusion gene [4, 5]. This alteration occurs in approximately 25% of childhood ALL diagnosed between the ages of 2 and 10 years, with a median age of 4 years [6-8]. Based on the excellent molecular response to treatment and beneficial clinical outcome, it was originally believed that this rearrangement is a rather favorable prognostic indicator [9, 10]. However, this notion was subsequently disputed as others found predominantly late relapses occurring in up to 20% of patients [7, 11]. Thus, the prognostic implication of the ETV6/RUNX1 (E/R) fusion continues to be debated. Over the past decades, remarkable advances have been made on the precise role of the E/R fusion gene product in childhood ALL. We review here some of the major findings that highlight the impact of the E/R fusion gene on childhood ALL at initiation and relapse.
PATHOGENESIS OF ETV6/RUNX1-POSITIVE CHILDHOOD ALL
The E/R fusion gene develops mainly as an early, prenatal event in childhood ALL. The expression of E/R results in the generation of a persistent pre-leukemic clone, which postnatally converts, at low frequency, to ALL after the acquisition of necessary secondary genetic abnormalities [12, 13]. Several studies have addressed the mechanism by which E/R contributes to leukemogenesis, including the necessary secondary genetic lesions, the cellular framework in which E/R initially arises and the maintenance of a pre-leukemic condition.
ETV6/RUNX1-positive childhood ALL: a “two-hit” disease
In considering the natural history and etiology of childhood ALL, a key problem is when and how the E/R fusion gene is generated and whether this is an early or initiating event [14]. The detection of the E/R fusion sequence in identical twins and in neonatal blood spots of children with ALL indicate that this gene fusion originates in the prenatal period [14-21]. A concordance rate for ALL of only 5% in twin children, the highly variable postnatal latency period (up to 14 years) of the disease, and evidence from transgenic animal models of E/R fusion all suggest that additional postnatal genetic events are required for the development of clinically overt leukemia [22-30]. Thus, although the E/R fusion gene constitutes the first step in the transformation of normal cells into malignant cells, a modest number of recurrent or “driver” copy number alterations (CNAs) are most likely necessary for leukemia to develop (Figure 1).
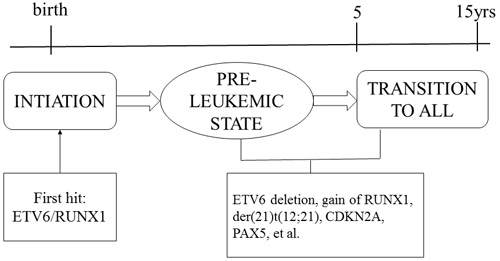
Figure 1: Two-hit model of the natural history of E/R-positive ALL.
Initially, most studies addressing secondary leukemia-promoting genetic changes have focused on the deletion of the normal, or non-rearranged, ETV6 allele [31]. This genetic abnormality accounts for approximately 70% of t(12;21)-positive ALL cases [32]. ETV6 can function as a tumor suppressor [33] and dimerize with E/R to reduce its transforming activity [34]. The other common genetic changes in the ETV6 and RUNX1 genes are an extra RUNX1 gene (23%) and/or an extra der(21)t(12;21) (10%) [35, 36]. Approximately 20% of the t(12;21)-positive patients have more than one additional genetic change in ETV6 or RUNX1 [35, 37]. These genomic changes are so prevalent that their existence can be taken as significant circumstantial evidence for a cryptic E/R fusion in pediatric ALL.
However, to fully understand the genetic evolution of E/R-positive ALL, identification of the entire spectrum of genetic changes that accompany this fusion gene is necessary. The genome of E/R-positive ALL has been well characterized at the copy number and cytogenetic level. A large number of secondary genetic abnormalities in E/R-positive ALL may be observed by conventional or molecular cytogenetics methods (Table 1) [38-48]. Each E/R-positive ALL patient has an average of 3.5 additional CNAs (range 0-14). Among these, several are recurrent deletions in genes controlling B-cell lineage differentiation, cell cycle, or nuclear hormone response [38, 47, 49]. In addition, the majority of E/R-positive cases display a unique pattern of CNAs, indicating that a large heterogeneity exists within this ALL subtype [47]. The frequent larger alterations include deletions of 12p, 6q, 9p, 11q, and 13q, losses of X, 8, and 13, and gains of 21, 4, 10, and 16 [38, 44, 50]. The deletion of 12p (39%) is the most common abnormality, its most likely pathogenetically important outcome being the loss of wild-type ETV6 allele [51]. The 9p deletion can be seen in up to 25% of E/R-positive patients, and it often affects the tumor suppressor CDKN2A/CDKN2B and the B-cell differentiation regulator PAX5 [35, 38, 40]. Genome-wide, high resolution CNAs analyses have revealed an increasing number of putative causative submicroscopic changes in diverse genes such as CD200, BTLA, TBL1XR1, GLRX2, NR3C2, NR3C1, TCF3, EBF1, LEF1, IKZF1, CCNC, ARMC2, BTG1, SPANXB, TP63, PTPRJ, ATF7IP, and MGA [13, 36, 42, 47, 52]. In addition, a rare case of an E/R-positive ALL patient that acquired the BCR-ABL1 gene rearrangement as a secondary change has been recently reported [53]. The genes listed above belong to many different classes and affect various pathways. For instance, the loss of PAX5, EBF1 and TCF3 can lead to impaired production of mature B-cells [49]; deletions of CD200 and BTLA contribute to a disrupted function of the immune system [47]; deletion of TBL1XR1, which is a key component of the SMRT and N-CoR compressor complexes, is associated with overexpression of genetic targets of the retinoic acid and thyroid hormone receptors [42]. Several studies have depicted the relationship between CNAs and their assumed temporal order of appearance with branching and distance-based oncogenetic trees [47, 54]. These tree models showed that the CDKN2A/B, ETV6, del(6q), PAX5 and +16 are probably among the earliest aberrations [47]. But further studies are required to confirm the sequential order of acquisition for different CNAs that can be observed directly.
These secondary genetic events are major rate-limiting events during the process of formation of E/R-positive ALL and may be expected to drive the evolution of overt leukemic cells, culminating in a clinical diagnosis of ALL. However, the mechanisms triggering these second hits have not been completely elucidated. A study reported that the expression of E/R was associated with increased ROS levels [55]. Elevated ROS could increase genetic instability and double-strand DNA breaks [56], thus leading to the accumulation of secondary mutations. Recently, Papaemmanuil et al. used exome and low-coverage whole-genome sequencing to characterize secondary events associated with leukemic transformation, confirming that aberrant RAG recombinase activity contributes critically to secondary events leading to leukemic transformation in E/R-positive ALL [13].
Table 1: Common (>5% frequency) secondary genetic changes observed in E/R-positive ALL
Chr |
Genetic abnormality |
Frequency (%) |
Possible candidate genes |
12 |
del(12p) del(12)(p11-13) del(12)(p13.2) del(12)(p21.3) |
18-69 |
ETV6, CDKN1B, BCL2L14, BTG1 |
6 |
del(6q) del(6)(q21-27) der(6)t(X;6) |
13-37 |
AIM1, PRDM1, FOXO3, CCNC, FYN |
9 |
del(9p) del(9)(p13-24) del(9)(p21.3) del(9)(p13.2) |
7-29 |
CDKN2A/B, PAX5, MTAP, JAK2, P14ARF, P16INK4a/ARF |
21 |
*gain(21) gain(21q) |
10-25 |
RUNX1 |
5 |
del(5)(q31.3-33.3) |
23 |
NR3C1, EBF1 |
14 |
del(14)(q32.33) del(14)(q11.2) |
3-21 |
DPF3 |
3 |
del(3)(p21) del(3)(q26.32) del(3)(q13.2) del(3)(p14.2) |
3-21 |
LIMD1, ARPP-21, ULK4, FHIT, CD200, BTLA, TBL1XR1 |
7 |
del(7)(q34) del(7)(p14.1) |
6-18 |
IKZF1 |
4 |
gain(4q) del(4)(q31.23) |
6-17 |
NR3C2, YIPF7, ARHGAP10 |
11 |
del(11q) del(11)(q22-23) del(11)(q23-25) del(11)(p13) |
6-13 |
CD44, RAG1, RAG2, BACL2, GNG3, HNRPUL2, TTC9C, ATM, KMT2A |
19 |
del(19)(q13.11) del(19)(q13.32) |
6-13 |
CEBPA, UHRF1, GRLF1, NPAS1, TMEM160 |
22 |
del(22)(q11.22) |
13 |
- |
X |
loss(X)(in females) gain(Xq)(in males) gain(Xp) |
4-11 |
SPANXB, HMGB3, FAM50A, HTATSF1 |
13 |
del(13q) del(13)(q14-34) del(13)(q12.2) |
5-10 |
BTG1, RB1, SERP2, DLEU2, STBP4, TRIM13, KCNRG, MIRN16-1, MIRN15A, DLEU1, DLEU7 |
1 |
del(1)(q31.3) |
10 |
TROVE2, GLRX2, CDC73, B3GALT2, PDE4B |
15 |
del(15)(q15.1) del(15)(q26.1) |
10 |
LTK, MIRN626 |
2 |
del(2)(p25.3) |
9 |
- |
8 |
del(8p) del(8)(p11-23) |
6-8 |
TOX |
16 |
gain (16) |
6 |
- |
Chr, chromosome;
*Additional copy of chromosome 21 or an additional copy of the der(21)t(12;21)(p13;q22)
Clonal origin of ETV6/RUNX1-positive childhood ALL
Understanding the pathogenesis of E/R-positive ALL requires knowledge of not only the specific contributory fusion and the secondary genetic hits but also the cellular framework in which they arise and function. Most of the leukemic cells in E/R-positive ALL can be characterized by phenotypes (CD19+, CD10+, TdT+ and RAG1/2+) and genotypes (ongoing clonal IgH rearrangements) indicative of a B precursor cell identity. However, the cell of origin in which the initial functional impact of the fusion gene occurs has been contentious. It may lie anywhere antecedent to B precursor cells in the lineage hierarchy [57]. IgH, TCR and ETV6/RUNX1 are leukemia-specific molecular markers in E/R-positive ALL. Analysis of these markers could illuminate when ETV6/RUNX1 gene fusion occurs during B cell development.
Confirmation of the leukemic cell of origin is fundamental to identify the cellular and molecular mechanisms that underlie leukemogenesis. A popular hypothesis is that completely transformed cancer clones are settled in a hierarchical pattern and sustained by a few “cancer stem cells” [58]. However, less is known about the “precancerous” cells upon which first-hit events occur [25]. In fact, it has been reported in other leukemias that the initiating events could occur upstream within the hematopoiesis hierarchy. For instance, early transformation events have been demonstrated to take place in CD34+/CD38− cells of Philadelphia chromosome-positive ALL [59]. In E/R-positive ALL, this fusion transcript can occur during B-cell development before rearrangement of IgH and TCR genes [60]. These rearrangements are features of B-cell precursor leukemia and commence at the pro-B cell stage of normal B-cell lineage development [61]. Thus, the E/R fusion premalignant clone may occur before the pro-B cell stage. Sequence analysis of the E/R fusion region further supports this possibility, as there are no binding motifs indicative of RAG1/2 or TdT activity [62, 63]. In accord with this, expansion of early multipotent progenitors has been detected after retroviral expression of the E/R fusion gene [64]. Besides, modeling data from murine experiments have also suggested a possible multipotential progenitor origin of the fusion gene [26, 65]. The activity of E/R in early progenitors or hematopoietic stem cells (HSCs) may account for the expression of the myeloid markers CD13 and CD33 in up to 48% of E/R-positive ALL patients [60]. The explanation for E/R’s requirement to be expressed in leukemic stem and/or progenitor cells is that lymphoid progenitors lack self-renewal and cannot live long enough to accumulate the mutations required for full transformation [26]. It is advantageous for E/R-positive pre-leukemic cells to preserve the regulatory control mechanisms of normal stem cells to persist in their niche and avoid exhaustion.
However, studies on clonal IgH and/or TCR gene rearrangements in E/R-positive ALL have demonstrated that the fetal cell type in which clonal advantage is elicited by E/R can be propagated after B-lineage commitment [25, 57, 66]. Consistent with this conclusion, retroviral transduction of the E/R fusion gene in murine and human models suggested that E/R could promote self-renewal of early B-cell precursors [25, 66]. Human modeling data showed that a small portion of CD34+CD38–/lowCD19+ cells were present in E/R-positive ALL patients [25, 67]. This kind of cell population, which may function as pre-leukemic stem cells, has significant self-renewal potential and contains a mixture of pro-B cell- and HSC–associated gene expression profiles [25]. Recently, a study analyzed all varieties of Ig and TCR gene rearrangements in five pairs of monozygotic twins concordant for ALL. They proved that the E/R fusion initially expanded in an early B-cell lineage-committed progenitor, most likely at the pro-B, or possibly at the pre-B, cell stage [57]. The effect of E/R at the pre-B or pro-B cell stage is intriguing, as it may prove that the E/R fusion gene can directly influence the phenotype of leukemic cells.
Understanding the cellular targets of primary mutations and the ensuing malignant transformation is fundamental to understanding the function of the E/R fusion. In addition, this is also relevant to cancer therapy, where specific targeting of tumor-propagating cells may be desirable. Thus, it seems safe to conclude that the jury is still out on this issue, and further research is still required.
ETV6/RUNX1 contribution to the pre-leukemic and leukemic states
Observations on normal cord blood and monozygotic twins demonstrated that the E/R fusion gene induces a pre-leukemic phenotype that can remain covert for up to 15 years [12]. Therefore, E/R is not only responsible for the initiation of leukemia but also essential for disease progression and maintenance. Although the expression of E/R interferes with critical regulatory functions [68], maintenance of a pre-leukemic condition by E/R fusion itself is not sufficient to explain the evolutionary and clinical emergence of ALL [69]. Instead, additional selective pressure and clonal advantage have been demonstrated to participate in this process [12, 70, 71].
Both ETV6 and RUNX1 are known regulators of hematopoiesis. The E/R fusion gene includes the N-terminal pointed (PNT) dimerization domain of ETV6 and the N-terminal Runt homology domain (RHD) and the C-terminal transactivation (TA) domain of RUNX1 [35]. The resulting hybrid fusion protein has been shown to maintain transcription factor properties [72]. It can bind to RUNX1 target genes and potentially convert RUNX1 to a negative transcriptional regulator [73, 74]. Maximal repression requires the binding of mSin3A, N-CoR and histone deacetylase-3 (HDAC3) to ETV6 [72]. Although the E/R chimeric protein is assumed to act as transcriptional repressor of RUNX1 target genes, several studies have reported similar levels of gene repression and activation for protein-coding and miRNA genes [68, 71, 75]. In addition, the E/R fusion gene product can disrupt the transcriptional repression of wild-type ETV6 by dimerization via the PNT domain [76].
Although the E/R rearrangement involves two well-characterized transcription factors, the molecular pathways affected by the translocation are also responsible for leukemia clone maintenance and propagation (Figure 2). First of all, E/R can reduce sensitivity to the inhibition of proliferation mediated by TGF-β signaling, suggesting that the dysregulated immune responses to infection might promote the malignant evolution of pre-leukemic clones [70]. The E/R fusion gene can also induce signal transducer and activator of transcription 3 (STAT3) activation, aiding to the self-renewal and leukemogenic activity of leukemia cells [77, 78]. STAT3 activation has been shown to transcriptionally induce MYC [77] and SMAD7 [78] gene expression in E/R leukemic cells. In addition, the E/R gene product can activate the phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) signaling pathway [79], resulting in proliferation and cell survival of leukemic cells. Reflecting the importance of PI3K/AKT/mTOR signaling cascade in many cellular tasks, several PI3K/AKT/mTOR-regulated processes, including cell adhesion and DNA damage response, are also affected in E/R-positive leukemia [79]. Moreover, E/R can directly bind to the EPOR promoter and increase cell survival through activation of the JAK2-STAT5-BCL-XL pathway [12] and the PI3K/AKT/mTOR pathway [80]. Last but not least, the chimeric E/R protein can upregulate MDM2 promoter activity. MDM2 functions as a crucial negative regulator of P53, and ultimately represses P53 signaling [68, 81]. The suppression of P53 signaling is a crucial event in the development of leukemic cell survival [81].
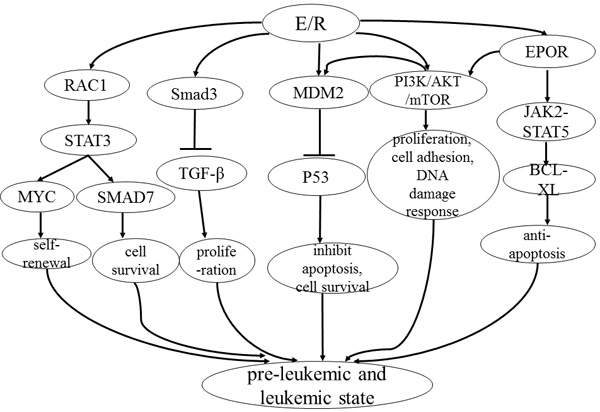
Figure 2: Model for pathways influenced by E/R. 1) E/R regulates the phosphorylation of STAT3 via activation of RAC1, resulting in the transcriptional induction of MYC. 2) E/R can bind to a principal TGF-β signaling target, Smad3, which has an inhibitory impact on the response to TGF-β signaling. 3) E/R can upregulate MDM2 promoter activity; MDM2 functions as a crucial negative regulator of p53, repressing its activity. 4) E/R activates PI3K/AKT/mTOR signaling and interferes with proliferation, cell adhesion and DNA damage response. Activation of PI3K/AKT/mTOR signaling also promotes the phosphorylation of MDM2 and its localization in the nucleus where it binds to p53. 5) E/R can directly bind the EPOR promoter and increase cell survival through activation of the JAK2-STAT5 pathway and upregulation of antiapoptotic BCL-XL. EPOR is also one of the upstream signaling components of the PI3K/AKT/mTOR signaling cascade. All these signal pathways facilitate the E/R pre-leukemic state.
Epigenetic alterations, including DNA methylation and histone modifications, have also been reported to contribute to ALL progression [82, 83]. Stephan et al. combined methylome and transcriptome approaches to identify epigenetic biomarkers specific for E/R-positive ALL, and showed the effects of methylation on the expression of 17 potential drivers of leukemogenesis [75]. Among these genes, EPOR has been shown to have a functional impact on promoting leukemic cell survival [12]. The oncofetal RNA-binding protein IGF2BP1 (IGF2 mRNA binding protein 1) is another hypomethylated and upregulated gene in E/R-positive ALL. As a powerful regulator of E/R mRNA stability, IGF2BP1 may be a prominent player in cell transformation events in E/R-associated leukemogenesis [84-86]. Several miRNA genes are also differentially methylated in ALL patients with the E/R fusion gene [75]. miRNA-320a was found to be both hypermethylated and downregulated, together with miRNA-494, in E/R-positive leukemia [71]. Both miRNAs are coupled to the repression of survivin and their inhibition may promote antiapoptotic signaling. miRNA200c is another hypermethylated miRNA gene that was shown to exhibit strong tumor suppressor properties, partially by interacting with the epigenetic regulator and polycomb group protein BMI1 [87]. In addition, studies on the association between E/R-positive ALL and histone modifications have shown that preserved global histone H4 acetylation [88] and mutations in NSD2 histone methyltransferase [89] may also promote leukemogenesis. Other miRNA-related mechanisms have also been demonstrated to provide survival advantage to E/R-positive leukemia cells. For instance, hsa-mir-125b-2 is highly expressed in E/R-positive leukemia, and can promote cell survival under growth inhibitory and apoptotic conditions in a P53-independent manner [90]. In contrast, mir-181a-1 is significantly reduced in E/R-positive cases, and a double-negative loop involving mir-181a-1 and E/R may contribute to E/R-driven arrest of differentiation [91].
Recently, a comprehensive analysis of the long non-coding RNA (lnc RNA) transcriptome has been conducted in E/R-positive ALL. Both lnc-RTN4R-1 and lnc-NKX2-3-1 can be regulated by the E/R fusion protein and might be functionally implicated in leukemogenesis [92]. In addition, E/R can downregulate mitotic arrest-deficient 2L1 (MAD2L1) to attenuate the mitotic checkpoint (MC) [93]. This may lead to near-triploidy, near-tetraploidy, and trisomy 21 as rather specific types of secondary changes. Furthermore, two recent studies showed a novel cooperative activity of E/R with the antiapoptotic protein BCL2 [94] and the protein tyrosine phosphatase 4A3 (PTP4A3) [95], although the impact on leukemia progression needs to be further explored.
MECHANISMS OF RELAPSE OF ETV6/RUNX1-POSITIVE CHILDHOOD ALL
Pediatric ETV6/RUNX1-positive leukemia generally has favorable long-term remission and survival rates [96], but as in ALL initial manifestation, ETV6/RUNX1 is the most common structural genetic aberration in childhood B-cell precursor (BCP) ALL at first relapse [97]. Only 10% of E/R-positive relapses occur early, and very early relapses are rare. In most patients relapses occur several years after cessation of treatment [98], and occasionally after 10-20 years [99]. Relapsed childhood ETV6/RUNX1-positive leukemia is a clinically and biologically heterogeneous disease. A variety of studies have focused on the relationship between the clones giving rise to the primary and recurrent leukemias, as well as the molecular abnormalities responsible for relapse.
Clonal origins of relapse in childhood ETV6/RUNX1-positive ALL
To characterize the clonal origins of E/R-positive ALL relapse, many studies have compared Ig/TCR rearrangements, genomic boundaries of unrearranged ETV6 allele, and CNA patterns of matched diagnostic and relapse samples [97, 100-103]. The simplest interpretation of relapse is the re-emergence of progeny derived from the dominant clone observed at diagnosis. This hypothesis was advanced by the discovery that the initial and the relapse clones shared identical clonotypic Ig/TCR markers in some cases [104, 105]. Late relapsing cases of ALL are chemosensitive with durable term remissions, possibly implying that initial survival and re-emergence share similar properties [106]. The cells from the original clone might keep smoldering for a long time [107]. Comparison of the genomic boundaries of unrearranged ETV6 allele indicated that the relapse clone, at least in some patients, derive not from a dominant clone but from a sibling clone present at diagnosis [101-103]. While leukemic clones both at initiation and relapse evolve from a shared ancestral clone [102, 103], the relapse clone seems to be already present at the time of diagnosis at rates of 1x10-4 to 5x10-3 cells [100]. During initial therapy, this minor population exhibits only moderate reduction with respect to the bulk of diagnostic dominant clones, but rapidly expands before clinical relapse. After relapse development, these initially slow-responding clones can be rapidly eradicated by the relapse treatment, emphasizing their incongruent biology at the two time points of leukemia manifestation [100].
Recently, research on the clonal origins of relapse in childhood ALL in general or E/R-positive ALL in particular [108, 109] has drawn the conclusion that, irrespective of time to relapse, relapses can emerge from either major or minor subclones already present at diagnosis [108-110]. These studies mainly compared SNP array–defined CNAs in relapse versus matched diagnosis samples. The relapse-associated CNAs, either present at diagnosis or acquired as new genomic alterations after initial therapy, allow mapping the evolutionary relationship between diagnosis and relapse [101, 109]. In most cases, the clones at diagnosis and relapse have some identical CNAs, indicating a shared clonal origin [109]. Some CNAs exclusively present in relapse are present at low levels at presentation, implying that relapses can indeed originate from a minor subclone at diagnosis [101, 108]. van Delft et al. have devised a scheme to define 4 types of CNAs, and for each of them they indicate possible clonal origins with respect to the matched diagnosis samples [109]. In type 1, the relapse CNAs resemble the CNAs at diagnosis, thus the clonal origins of relapse may be linked to the dominant clone at diagnosis. In type 2, the clone at relapse acquires extra CNAs, and may derive from a derivative minor clone (from the major clone) at diagnosis. In types 3 and 4, if the clone at relapse shows losses and gains of CNAs, or loses all CNAs present at diagnosis and acquires a completely new set of alterations, suggesting that it may derive from a minor clone at diagnosis. Some researchers have equated minor subclones to ancestral clones in a linear clonal succession model [101, 108], whereas recent studies indicate a rather complex, nonlinear, branching clonal structure [30, 109]. In addition, there is no discernable relationship between the size of the subclone at diagnosis and the time to relapse [109].
Thus, up to date, it can be concluded that the relapse clone of E/R-positive leukemia can originate from either a major or minor clone at presentation (Figure 3). The subclonal diversity at diagnosis provides a variable basis for intraclonal origins of relapse in E/R-positive ALL.
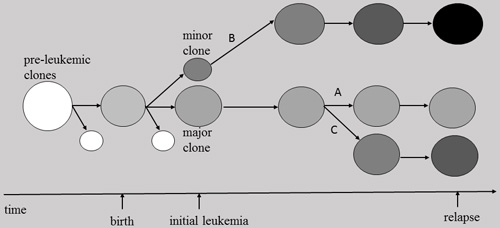
Figure 3: Models for the relapse of ETV6/RUNX1-positive leukemia. Circles with different gray shades represent E/R (+) clones. Their respective location within the spectrum designates their pre-leukemic or leukemic nature. The relapse clones originate from either a major or minor clone at presentation. A, the relapse CNAs resemble the CNAs at diagnosis, hence the relapse clone derives from the dominant clone at diagnosis. B, the clone at relapse acquires extra CNAs, thus it derives from a derivative minor clone at diagnosis. C, the clone at relapse acquires a completely new set of CNAs, thus it may derive from a minor clone at diagnosis.
CNAs in ETV6/RUNX1-positive ALL relapse
Although the majority of E/R-positive relapses can achieve a second continuous complete remission [111], a considerable proportion exhibits drug resistance; these patients eventually experience a subsequent relapse, or even qualify for stem cell transplantation [97]. The clinical heterogeneity among first relapses implies that further genetic abnormalities exist in leukemic cells. Thus, some studies compared diagnostic and relapse CNA patterns to gain further insights into the molecular mechanisms of disease recurrence in E/R-positive ALL [97, 108, 112].
A substantial number of CNAs in relapsed E/R-positive ALL occur in recurrently affected regions (Table 2) [30, 97, 108, 109, 112]. Also, an increase in the number of CNAs per case has been reported between diagnosis and relapse [101, 108, 109]. The pathways primarily affected by these CNAs are those affecting cell cycle regulation, B-cell development, drug metabolism and drug response [101, 108, 113]. Consistent with the CNAs of E/R-positive ALL at diagnosis, chromosomal losses at relapse clearly outnumber gains, and generally involve small genomic regions [97]. Also, additional alterations on chromosomes 12 and 21 are often detected in leukemic cells [36]. The extra der(21)t(12;21) has been reported to be more frequently present in relapsed cases. Patients with this chromosomal change probably have unfavorable long-term clinical outcomes [35]. The recurrent losses at relapse affect chromosomal regions 12p13, 6q21, 6q16.2-3, 8q24, 15q15.1, 9p21, 3p21, 5q, and 3p14.2, whereas gains occur in regions 21q22, 12p and 16 [97, 109]. The genes influenced by loss of 12q13 are ETV6, BCL2L14, and CDKN1B [97]. Loss of ETV6 is the most frequent CNA, observed in about 50% of the relapses [36, 97, 108]. Concomitant deletions of the BCL2L14 and/or CDKN1B genes can affect apoptosis and drug response. These lesions are associated with prominently shorter remissions in relapse patients [97, 112]. The combined loss of 12p13 and 6q21 is common in relapse, resulting in co-inactivation of both FOXO3A (6q21) and CDKN1B (12p13) genes, which may have an adverse prognostic impact [97, 108]. In addition, deletions of CDKN2A/B, which are cell cycle regulators, are associated with poor minimal residual disease response and early emerging relapses [112]. Thus, the deletions of BCL2L14, CDKN1B, CDKN2A/B and FOXO3A may create a genetic background that renders an E/R-positive leukemic cell population drug-resistant and favors the development of early relapses. mir-650 may be associated with time to relapse, as it shows higher expression in early relapse cases [112]. Moreover, several B-lineage transcription factors, among them PAX5, EBF1, and IKZF1, are associated with B-lymphocyte differentiation and are affected by CNAs in E/R-positive relapse as well [97, 108, 113]. CNAs of the X-chromosome and SLX4IP show a gender-specific distribution at relapse; gain of Xq and deletion of SLX4IP are restricted to male patients [97, 112], while loss of Xq is confined to female patients [97]. In a recent study, Yanliang Jin et al. found that relapses occur more frequently in E/R-positive patients who have a three-way translocation pattern, comprising the classic E/R translocation plus a fusion of the remaining parts of ETV6 and RUNX1 with another chromosome. This pattern might be an important risk factor for relapse in patients with the E/R fusion gene [114].
Glucocorticoids (GC) represent an essential component in frontline and relapse treatment protocols for ALL. GC resistance seems to be associated with ineffective blast cell clearance, disease recurrence and treatment failure in childhood ALL [112, 115]. The GC receptor gene NR3C1 (5q31.3) encodes the human GC receptor, and its deletion is linked with impaired response to GC [97, 108, 112]. NR3C1 deletions prevail at relapse and possibly account for the adverse prognosis of some E/R-positive relapses [112]. Other deletions associated with GC-mediated apoptosis target the BCL2 modifying factor (BMF) and components of the mismatch repair (MMR) pathways [108]. BMF plays an important role in GC-mediated induction of apoptosis, and its downregulation leads to GC resistance by E/R-expressing leukemic cells [108, 116]. Defects in the MMR pathway can alter the apoptotic response to DNA-damaging agents and cause resistance to drugs (e.g. mercaptopurine and prednisone) used in the treatment of childhood ALL [108, 113, 117]. These findings indicate that GC-associated drug resistance may play a role in E/R-positive relapse pathogenesis, an observation that may contribute to guide future therapies.
Table 2: Common (>5% frequency) secondary genetic changes observed in E/R-positive relapse ALL
Chr |
Genetic abnormality |
Frequency (%) |
Possible candidate genes |
12 |
del(12p) del(12)(q22) del(12)(p13.1-p12) del(12)(p12) del(12)(p13.1) del(12)(p13.3) del(12)(q21.33) gain(12)(p13.2-13.33) gain(12)(p13.33-13.31) |
22-67 |
ETV6, CDKN1B, BCL2L14, BTG1, ATF7IP, VWF, GABARAPL1 |
22 |
del(22)(q11.22) |
61 |
VPREB1 |
9 |
gain(9)(p13.3) del(9)(p21.3) del(9)(p13.2) |
8-44 |
CDKN2A/B, PAX5, MTAP, ZCCHC7, OPRS1, IL11RA, CCL27, IFN cluster |
21 |
gain(21q) gain(21)(q21.30-22.12) |
29-33 |
RUNX1 |
6 |
del(6q21) |
7-33 |
FOXO3A, AIM1, ARMC2, SESN1 |
11 |
del(11p12) |
9-32 |
RAG1, RAG2 |
3 |
del(3)(p21.21) del(3)(q26.32) del(3)(q13.2) del(3)(p14.2) gain(3)(p21.21) |
12-29 |
FHIT, SMARCC1, MAP4, TBL1XR1, CD200/BTLA, CISH, HEMK1, RASSF1, HYAL2 |
20 |
del(20)(p12.2) (in males) |
26 |
SLX4IP |
5 |
del(5)(q31.3) del(5)(q33.3) |
6-21 |
NR3C1, EBF1 |
15 |
del(15)(q15.1) del(15)(q14) |
10-20 |
LTK, ITPKA, TY-RO3, BMF |
16 |
gain(16) gain(16)(q22.1) |
16-19 |
HSF4, E24F, CTCF, FAM65A |
X |
gain(Xq26.2-28) (in males) gain(Xq27.3-28) (in males) |
9-14 |
SPANX1 |
7 |
del(7)(q34) del(7)(p12.1-12.2) |
7-14 |
IKZF1, TTC26 |
4 |
del(4)(q31.23-31.3) |
8-14 |
NR3C2 |
19 |
del(19)(q13.11) del(19)(p13.3) |
6-12 |
GPI, PDCD2L, UBA2, WTIP, TCF3 |
13 |
del(13q14.2) |
10 |
RB1 |
8 |
gain(8)(q23.1-24.3) |
10 |
cMYC |
10 |
del(10)(q24.1) |
10 |
BLNK(SLP-65) |
2 |
del(2)(p21) |
7 |
MSH2 |
Chr, chromosome;
CONCLUSIONs
The E/R fusion gene is the most frequent genetic abnormality in pediatric ALL. Although multiple studies have identified a variety of second genetic hits, their correlation with E/R and their effects on the pathogenesis of leukemia need to be further explored. The cell of origin in which the initial functional impact of the E/R fusion gene occurs has been contentious. It may lie antecedent to B precursor cells or after B-lineage commitment. Further investigation into the precise cell type of origin has prominent therapeutic significance, as these cells may be the most relevant targets for treatment. In addition, functional studies are needed to unveil the key mechanisms channeling the evolution of E/R-positive pre-leukemic cells into B-ALL. E/R fusion drives not only the leukemic transformation process but also the maintenance and propagation of leukemia cells. The specific molecular alterations, including components of affected signaling pathways and epigenetic regulators, constitute ideal therapeutic targets. For instance, inhibitions of the aberrantly activated PI3K/AKT/mTOR pathway, STAT3 signaling and/or MDM2/P53 interaction represent promising therapeutic strategies.
Analyses of CNAs and Ig/TCR rearrangements on matched diagnostic and relapse samples showed that relapse in E/R-positive ALL originates from either major or minor clones present at diagnosis. The CNAs specific to relapse contribute to not only the development of relapse but also to drug resistance. GC-associated drug resistance is important in the pathogenesis of E/R-positive ALL relapse. Consideration of the course of childhood ALL implies the need for further biological risk markers that enable a more detailed understanding of poor treatment response. These risk markers may contribute to rationalize specific treatment intensification before stem cell transplantation in poor responding E/R-positive relapses.
Abbreviations
ALL-acute lymphoblastic leukemia; E/R-ETV6/RUNX1; CNAs-copy number alterations; HSC-hematopoietic stem cell; STAT-signal transducer and activator of transcription; IGF2BP1-IGF2 mRNA binding protein 1; MAD2L1-mitotic arrest-deficient 2L1; MC-mitotic checkpoint; GC-glucocorticoids; BMF-Bcl2 modifying factor; MMR-mismatch repair.
Author contributions
Congcong Sun mainly prepared and wrote the manuscript. Lixian Chang reviewed the manuscript. Dr. Xiaofan Zhu reviewed the manuscript and approved the final version.
ACKNOWLEDGMENTS
This study was supported by National Natural Science Funds of China (No. 81470339).
CONFLICTs OF INTEREST
The authors declare that they have no competing interests.
FUNDING
This study was supported by National Natural Science Funds of China (No. 81470339).
REFERENCES
1. Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008; 371: 1030-43. doi: 10.1016/s0140-6736(08)60457-2.
2. Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006; 354: 166-78. doi: 10.1056/NEJMra052603.
3. Stanulla M, Schrappe M. Treatment of childhood acute lymphoblastic leukemia. Semin Hematol. 2009; 46: 52-63. doi: 10.1053/j.seminhematol.2008.09.007.
4. Romana SP, Mauchauffe M, Le Coniat M, Chumakov I, Le Paslier D, Berger R, Bernard OA. The t(12;21) of acute lymphoblastic leukemia results in a tel-AML1 gene fusion. Blood. 1995; 85: 3662-70.
5. Shurtleff SA, Buijs A, Behm FG, Rubnitz JE, Raimondi SC, Hancock ML, Chan GC, Pui CH, Grosveld G, Downing JR. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia. 1995; 9: 1985-9.
6. Golub TR, Barker GF, Stegmaier K, Gilliland DG. Involvement of the TEL gene in hematologic malignancy by diverse molecular genetic mechanisms. Curr Top Microbiol Immunol. 1996; 211: 279-88.
7. Harbott J, Viehmann S, Borkhardt A, Henze G, Lampert F. Incidence of TEL/AML1 fusion gene analyzed consecutively in children with acute lymphoblastic leukemia in relapse. Blood. 1997; 90: 4933-7.
8. McLean TW, Ringold S, Neuberg D, Stegmaier K, Tantravahi R, Ritz J, Koeffler HP, Takeuchi S, Janssen JW, Seriu T, Bartram CR, Sallan SE, Gilliland DG, et al. TEL/AML-1 dimerizes and is associated with a favorable outcome in childhood acute lymphoblastic leukemia. Blood. 1996; 88: 4252-8.
9. Uckun FM, Pallisgaard N, Hokland P, Navara C, Narla R, Gaynon PS, Sather H, Heerema N. Expression of TEL-AML1 fusion transcripts and response to induction therapy in standard risk acute lymphoblastic leukemia. Leuk Lymphoma. 2001; 42: 41-56. doi: 10.3109/10428190109097675.
10. Rubnitz JE, Behm FG, Wichlan D, Ryan C, Sandlund JT, Ribeiro RC, Rivera GK, Hancock ML, Relling MV, Evans WE, Pui CH, Downing JR. Low frequency of TEL-AML1 in relapsed acute lymphoblastic leukemia supports a favorable prognosis for this genetic subgroup. Leukemia. 1999; 13: 19-21.
11. Seeger K, Adams HP, Buchwald D, Beyermann B, Kremens B, Niemeyer C, Ritter J, Schwabe D, Harms D, Schrappe M, Henze G. TEL-AML1 fusion transcript in relapsed childhood acute lymphoblastic leukemia. The Berlin-Frankfurt-Munster Study Group. Blood. 1998; 91: 1716-22.
12. Torrano V, Procter J, Cardus P, Greaves M, Ford AM. ETV6-RUNX1 promotes survival of early B lineage progenitor cells via a dysregulated erythropoietin receptor. Blood. 2011; 118: 4910-8. doi: 10.1182/blood-2011-05-354266.
13. Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, Alexandrov LB, Van Loo P, Cooke SL, Marshall J, Martincorena I, Hinton J, Gundem G, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet. 2014; 46: 116-25. doi: 10.1038/ng.2874.
14. Ford AM, Bennett CA, Price CM, Bruin MC, Van Wering ER, Greaves M. Fetal origins of the TEL-AML1 fusion gene in identical twins with leukemia. Proc Natl Acad Sci U S A. 1998; 95: 4584-8.
15. Wiemels JL, Ford AM, Van Wering ER, Postma A, Greaves M. Protracted and variable latency of acute lymphoblastic leukemia after TEL-AML1 gene fusion in utero. Blood. 1999; 94: 1057-62.
16. Maia AT, Ford AM, Jalali GR, Harrison CJ, Taylor GM, Eden OB, Greaves MF. Molecular tracking of leukemogenesis in a triplet pregnancy. Blood. 2001; 98: 478-82.
17. Wiemels JL, Cazzaniga G, Daniotti M, Eden OB, Addison GM, Masera G, Saha V, Biondi A, Greaves MF. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet. 1999; 354: 1499-503.
18. Hjalgrim LL, Madsen HO, Melbye M, Jorgensen P, Christiansen M, Andersen MT, Pallisgaard N, Hokland P, Clausen N, Ryder LP, Schmiegelow K, Hjalgrim H. Presence of clone-specific markers at birth in children with acute lymphoblastic leukaemia. Br J Cancer. 2002; 87: 994-9. doi: 10.1038/sj.bjc.6600601.
19. Zuna J, Madzo J, Krejci O, Zemanova Z, Kalinova M, Muzikova K, Zapotocky M, Starkova J, Hrusak O, Horak J, Trka J. ETV6/RUNX1 (TEL/AML1) is a frequent prenatal first hit in childhood leukemia. Blood. 2011; 117: 368-9; author reply 70-1. doi: 10.1182/blood-2010-09-309070.
20. Lausten-Thomsen U, Madsen HO, Vestergaard TR, Hjalgrim H, Nersting J, Schmiegelow K. Prevalence of t(12;21)[ETV6-RUNX1]-positive cells in healthy neonates. Blood. 2011; 117: 186-9. doi: 10.1182/blood-2010-05-282764.
21. Olsen M, Hjalgrim H, Melbye M, Madsen HO, Schmiegelow K. RT-PCR screening for ETV6-RUNX1-positive clones in cord blood from newborns in the Danish National Birth Cohort. J Pediatr Hematol Oncol. 2012; 34: 301-3. doi: 10.1097/MPH.0b013e3182332268.
22. Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, Hows JM, Navarrete C, Greaves M. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A. 2002; 99: 8242-7. doi: 10.1073/pnas.112218799.
23. Maia AT, Koechling J, Corbett R, Metzler M, Wiemels JL, Greaves M. Protracted postnatal natural histories in childhood leukemia. Genes Chromosomes Cancer. 2004; 39: 335-40. doi: 10.1002/gcc.20003.
24. Broadfield ZJ, Hain RD, Harrison CJ, Reza Jalali G, McKinley M, Michalova K, Robinson HM, Zemanova Z, Martineau M. Complex chromosomal abnormalities in utero, 5 years before leukaemia. Br J Haematol. 2004; 126: 307-12. doi: 10.1111/j.1365-2141.2004.05036.x.
25. Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, Green J, Colman S, Piacibello W, Buckle V, Tsuzuki S, Greaves M, Enver T. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science. 2008; 319: 336-9. doi: 10.1126/science.1150648.
26. Schindler JW, Van Buren D, Foudi A, Krejci O, Qin J, Orkin SH, Hock H. TEL-AML1 corrupts hematopoietic stem cells to persist in the bone marrow and initiate leukemia. Cell Stem Cell. 2009; 5: 43-53. doi: 10.1016/j.stem.2009.04.019.
27. van der Weyden L, Giotopoulos G, Rust AG, Matheson LS, van Delft FW, Kong J, Corcoran AE, Greaves MF, Mullighan CG, Huntly BJ, Adams DJ. Modeling the evolution of ETV6-RUNX1-induced B-cell precursor acute lymphoblastic leukemia in mice. Blood. 2011; 118: 1041-51. doi: 10.1182/blood-2011-02-338848.
28. Li M, Jones L, Gaillard C, Binnewies M, Ochoa R, Garcia E, Lam V, Wei G, Yang W, Lobe C, Hermiston M, Passegue E, Kogan SC. Initially disadvantaged, TEL-AML1 cells expand and initiate leukemia in response to irradiation and cooperating mutations. Leukemia. 2013; 27: 1570-3. doi: 10.1038/leu.2013.15.
29. Sabaawy HE, Azuma M, Embree LJ, Tsai HJ, Starost MF, Hickstein DD. TEL-AML1 transgenic zebrafish model of precursor B cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2006; 103: 15166-71. doi: 10.1073/pnas.0603349103.
30. Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, Kempski H, Moorman AV, Titley I, Swansbury J, Kearney L, Enver T, Greaves M. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011; 469: 356-61. doi: 10.1038/nature09650.
31. Raynaud S, Cave H, Baens M, Bastard C, Cacheux V, Grosgeorge J, Guidal-Giroux C, Guo C, Vilmer E, Marynen P, Grandchamp B. The 12;21 translocation involving TEL and deletion of the other TEL allele: two frequently associated alterations found in childhood acute lymphoblastic leukemia. Blood. 1996; 87: 2891-9.
32. Cave H, Cacheux V, Raynaud S, Brunie G, Bakkus M, Cochaux P, Preudhomme C, Lai JL, Vilmer E, Grandchamp B. ETV6 is the target of chromosome 12p deletions in t(12;21) childhood acute lymphocytic leukemia. Leukemia. 1997; 11: 1459-64.
33. Fenrick R, Wang L, Nip J, Amann JM, Rooney RJ, Walker-Daniels J, Crawford HC, Hulboy DL, Kinch MS, Matrisian LM, Hiebert SW. TEL, a putative tumor suppressor, modulates cell growth and cell morphology of ras-transformed cells while repressing the transcription of stromelysin-1. Mol Cell Biol. 2000; 20: 5828-39.
34. Lopez RG, Carron C, Oury C, Gardellin P, Bernard O, Ghysdael J. TEL is a sequence-specific transcriptional repressor. J Biol Chem. 1999; 274: 30132-8.
35. Stams WA, Beverloo HB, den Boer ML, de Menezes RX, Stigter RL, van Drunen E, Ramakers-van-Woerden NL, Loonen AH, van Wering ER, Janka-Schaub GE, Pieters R. Incidence of additional genetic changes in the TEL and AML1 genes in DCOG and COALL-treated t(12;21)-positive pediatric ALL, and their relation with drug sensitivity and clinical outcome. Leukemia. 2006; 20: 410-6. doi: 10.1038/sj.leu.2404083.
36. Peter A, Heiden T, Taube T, Korner G, Seeger K. Interphase FISH on TEL/AML1 positive acute lymphoblastic leukemia relapses--analysis of clinical relevance of additional TEL and AML1 copy number changes. Eur J Haematol. 2009; 83: 420-32. doi: 10.1111/j.1600-0609.2009.01315.x.
37. Fears S, Vignon C, Bohlander SK, Smith S, Rowley JD, Nucifora G. Correlation between the ETV6/CBFA2 (TEL/AML1) fusion gene and karyotypic abnormalities in children with B-cell precursor acute lymphoblastic leukemia. Genes Chromosomes Cancer. 1996; 17: 127-35. doi: 10.1002/(sici)1098-2264(199610)17:2<127::aid-gcc8>3.0.co;2-7.
38. Borst L, Wesolowska A, Joshi T, Borup R, Nielsen FC, Andersen MK, Jonsson OG, Wehner PS, Wesenberg F, Frost BM, Gupta R, Schmiegelow K. Genome-wide analysis of cytogenetic aberrations in ETV6/RUNX1-positive childhood acute lymphoblastic leukaemia. Br J Haematol. 2012; 157: 476-82. doi: 10.1111/j.1365-2141.2012.09083.x.
39. Lilljebjorn H, Heidenblad M, Nilsson B, Lassen C, Horvat A, Heldrup J, Behrendtz M, Johansson B, Andersson A, Fioretos T. Combined high-resolution array-based comparative genomic hybridization and expression profiling of ETV6/RUNX1-positive acute lymphoblastic leukemias reveal a high incidence of cryptic Xq duplications and identify several putative target genes within the commonly gained region. Leukemia. 2007; 21: 2137-44. doi: 10.1038/sj.leu.2404879.
40. Forestier E, Andersen MK, Autio K, Blennow E, Borgstrom G, Golovleva I, Heim S, Heinonen K, Hovland R, Johannsson JH, Kerndrup G, Nordgren A, Rosenquist R, et al. Cytogenetic patterns in ETV6/RUNX1-positive pediatric B-cell precursor acute lymphoblastic leukemia: A Nordic series of 245 cases and review of the literature. Genes Chromosomes Cancer. 2007; 46: 440-50. doi: 10.1002/gcc.20423.
41. Tsuzuki S, Karnan S, Horibe K, Matsumoto K, Kato K, Inukai T, Goi K, Sugita K, Nakazawa S, Kasugai Y, Ueda R, Seto M. Genetic abnormalities involved in t(12;21) TEL-AML1 acute lymphoblastic leukemia: analysis by means of array-based comparative genomic hybridization. Cancer Sci. 2007; 98: 698-706. doi: 10.1111/j.1349-7006.2007.00443.x.
42. Parker H, An Q, Barber K, Case M, Davies T, Konn Z, Stewart A, Wright S, Griffiths M, Ross FM, Moorman AV, Hall AG, Irving JA, et al. The complex genomic profile of ETV6-RUNX1 positive acute lymphoblastic leukemia highlights a recurrent deletion of TBL1XR1. Genes Chromosomes Cancer. 2008; 47: 1118-25. doi: 10.1002/gcc.20613.
43. Tsang KS, Li CK, Chik KW, Shing MM, Tsoi WC, Ng MH, Lau TT, Leung Y, Yuen PM. TEL/AML1 rearrangement and the prognostic significance in childhood acute lymphoblastic leukemia in Hong Kong. Am J Hematol. 2001; 68: 91-8.
44. Attarbaschi A, Mann G, Strehl S, Konig M, Steiner M, Jeitler V, Lion T, Dworzak MN, Gadner H, Haas OA. Deletion of 11q23 is a highly specific nonrandom secondary genetic abnormality of ETV6/RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia. 2007; 21: 584-6. doi: 10.1038/sj.leu.2404507.
45. Betts DR, Stanchescu R, Niggli FK, Cohen N, Rechavi G, Amariglio N, Trakhtenbrot L. SKY reveals a high frequency of unbalanced translocations involving chromosome 6 in t(12;21)-positive acute lymphoblastic leukemia. Leuk Res. 2008; 32: 39-43. doi: 10.1016/j.leukres.2007.03.010.
46. Emerenciano M, Bungaro S, Cazzaniga G, Dorea MD, Coser VM, Magalhaes IQ, Biondi A, Pombo-de-Oliveira MS. ETV6-RUNX1 fusion gene and additional genetic changes in infant leukemia: a genome-wide analysis. Cancer Genet Cytogenet. 2009; 193: 86-92. doi: 10.1016/j.cancergencyto.2009.04.021.
47. Lilljebjorn H, Soneson C, Andersson A, Heldrup J, Behrendtz M, Kawamata N, Ogawa S, Koeffler HP, Mitelman F, Johansson B, Fontes M, Fioretos T. The correlation pattern of acquired copy number changes in 164 ETV6/RUNX1-positive childhood acute lymphoblastic leukemias. Hum Mol Genet. 2010; 19: 3150-8. doi: 10.1093/hmg/ddq224.
48. Kjeldsen E. Oligo-based aCGH analysis reveals cryptic unbalanced der (6) t (X; 6) in pediatric t (12; 21)-positive acute lymphoblastic leukemia. Experimental and molecular pathology. 2016; 101: 38-43.
49. Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, Pui CH, Relling MV, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007; 446: 758-64. doi: 10.1038/nature05690.
50. Zakaria Z, Ahid MF, Ismail A, Keoh TS, Nor NM, Kamaluddin NR, Esa E, Yuen LK, Rahman EJ, Osman R. Chromosomal Aberrations in ETV6/RUNX1-positive Childhood Acute Lymphoblastic Leukemia using 244K Oligonucleotide Array Comparative Genomic Hybridization. Mol Cytogenet. 2012; 5: 41. doi: 10.1186/1755-8166-5-41.
51. Lee JW, Kim SK, Jang PS, Chung NG, Jeong DC, Kim M, Cho B, Kim HK. Outcome and Prognostic Factors for ETV6/RUNX1 Positive Pediatric Acute Lymphoblastic Leukemia Treated at a Single Institution in Korea. Cancer Res Treat. 2016; 49:446-453. doi: 10.4143/crt.2016.211.
52. Ellinghaus E, Stanulla M, Richter G, Ellinghaus D, te Kronnie G, Cario G, Cazzaniga G, Horstmann M, Panzer Grumayer R, Cave H, Trka J, Cinek O, Teigler-Schlegel A, et al. Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia. 2012; 26: 902-9. doi: 10.1038/leu.2011.302.
53. Dun KA, Vanhaeften R, Batt TJ, Riley LA, Diano G, Williamson J. BCR-ABL1 gene rearrangement as a subclonal change in ETV6-RUNX1–positive B-cell acute lymphoblastic leukemia. Blood Advances. 2016; 1: 132-8.
54. Desper R, Jiang F, Kallioniemi OP, Moch H, Papadimitriou CH, Schaffer AA. Distance-based reconstruction of tree models for oncogenesis. J Comput Biol. 2000; 7: 789-803. doi: 10.1089/10665270050514936.
55. Kantner HP, Warsch W, Delogu A, Bauer E, Esterbauer H, Casanova E, Sexl V, Stoiber D. ETV6/RUNX1 induces reactive oxygen species and drives the accumulation of DNA damage in B cells. Neoplasia. 2013; 15: 1292-300.
56. Ziech D, Franco R, Pappa A, Panayiotidis MI. Reactive oxygen species (ROS)--induced genetic and epigenetic alterations in human carcinogenesis. Mutat Res. 2011; 711: 167-73. doi: 10.1016/j.mrfmmm.2011.02.015.
57. Alpar D, Wren D, Ermini L, Mansur MB, van Delft FW, Bateman CM, Titley I, Kearney L, Szczepanski T, Gonzalez D, Ford AM, Potter NE, Greaves M. Clonal origins of ETV6-RUNX1(+) acute lymphoblastic leukemia: studies in monozygotic twins. Leukemia. 2015; 29: 839-46. doi: 10.1038/leu.2014.322.
58. O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007; 445: 106-10. doi: 10.1038/nature05372.
59. Cobaleda C, Gutierrez-Cianca N, Perez-Losada J, Flores T, Garcia-Sanz R, Gonzalez M, Sanchez-Garcia I. A primitive hematopoietic cell is the target for the leukemic transformation in human philadelphia-positive acute lymphoblastic leukemia. Blood. 2000; 95: 1007-13.
60. Pine SR, Wiemels JL, Jayabose S, Sandoval C. TEL-AML1 fusion precedes differentiation to pre-B cells in childhood acute lymphoblastic leukemia. Leuk Res. 2003; 27: 155-64.
61. van Zelm MC, van der Burg M, de Ridder D, Barendregt BH, de Haas EF, Reinders MJ, Lankester AC, Revesz T, Staal FJ, van Dongen JJ. Ig gene rearrangement steps are initiated in early human precursor B cell subsets and correlate with specific transcription factor expression. J Immunol. 2005; 175: 5912-22.
62. Wiemels JL, Greaves M. Structure and possible mechanisms of TEL-AML1 gene fusions in childhood acute lymphoblastic leukemia. Cancer Res. 1999; 59: 4075-82.
63. Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, Alexandrov LB. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. 2014; 46: 116-25. doi: 10.1038/ng.2874.
64. Tsuzuki S, Seto M, Greaves M, Enver T. Modeling first-hit functions of the t(12;21) TEL-AML1 translocation in mice. Proc Natl Acad Sci U S A. 2004; 101: 8443-8. doi: 10.1073/pnas.0402063101.
65. Fischer M, Schwieger M, Horn S, Niebuhr B, Ford A, Roscher S, Bergholz U, Greaves M, Lohler J, Stocking C. Defining the oncogenic function of the TEL/AML1 (ETV6/RUNX1) fusion protein in a mouse model. Oncogene. 2005; 24: 7579-91. doi: 10.1038/sj.onc.1208931.
66. Morrow M, Horton S, Kioussis D, Brady HJ, Williams O. TEL-AML1 promotes development of specific hematopoietic lineages consistent with preleukemic activity. Blood. 2004; 103: 3890-6. doi: 10.1182/blood-2003-10-3695.
67. Castor A, Nilsson L, Astrand-Grundstrom I, Buitenhuis M, Ramirez C, Anderson K, Strombeck B, Garwicz S, Bekassy AN, Schmiegelow K, Lausen B, Hokland P, Lehmann S, et al. Distinct patterns of hematopoietic stem cell involvement in acute lymphoblastic leukemia. Nat Med. 2005; 11: 630-7. doi: 10.1038/nm1253.
68. Fuka G, Kauer M, Kofler R, Haas OA, Panzer-Grumayer R. The leukemia-specific fusion gene ETV6/RUNX1 perturbs distinct key biological functions primarily by gene repression. PLoS One. 2011; 6: e26348. doi: 10.1371/journal.pone.0026348.
69. Zaliova M, Madzo J, Cario G, Trka J. Revealing the role of TEL/AML1 for leukemic cell survival by RNAi-mediated silencing. Leukemia. 2011; 25: 313-20. doi: 10.1038/leu.2010.277.
70. Ford AM, Palmi C, Bueno C, Hong D, Cardus P, Knight D, Cazzaniga G, Enver T, Greaves M. The TEL-AML1 leukemia fusion gene dysregulates the TGF-beta pathway in early B lineage progenitor cells. J Clin Invest. 2009; 119: 826-36. doi: 10.1172/jci36428.
71. Diakos C, Zhong S, Xiao Y, Zhou M, Vasconcelos GM, Krapf G, Yeh RF, Zheng S, Kang M, Wiencke JK, Pombo-de-Oliveira MS, Panzer-Grumayer R, Wiemels JL. TEL-AML1 regulation of survivin and apoptosis via miRNA-494 and miRNA-320a. Blood. 2010; 116: 4885-93. doi: 10.1182/blood-2009-02-206706.
72. Loh ML, Rubnitz JE. TEL/AML1-positive pediatric leukemia: prognostic significance and therapeutic approaches. Curr Opin Hematol. 2002; 9: 345-52.
73. Zelent A, Greaves M, Enver T. Role of the TEL-AML1 fusion gene in the molecular pathogenesis of childhood acute lymphoblastic leukaemia. Oncogene. 2004; 23: 4275-83. doi: 10.1038/sj.onc.1207672.
74. Teppo S, Laukkanen S, Liuksiala T, Nordlund J, Oittinen M, Teittinen K, Grönroos T, St-Onge P, Sinnett D, Syvänen AC. Genome-wide repression of eRNA and target gene loci by the ETV6-RUNX1 fusion in acute leukemia. Genome Research. 2016; 26: 1468-77.
75. Busche S, Ge B, Vidal R, Spinella JF, Saillour V, Richer C, Healy J, Chen SH, Droit A, Sinnett D, Pastinen T. Integration of high-resolution methylome and transcriptome analyses to dissect epigenomic changes in childhood acute lymphoblastic leukemia. Cancer Res. 2013; 73: 4323-36. doi: 10.1158/0008-5472.can-12-4367.
76. Gunji H, Waga K, Nakamura F, Maki K, Sasaki K, Nakamura Y, Mitani K. TEL/AML1 shows dominant-negative effects over TEL as well as AML1. Biochem Biophys Res Commun. 2004; 322: 623-30. doi: 10.1016/j.bbrc.2004.07.169.
77. Mangolini M, de Boer J, Walf-Vorderwulbecke V, Pieters R, den Boer ML, Williams O. STAT3 mediates oncogenic addiction to TEL-AML1 in t(12;21) acute lymphoblastic leukemia. Blood. 2013; 122: 542-9. doi: 10.1182/blood-2012-11-465252.
78. Sundaresh A, Gasparoli L, Mangolini M, Edwards D, Hubank M, Brooks T, Bartram J, Goulden N, Ancliff P, de Boer J. Aberrant transcriptional pathways in t (12; 21) Acute Lymphoblastic Leukemia. Klinische Pädiatrie. 2016; 228: A12.
79. Fuka G, Kantner HP, Grausenburger R, Inthal A, Bauer E, Krapf G, Kaindl U, Kauer M, Dworzak MN, Stoiber D, Haas OA, Panzer-Grumayer R. Silencing of ETV6/RUNX1 abrogates PI3K/AKT/mTOR signaling and impairs reconstitution of leukemia in xenografts. Leukemia. 2012; 26: 927-33. doi: 10.1038/leu.2011.322.
80. Inthal A, Krapf G, Beck D, Joas R, Kauer MO, Orel L, Fuka G, Mann G, Panzer-Grumayer ER. Role of the erythropoietin receptor in ETV6/RUNX1-positive acute lymphoblastic leukemia. Clin Cancer Res. 2008; 14: 7196-204. doi: 10.1158/1078-0432.CCR-07-5051.
81. Kaindl U, Morak M, Portsmouth C, Mecklenbrauker A, Kauer M, Zeginigg M, Attarbaschi A, Haas OA, Panzer-Grumayer R. Blocking ETV6/RUNX1-induced MDM2 overexpression by Nutlin-3 reactivates p53 signaling in childhood leukemia. Leukemia. 2014; 28: 600-8. doi: 10.1038/leu.2013.345.
82. Chen J, Odenike O, Rowley JD. Leukaemogenesis: more than mutant genes. Nat Rev Cancer. 2010; 10: 23-36. doi: 10.1038/nrc2765.
83. Milani L, Lundmark A, Kiialainen A, Nordlund J, Flaegstad T, Forestier E, Heyman M, Jonmundsson G, Kanerva J, Schmiegelow K, Soderhall S, Gustafsson MG, Lonnerholm G, et al. DNA methylation for subtype classification and prediction of treatment outcome in patients with childhood acute lymphoblastic leukemia. Blood. 2010; 115: 1214-25. doi: 10.1182/blood-2009-04-214668.
84. Stoskus M, Gineikiene E, Valceckiene V, Valatkaite B, Pileckyte R, Griskevicius L. Identification of characteristic IGF2BP expression patterns in distinct B-ALL entities. Blood Cells Mol Dis. 2011; 46: 321-6. doi: 10.1016/j.bcmd.2011.02.005.
85. Stoskus M, Vaitkeviciene G, Eidukaite A, Griskevicius L. ETV6/RUNX1 transcript is a target of RNA-binding protein IGF2BP1 in t(12;21)(p13;q22)-positive acute lymphoblastic leukemia. Blood Cells Mol Dis. 2016; 57: 30-4. doi: 10.1016/j.bcmd.2015.11.006.
86. Stoskus M, Eidukaite A, Griskevicius L. Defining the significance of IGF2BP1 overexpression in t(12;21)(p13;q22)-positive leukemia REH cells. Leuk Res. 2016; 47: 16-21. doi: 10.1016/j.leukres.2016.05.009.
87. Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009; 138: 592-603.
88. Janczar K, Janczar S, Pastorczak A, Mycko K, Paige AJ, Zalewska-Szewczyk B, Wagrowska-Danilewicz M, Danilewicz M, Mlynarski W. Preserved global histone H4 acetylation linked to ETV6-RUNX1 fusion and PAX5 deletions is associated with favorable outcome in pediatric B-cell progenitor acute lymphoblastic leukemia. Leuk Res. 2015; 39: 1455-61. doi: 10.1016/j.leukres.2015.10.006.
89. Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang HE, Taylor JE, Hu M, Englund NP, Yan F, Wang Z, Robert McDonald E 3rd, et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet. 2013; 45: 1386-91. doi: 10.1038/ng.2777.
90. Gefen N, Binder V, Zaliova M, Linka Y, Morrow M, Novosel A, Edry L, Hertzberg L, Shomron N, Williams O, Trka J, Borkhardt A, Izraeli S. Hsa-mir-125b-2 is highly expressed in childhood ETV6/RUNX1 (TEL/AML1) leukemias and confers survival advantage to growth inhibitory signals independent of p53. Leukemia. 2010; 24: 89-96. doi: 10.1038/leu.2009.208.
91. Yang YL, Yen CT, Pai CH, Chen HY, Yu SL, Lin CY, Hu CY, Jou ST, Lin DT, Lin SR, Lin SW. A Double Negative Loop Comprising ETV6/RUNX1 and MIR181A1 Contributes to Differentiation Block in t(12;21)-Positive Acute Lymphoblastic Leukemia. PLoS One. 2015; 10: e0142863. doi: 10.1371/journal.pone.0142863.
92. Ghazavi F, De Moerloose B, Van Loocke W, Wallaert A, Helsmoortel HH, Ferster A, Bakkus M, Plat G, Delabesse E, Uyttebroeck A, Van Nieuwerburgh F, Deforce D, Van Roy N, et al. Unique long non-coding RNA expression signature in ETV6/RUNX1-driven B-cell precursor acute lymphoblastic leukemia. Oncotarget. 2016; 7:73769-73780. doi: 10.18632/oncotarget.12063.
93. Krapf G, Kaindl U, Kilbey A, Fuka G, Inthal A, Joas R, Mann G, Neil JC, Haas OA, Panzer-Grumayer ER. ETV6/RUNX1 abrogates mitotic checkpoint function and targets its key player MAD2L1. Oncogene. 2010; 29: 3307-12. doi: 10.1038/onc.2010.53.
94. Bauer E, Schlederer M, Scheicher R, Horvath J, Aigner P, Schiefer AI, Kain R, Regele H, Hoermann G, Steiner G. Cooperation of ETV6/RUNX1 and BCL2 enhances immunoglobulin production and accelerates glomerulonephritis in transgenic mice. Oncotarget. 2016; 7: 12191-205. doi: 10.18632/oncotarget.7687.
95. Gronroos T, Teppo S, Mehtonen J, Laukkanen S, Liuksiala T, Nykter M, Heinaniemi M, Lohi O. Overexpression of PTP4A3 in ETV6-RUNX1 acute lymphoblastic leukemia. Leuk Res. 2016; 54: 1-6. doi: 10.1016/j.leukres.2016.12.005.
96. Loh ML, Goldwasser MA, Silverman LB, Poon WM, Vattikuti S, Cardoso A, Neuberg DS, Shannon KM, Sallan SE, Gilliland DG. Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01. Blood. 2006; 107: 4508-13. doi: 10.1182/blood-2005-08-3451.
97. Bokemeyer A, Eckert C, Meyr F, Koerner G, von Stackelberg A, Ullmann R, Turkmen S, Henze G, Seeger K. Copy number genome alterations are associated with treatment response and outcome in relapsed childhood ETV6/RUNX1-positive acute lymphoblastic leukemia. Haematologica. 2014; 99: 706-14. doi: 10.3324/haematol.2012.072470.
98. Forestier E, Heyman M, Andersen MK, Autio K, Blennow E, Borgstrom G, Golovleva I, Heim S, Heinonen K, Hovland R, Johannsson JH, Kerndrup G, Nordgren A, et al. Outcome of ETV6/RUNX1-positive childhood acute lymphoblastic leukaemia in the NOPHO-ALL-1992 protocol: frequent late relapses but good overall survival. Br J Haematol. 2008; 140: 665-72. doi: 10.1111/j.1365-2141.2008.06980.x.
99. Chow CD, Dalla-Pozza L, Gottlieb DJ, Hertzberg MS. Two cases of very late relapsing ALL carrying the TEL:AML1 fusion gene. Leukemia. 1999; 13: 1893-4.
100. Konrad M, Metzler M, Panzer S, Ostreicher I, Peham M, Repp R, Haas OA, Gadner H, Panzer-Grumayer ER. Late relapses evolve from slow-responding subclones in t(12;21)-positive acute lymphoblastic leukemia: evidence for the persistence of a preleukemic clone. Blood. 2003; 101: 3635-40. doi: 10.1182/blood-2002-10-3252.
101. Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, Downing JR. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008; 322: 1377-80. doi: 10.1126/science.1164266.
102. Panzer-Grumayer ER, Cazzaniga G, van der Velden VH, del Giudice L, Peham M, Mann G, Eckert C, Schrauder A, Germano G, Harbott J, Basso G, Biondi A, van Dongen JJ, et al. Immunogenotype changes prevail in relapses of young children with TEL-AML1-positive acute lymphoblastic leukemia and derive mainly from clonal selection. Clin Cancer Res. 2005; 11: 7720-7. doi: 10.1158/1078-0432.CCR-05-1239.
103. Zuna J, Ford AM, Peham M, Patel N, Saha V, Eckert C, Kochling J, Panzer-Grumayer R, Trka J, Greaves M. TEL deletion analysis supports a novel view of relapse in childhood acute lymphoblastic leukemia. Clin Cancer Res. 2004; 10: 5355-60. doi: 10.1158/1078-0432.ccr-04-0584.
104. Szczepanski T, Willemse MJ, Brinkhof B, van Wering ER, van der Burg M, van Dongen JJ. Comparative analysis of Ig and TCR gene rearrangements at diagnosis and at relapse of childhood precursor-B-ALL provides improved strategies for selection of stable PCR targets for monitoring of minimal residual disease. Blood. 2002; 99: 2315-23.
105. Levasseur M, Maung ZT, Jackson GH, Kernahan J, Proctor SJ, Middleton PG. Relapse of acute lymphoblastic leukaemia 14 years after presentation: use of molecular techniques to confirm true re-emergence. Br J Haematol. 1994; 87: 437-8.
106. Lawson SE, Harrison G, Richards S, Oakhill A, Stevens R, Eden OB, Darbyshire PJ. The UK experience in treating relapsed childhood acute lymphoblastic leukaemia: a report on the medical research council UKALLR1 study. Br J Haematol. 2000; 108: 531-43.
107. Ly-Sunnaram B, Henry C, Gandemer V, Mee FL, Burtin F, Blayau M, Cayuela JM, Oster M, Clech P, Rambeau M, Marie C, Pampin C, Edan C, et al. Late ovarian relapse of TEL/AML1 positive ALL confirming that TEL deletion is a secondary event in leukemogenesis. Leuk Res. 2005; 29: 1089-94. doi: 10.1016/j.leukres.2004.11.027.
108. Lilian Kuster RG. ETV6/RUNX1-positive relapses evolve from an ancestral clone and frequently acquire deletions of genes implicated in glucocorticoid signaling. Blood Cancer J. 2011; 117:2658-67. doi: 10.1182/blood-2010-03275347.
109. van Delft FW, Horsley S, Colman S, Anderson K, Bateman C, Kempski H, Zuna J, Eckert C, Saha V, Kearney L, Ford A, Greaves M. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood. 2011; 117: 6247-54. doi: 10.1182/blood-2010-10-314674.
110. Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013; 14: e205-17. doi: 10.1016/s1470-2045(12)70580-6.
111. Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol. 2011; 29: 551-65. doi: 10.1200/jco.2010.30.7405.
112. Grausenburger R, Bastelberger S, Eckert C, Kauer M, Stanulla M, Frech C, Bauer E, Stoiber D, von Stackelberg A, Attarbaschi A, Haas OA, Panzer-Grumayer R. Genetic alterations in glucocorticoid signaling pathway components are associated with adverse prognosis in children with relapsed ETV6/RUNX1-positive acute lymphoblastic leukemia. Leuk Lymphoma. 2015: 1-11. doi: 10.3109/10428194.2015.1088650.
113. Yang JJ, Bhojwani D, Yang W, Cai X, Stocco G, Crews K, Wang J, Morrison D, Devidas M, Hunger SP, Willman CL, Raetz EA, Pui CH, et al. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood. 2008; 112: 4178-83. doi: 10.1182/blood-2008-06-165027.
114. Jin Y, Wang X, Hu S, Tang J, Li B, Chai Y. Determination of ETV6-RUNX1 genomic breakpoint by next-generation sequencing. Cancer Med. 2016; 5: 337-51. doi: 10.1002/cam4.579.
115. Bachmann PS, Gorman R, Papa RA, Bardell JE, Ford J, Kees UR, Marshall GM, Lock RB. Divergent mechanisms of glucocorticoid resistance in experimental models of pediatric acute lymphoblastic leukemia. Cancer Res. 2007; 67: 4482-90. doi: 10.1158/0008-5472.can-06-4244.
116. Pinon JD, Labi V, Egle A, Villunger A. Bim and Bmf in tissue homeostasis and malignant disease. Oncogene. 2008; 27 Suppl 1: S41-52. doi: 10.1038/onc.2009.42.
117. Matheson EC, Hall AG. Assessment of mismatch repair function in leukaemic cell lines and blasts from children with acute lymphoblastic leukaemia. Carcinogenesis. 2003; 24: 31-8.