
INTRODUCTION
More than 40 years ago we showed, for the first time, that in vivo treatment of leukemia bearing mice with the antitumor agent dacarbazine (dimethyltriazene-imidazole-4-carboxamide, DTIC) was able to induce the appearance of novel transplantation antigens (Ags) in malignant cells [1]. This phenomenon was successively termed “chemical xenogenization”(CX) based on analogous definition proposed by Hiroshi Kobayashi in 1969 describing the presence of transplantation Ags induced by Friend virus infection in rat tumors (i.e. “viral xenogenization”, [2]).
After about 15 years of investigations (reviewed in [3]), CX was slowly relegated to oblivion. This was probably due to the lack of instruments that could translate into clinical benefits the appearance of drug-induced neoantigens in patients essentially unable to mount an adequate antitumor immune response.
Few years ago a monoclonal antibody (mAb), Ipilimumab, came to the worldwide attention as a potent inducer of cell-mediated immunity through down-regulation of Cytotoxic T Lymphocyte Antigen-4 (CTLA-4)-mediated T cell suppression [4]. In particular, Ipilimumab was found to substantially increase the survival of patients with advanced melanoma, essentially resistant to classical antitumor drugs. Therefore, on March 25th, 2011 the US Food and Drug Administration approved Ipilimumab for the management of advanced melanoma. This approval was a landmark event in the history of cancer immunotherapy, since for the first time an unusually potent amplifier of T cell-mediated cytotoxic responses was available to oncologists.
This event and the successive appearance in the cancer immunotherapy scenario of a growing number of immune checkpoint inhibitors (ICpI, reviewed in [5, 6]) have provided the ground to bring CX back to life. There is no doubt that drug-induced neoantigens could be considered novel “pharmacologically driven” targets of amplified host’s antitumor T-cell responses with great potential therapeutic value.
Up to now, the remarkable progress that has been made in the development of antitumor targeted therapy has not provided a concrete answer to long-term cancer control, especially in solid malignancies. From anti-infective therapy we have learned that, in the absence of adequate host’s immune responses, no cure can be attained in spite of the use of insuperably “targeted” agents (e.g. penicillin) in immuno-compromised patients. Therefore, the (re)appearance on the scene of successfully active anti-tumor immunity have disclosed novel and exciting perspectives in cancer management.
Drug-induced appearance of non-preexisting tumor Ags underlies CX phenomenon
Evidence that in vivo treatment with triazene compounds (hereafter referred to as triazenes) including DTIC, is able to induce the appearance of novel transplantations Ags required a long series of investigations.
It was demonstrated that the high doses of DTIC and of the other imidazole or aryltriazenes utilized to induce CX, inhibit severely T-cell dependent graft responses in mice [7]. Therefore, it was necessary to rule out that CX could be due to the emergence of immunogenic sublines in mice immunodepressed by triazenes, and therefore not competent to suppress spontaneously developing immunogenic clones. Two leukemia cell lines were passaged in untreated or DTIC-treated athymic H-2d/H-2d nu/nu BALB/c mice not able to reject allogeneic or xenogeneic cells [8]. In no case, leukemic cells passaged in untreated nude mice became immunogenic for euthymic histocompatible hosts. On the other hand, DTIC treatment of leukemia-bearing nude mice generated highly immunogenic sublines similar to those obtainable in conventional euthymic hosts [8].
In order to consolidate the concept that triazenes induce novel non-preexisting Ags, tolerance studies in vivo were performed in BALB/c mice challenged with the Moloney-Leukemia-Virus-induced lymphoma cell line LSTRA, positive for virus-derived Ags. The results showed that mice rendered tolerant to the Ags of the LSTRA cell line, were able to reject DTIC-treated but not untreated LSTRA cells [9].
The final molecular evidence showing that CX is the result of induction of novel Ags was obtained by Grohmann et al. in the 1990s. Through an original and highly accurate investigation [10], the authors were able to identify mutated peptides derived from endogenous retroviral env sequences detectable in the immunogenic “D” clone originated from xenogenized L5178Y/DTIC cell line. No similar mutated peptides were found in parental, non-xenogenized cells. Transfection experiments showed that products of mutated env gp70 subgenic fragments render target cells susceptible to lysis by D-cell primed, H-2Kd or H-2Ld-restricted cytotoxic T lymphocytes (CTL, [10]).
In collaboration with Michel Moore’s group, D’Atri et al. carried out a series of investigations in order to establish whether CX could be induced in human neoplasms [11]. The human lung cancer cell line H-125, treated with an in vitro active triazene for a number of cycles, was co-cultured with peripheral blood mononuclear cells of a healthy donor to generate allo-CTL. Thereafter, selected CTL clones able to specifically kill triazene-treated cells but not parental cells were identified. This study supported the hypothesis that CX could be generated also in human tumor cells. However, since no detailed analysis was performed in order to identify possible HLA restriction elements, these results appear to be incomplete and require further investigations.
Kinetics of triazene-induced CX and immunogenicity of drug-treated cells at clonal level
In most of published studies, fully immunogenic xenogenized cell lines were generated following 5-7 transplant generations of treatment with high daily doses of triazenes (see Figure 1A). The magnitude of graft response of histocompatible mice against triazene-treated cells was found to be comparable to that detectable in mice challenged with major histocompatibility complex (MHC)-incompatible malignant cells [12]. Actually, the status of “fully immunogenic” xenogenized cells is revealed by the rejection of at least 105 (and sometimes up to 107) triazene-treated cells by intact, wholly histocompatible recipients. It is noteworthy that in the same host/tumor systems, even 1 murine leukemia cell (e.g. L1210 leukemia in DBA/2 or CD2F1 mice) is often able to kill the untreated host with generalized leukemia within 15-17 days (see Figure 1B).
A typical kinetics of the xenogenization process occurring in CD2F1 mice challenged with L5178Y leukemia (105 cells ip) and treated with DTIC [13] is illustrated in Figure 1A. At transplant generation “0” (i.e. at the beginning of the process), DTIC-treated mice showed a median survival time (MST) longer compared to controls, although no animal survived beyond the 60-day observation period. At transplant generation 1 and 2 no significant difference in MSTs was noticed between control and DTIC-treated mice, probably indicating the onset of drug resistance in leukemic cells exposed in vivo to the alkylating agent. Progressively, from transplant generation 3 onward, control mice survived significantly longer than DTIC-treated recipients, and at transplant generation 5 all controls were long-term survivors, whereas all DTIC treated mice died, with an MST of 10 days. This phenomenon was interpreted as a result of host’s graft response against highly immunogenic DTIC-treated cells. Indeed, intact mice rejected the tumor, whereas DTIC-treated hosts, which were severely immunodepressed by the compound [7, 14], or mice pretreated with cyclophosphamide succumbed with generalized leukemia. This finding ruled out the possibility that DTIC-treated L5178Y was a leukemia subline dependent on DTIC for growth.
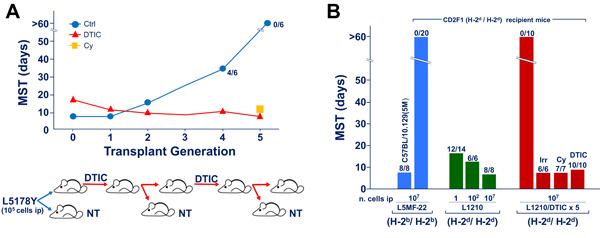
Figure 1: Drug-induced xenogenization (DIX). Figure 1A. Typical DIX pattern indicating the kinetics of the appearance of leukemia cell immunogenicity in CD2F1 mice exposed to DTIC (50 mg/Kg/day ip, from day 1 through day 10) after ip challenge with 105 L5178Y leukemia cells of DBA/2 origin. At transplant generation 4, 2 out of 6 control mice not subjected to DTIC treatment, survived longer than the 60 days of observation period. At transplant generation 5, all untreated mice survived beyond the 60 day-observation period. On the other hand, all CD2F1 recipients treated with DTIC or immunodepressed by pre-treatment with cyclophosphamide (Cy, 180 mg/Kg, 6 h before tumor challenge) died with evidence of generalized leukemia at the autopsy. Figure 1B. Extent of immunogenicity levels of xenogenized leukemia cells (L1210/DTIC x 5, i.e. L1210 leukemia of DBA/2 origin, treated with DTIC 100 mg/Kg/day for 10 days, for 5 transplant generations). The number of dead mice over the total injected are indicated on the top of each column. Two types of recipient mice were used, i.e. the H-2b homozygous C57BL/10.129(5M) mice syngeneic with L5MF-22 leukemia, and the CD2F1 hosts (H-2d/H-2d), fully histocompatible with L1210 leukemia. The degree of full compatibility between host and tumor is indicated by the observation that almost all CD2F1 mice died for leukemia even after challenge with as low as 1 L1210 cell ip. In addition all C57BL/10.129(5M) mice died with generalized leukemia after injection of 107 cells of the syngeneic L5MF-22 leukemia. The degree of immunogenicity of L1210/DTIC x 5 cells for the H-2-compatible CD2F1 hosts appears to be comparable to that of the H-2-incompatible L5MF-22 for the same CD2F1 recipients. In fact, up to 107 cells of both leukemias were completely rejected by CD2F1 mice that survived beyond the 60 day observation period. On the other hand, all immunodepressed recipients, either irradiated (Irr, 4 Gy delivered on day -1 before challenge), pretreated with cyclophosphamide (Cy, 180 mg/Kg administered 6 h before tumor transplantation) or treated with DTIC (DTIC 100 mg/Kg/day for 10 days) died after challenge with the same number of xenogenized L1210 leukemia cells.
Similar results were obtained with a number of mouse leukemias and the degree of immunogenicity of drug-treated cells for the histocompatible host was often similar to that detectable in target cells incompatible for the entire H-2 haplotype. In fact, the results illustrated in Figure 1B show that all intact CD2F1 mice that were able to reject 107 cells of the H-2-incompatible L5MF-22 leukemia, were also able to reject 107 cells of the histocompatible L1210 cells subjected to 5 transplant generations of DTIC treatment. On the other hand, all mice immunodepressed by total-body irradiation, or by pretreatment with DTIC or cyclophosphamide succumbed with generalized leukemia following challenge with the “xenogenized” L1210 leukemia cells.
Of particular interest are the findings that CX is also inducible in vitro [15] and that the highly immunogenic triazene-treated cells obtained either in vivo or in vitro, retain their immunogenic properties after up to 90 passages in immunodepressed mice not exposed to triazenes [13]. It is obvious that these tumor neoantigens are heritable after a number of malignant cell divisions and can thus be considered immunological targets even after triazene withdrawal. Therefore the presence of drug-induced neoantigens in tumor cell population could be of therapeutic advantage for different immunotherapeutic strategies in cancer treatment.
Further experiments were conducted in order to explore whether a limited degree of immunogenicity could be revealed during the initial 1-3 transplant generations of triazene treatment using a protocol based on immuno-chemotherapy synergism [16]. Figures 2A and 2B show the survival times of mice challenged with L1210 leukemia of DBA/2 origin. No difference in survival times was detected between fully histocompatible CD2F1 mice and H-2d-compatible BALB/c mice incompatible for minor histocompatibility loci. However, while treatment with a low dose of bis-chloroethyl-nitrosourea (BCNU) was minimally active in CD2F1 hosts, in the majority of experiments, it was able to “cure”, all allogeneic BALB/c mice [17] that are thought to be able to mount a weak allograft response. In this model, therefore, synergism between weak graft response and chemotherapy reveals antitumor immune reactions not easily detectable without drug treatment.
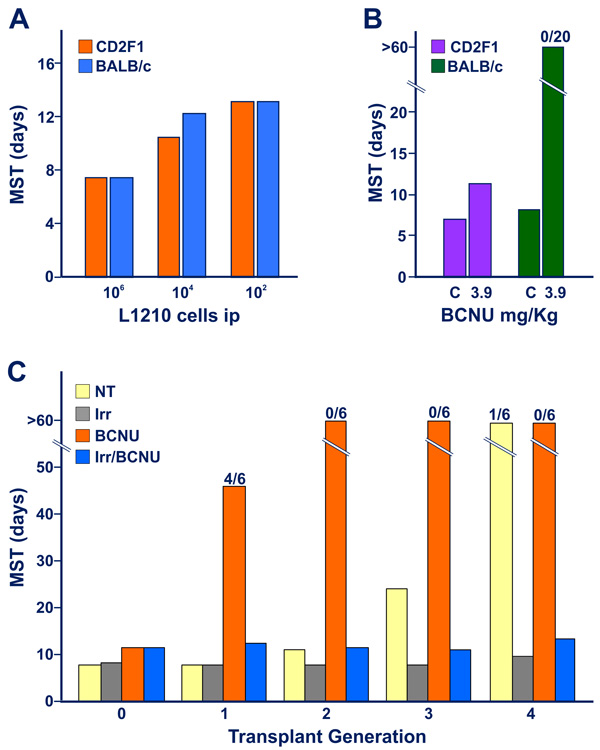
Figure 2: Appearance of limited degree of immunogenicity in L5178Y DBA/2 leukemia cells exposed to early transplant generations of DTIC treatment, revealed by immuno-chemotherapy synergism. When not specified in terms of number of dead mice over the total tested indicated on the top of the columns, all mice (6-8 animals for group) died with generalized leukemia. Figure 2A. Graded numbers of L1210 leukemia cells were inoculated into fully histocompatible CD2F1 mice, or into H-2d-compatible BALB/c mice, incompatible for multiple minor histocompatibility antigens (see Ref 17). The marginal allograft response of BALB/c hosts was not adequate to restrain the growth of leukemic cells, as evidenced by the finding that no substantial difference in median survival time was detected between CD2F1 and BALB/c mice inoculated with as low as 102 L1210 cells ip. Figure 2B. CD2F1 and BALB/c mice were inoculated with 105 L1210 cells ip. In this case, CD2F1 mice treated with a low dose of BCNU, (3.9 mg/Kg ip, administered on day 3 after tumor transplantation) showed a limited increase in MST with respect to that of untreated controls (C). In contrast, all BCNU-treated allogeneic BALB/c mice, survived beyond the 60 day-observation period, thus confirming the possibility of revealing a marginal antitumor graft response of the host through an immune-chemotherapy synergistic effect. Figure 2C. (data from Ref 13). The strategy of immune-chemotherapy synergism indicates that murine leukemia cells exposed in vivo to DTIC acquire appreciable levels of immunogenicity already at transplant generation “1”. Malignant cell immunogenicity progressively increases at the successive generations of treatment with the triazene compound. This figure illustrates the results of a typical experiment performed to evaluate the immunogenic properties of L5178Y leukemia cells in the course of the first 4 transplant generations of DTIC treatment (DTIC 100 mg/Kg/day ip for 10 days) in CD2F1 mice. Blasts (105 cells) obtained from non-treated leukemic donors (i.e. at transplant generation “0”) or from DTIC-treated leukemic donors (at transplant generations 1 through 4) were inoculated into 4 groups of mice, i.e. non-treated (NT), immunodepressed through exposure to total-body irradiation (Irr, 4 Gy X rays, on day -1), treated with BCNU (10 mg/Kg ip), immunodepressed (i.e. pre-irradiated) and treated with BCNU (Irr/BCNU). An additional group of mice was treated with DTIC to obtain a further generation of treatment with the triazene compound. At transplant generation “0” the intact L5178Y cells did not show appreciable immunogenicity, since all non-immunodepressed or irradiated recipients treated with BCNU showed a similar modest increase of MST over that of non-treated controls. Remarkably, at transplant generation “1” instead, L5178Y cells obtained from DTIC-treated donors showed immunogenicity strength similar to that conferred by products of minor histocompatibility loci. This is evidenced by the consistent increase of survival times of BCNU-treated animals respect to those of mice not subjected to chemotherapy, or treated with BCNU but immunodepressed by means of total-body irradiation.
As illustrated in Figure 2C, CD2F1 mice were inoculated with leukemic cells obtained from DTIC-treated donors during initial transplant generations of DTIC treatment, before the appearance of strong immunogenicity in L5178Y/DTIC leukemia. Animals were then treated with low-dose BCNU and survival time analysis showed longer overall survival than those subjected to the same treatment, but immunodepressed with total-body irradiation delivered one day before tumor challenge. This observation suggests a stepwise increase in immunogenicity of leukemic blasts exposed in vivo to daily pulses of DTIC, possibly as a result of a progressive rise of mutation load, in line with the hypothesis illustrated in Figure 3 (see below).
The appearance of relatively weak immunogenicity after a single exposure to DTIC was confirmed in CD2F1 mice inoculated ip with 108 L1210 cells and injected 1 h later with a single high-dose of DTIC (300 mg/Kg, [18]). Again, almost all intact CD2F1 mice challenged with 106 leukemic cells collected from the DTIC-treated donors were “cured” by a single dose of BCNU (10 mg/Kg), whereas most recipient mice exposed to the same treatment, but immunodepressed with cyclophosphamide, died with generalized leukemia.
After the discovery of CX [1], in view of its possible biological and clinical relevance, some fundamental cellular and molecular aspects of the xenogenization process were investigated by different groups. Great priority was given to studies aimed at establishing whether different xenogenized neoplastic cells present cross-reacting neoantigens, or if each triazene-treated tumor is composed of a homogeneous cell population containing a single set of neoantigens, or of a number of clones showing different non-cross-reacting neoantigens. In 1986 the group of Angelo Nicolin [19] cloned a xenogenized L1210 leukemia cell line and analyzed the single clones for immunogenicity and cross-reactivity in terms of CTL generation and susceptibility to lysis. They concluded that the xenogenized leukemia contained a heterogeneous but limited number of different antigenically cross-reacting and non-cross reacting cell clones. Later, Marelli et al.[20] xenogenized a homogeneous mouse leukemia cell population starting from a single L1210 clone, cloned the DTIC-treated cell line and evaluated the immunogenic properties and cross-reactivity of the clones. The authors concluded that at least one common specific neoantigen is reproducibly elicited by DTIC treatment within an identical malignant cell population. In the light of the mutational mechanism underlying CX (see below), it is reasonable to hypothesize that multiple point mutational events generated by a triazene-targeted hot spot could result in the appearance of common non-self peptide(s) in leukemic cell population. Notably, no CX affecting normal bone marrow cells was detected in DTIC-treated mice (Bonmassar E et al., unpublished data).
CX is the result of drug-induced mutational mechanisms leading to the appearance of non-self immunogenic peptides
The first report describing CX [1] already contained the hypothesis that this phenomenon could have been generated by somatic mutations, since DTIC was classified as a carcinogenic compound able to alkylate DNA [21, 22]. Further studies established that Quinacrine, an antimalarial drug with anti-mutagenic activity [23], suppresses entirely CX without impairing the antitumor and the immunosuppressant activity of DTIC [24]. However, direct evidence that a mutational mechanism was involved in triazene-induced appearance of neoantigens was not obtained until the molecular investigations performed by Grohmann et al. in the 1990s [10, 25, 26]. As previously mentioned, the authors found that CX was the result of point mutations provoked mainly by triazene-induced methyl adducts to the oxygen 6 of DNA guanine, affecting retroviral sequences normally present in mouse genome [27], followed by the appearance of MHC-restricted highly immunogenic non-self peptides.
Seven years after the discovery of DTIC-induced CX, the group of Thierry Boon found that selected clones obtained from a mouse teratocarcinoma cell line treated in vitro with the mutagen/carcinogen N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) were rejected by histocompatible recipients through an immunomediated mechanism [28]. MNNG is a classical mutagen that adds methyl groups to a number of nucleophilic sites on DNA bases, including the oxygen-6 of guanine [29]. In particular, if not repaired (see below), the O6-methylguanine (O6-MeG) is responsible of point mutations leading to the appearance of specific Ags able to elicit cell-mediated immune responses and graft rejection. Boon and his group identified several MNNG-treated tumor cell sublines that were termed tum- variants since they were unable to grow into immunocompetent histocompatible mice, but grew rapidly in immunodepressed recipients and elicited cytotoxic T-cell responses [28, 30]. In addition, the mutational mechanism underlying the biological features of tum- variants was confirmed by molecular investigations of the same group between 1988 and 1990 [31, 32]. In a mouse mastocytoma P815 model they identified genome sequences targets of MNNG-induced mutations, responsible for malignant cell immunogenicity. In the meantime, Altevogt et al. [33] found that a similar mechanism underlies the immunogenicity of the mouse lymphoma cell line Eb (a subline of L5178YE) exposed to MNNG. Also in MNNG-induced xenogenization, tumor cell immunogenicity was based on the presence of mutation-generated MHC-restricted non-self peptides [34].
All these findings indicate that malignant cell xenogenization can be obtained with two distinct classes of compounds, i.e. triazenes of large clinical application and MNNG, a mutagenic compound of no use in cancer chemotherapy (see Figure 3). Therefore, in the present review we have decided to maintain the term “CX” for mutagen-induced xenogenization and to adopt the new term “Drug-Induced Xenogenization” (DIX) to describe the induction of novel antigenic specificities by exposure to pharmacological agents.
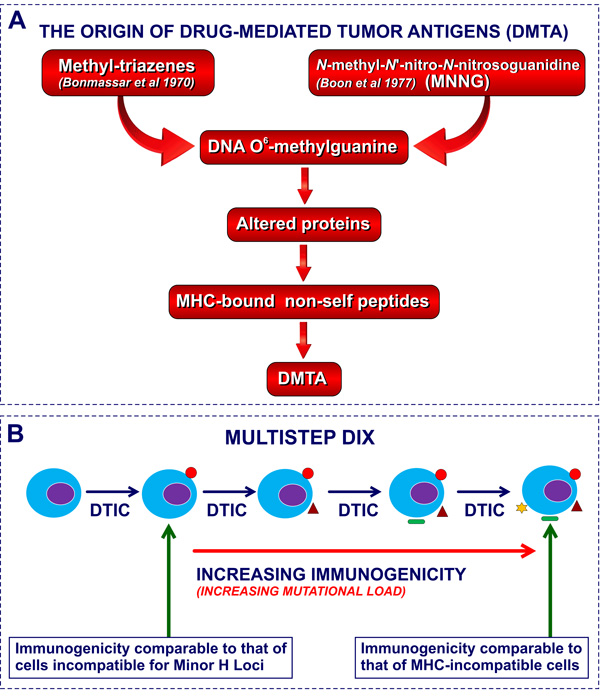
Figure 3: Molecular basis of DIX and CX: DNA O6-methyl-guanine adducts. Figure 3A. The mutation-dependent origin of DIX and CX. DMTA, drug-mediated tumor antigens, i.e neoantigens induced by drug-treatment. Figure 3B. The hypothesis of increased mutational load as the mechanism underlying the progressive increase of immunogenicity in the course of sequential transplant generations of DTIC treatment
Neoantigens generated by DIX elicit cell-mediated and humoral immunity in vivo and in vitro
Immune responses against xenogenized leukemia cells were found to be extremely complex, including T-cell dependent graft responses (as detailed in the previous sections), T-cell independent, radio-resistant graft response [35], H-2-restricted T-cell mediated cytotoxicity [10, 19, 20, 25, 26, 36-38] and weak humoral responses [39-43]. The T-cell dependency of the classical graft rejection found in histocompatible mice was confirmed by the finding that BALB/c nu/nu mice were not competent for rejecting xenogenized leukemia cells [8]. In addition, the growth kinetics and rejection pattern of xenogenized cells in the peritoneal cavity of mice were similar to those of untreated leukemic cells transplanted into H-2-incompatible recipients [44]. Significant impairment of xenogenized blast growth limited to the splenic territory, was found in lethally-irradiated histocompatible mice [35], suggesting that triazene-treated cells could be susceptible to radioresistant natural immunity of “hybrid resistance (Hh)” type [45, 46].
DIX/CX and role of DNA repair enzymes
The antitumor activity and the DIX property of triazenes are crucially dependent on the function of different DNA repair enzymes (reviewed in [47] and briefly summarized in Figure 4). In particular, triazenes and MNNG are mutagenic since they both induce methyl adducts to the oxygen-6 of DNA guanine (reviewed in [48]), This type of biochemical lesion is also responsible for the antitumor activity of triazenes as evidenced by Catapano et al. [49]. The cytotoxic and xenogenizing activity of these DNA mono-methylating compounds is antagonized by the DNA repair enzyme O6-methylguanine-DNA methyltransferase (MGMT). If the drug-treated cell does not eliminate the adducts through adequate nuclear levels of MGMT, O6-MeG of DNA loses pairing compatibility with C and triggers a Mismatch Repair (MMR) response [47, 50]. Since the MMR system is competent to replace an appropriate nucleotide in the newly formed strand but not in the “old” strand during DNA synthesis, no repair by this enzymatic system is feasible. It follows that after several “futile” unsuccessful repair attempts, apoptotic signal can be generated and, if the apoptosis machinery is correctly working, the damaged cell dies. In the absence of a functional MMR system, proliferation is not impaired. Frequent G:C to A:T transitions occur since during the DNA duplication process O6-MeG preferably pairs with T rather than with C, and at the second round of duplication, the new strand will contain A instead of G (Figure 4). Therefore, the molecular mainstay of DIX relies on MGMT and MMR deficiency that assures the highest mutation load attainable following triazene treatment. In contrast, the cytotoxic activity of these agents requires low levels of MGMT but a fully efficient MMR system [47]. This seems to be in line with previous findings illustrated by Fioretti et al. [51] that demonstrated a clear dissociation between the onset of resistance to DTIC treatment and emergence of DIX, although no data on MMR status of target leukemia cells was mentioned in that report.
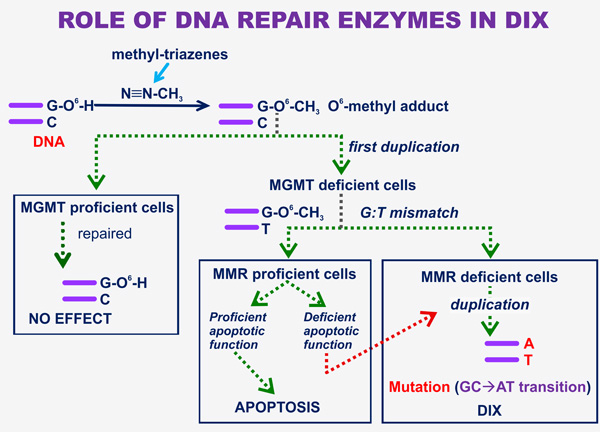
Figure 4: The role of DNA repair enzymes in DIX. The end-metabolic product of triazene compounds of clinical use (i.e. DTIC and temozolomide) is diazomethane [46] that reacts with the oxygen-6 of DNA guanine, generating an O6-methyl adduct. If target tumor cells express high levels of the DNA repair MGMT that acts as methyl acceptor molecule, all adducts are rapidly repaired and triazene treatment is totally ineffective. On the other hand, if MGMT levels are low or are down-regulated by MGMT inhibitors (e.g. Lomeguatrib, [202]), the adduct is stable and a mismatch signal is generated by C:O6-MeG, and after one duplication by frequent G:T mismatches. If the cells express a fully active MMR system and the apoptotic function is not compromised, triazene treatment results in target cell apoptotic death. Alternatively, if neoplastic cells are MMR deficient or their apoptotic pathways do not work properly, malignant growth normally goes on and mutation of G:C -> A:T transition type frequently occurs, leading to DIX appearance.
Preclinical studies on DIX-mediated antitumor immuno-chemotherapy
Since host’s antitumor responses appear to be mandatory for a successful treatment of cancer, numerous attempts have been made to exploit DIX for treatment of primary recipient mice bearing a non-treated parental leukemia. Therefore, histocompatible mice bearing L1210 were treated with a xenogenizing and immunosuppressive dose of DTIC (50 mg/Kg/day for 5 days) to induce a limited but significant DIX. Thereafter, recipient mice were infused with intact syngeneic splenocytes to restore immune reactivity compromised by DTIC administration, followed by treatment with BCNU in order to obtain synergistic effects between host’s graft response against xenogenized leukemia and chemotherapy. However, this protocol did not work until it was modified by adding a single limited dose of cyclophosphamide (100 mg/Kg) administered before spleen cell injection [52]. In this case a relatively high number of long-term survivors was observed, demonstrating in a preclinical model that DIX strategy can be a realistic approach for cancer immuno-chemotherapy. The role of cyclophosphamide in this study has not been definitely established although it could be ascribed to regulatory T cell (Treg) suppression and/or dendritic cell maturation [53, 54]. In any case, several antitumor agents, including cyclophosphamide and irradiation have shown a number of immunoenhancing effects (reviewed in [55]). In a mouse model, cyclophosphamide produces an initial lymphoid organ depletion which enhances the activity of adoptively transferred antitumor immune cells. This effect appears to be driven mainly by a cyclophosphamide-induced “cytokine storm” within 48 h from the injection [56], consisting in the up-regulation of interleukin (IL)-1, IL-2, IL-7 and IL-21, essential for T cell homeostatic proliferation.
Consistent with these findings, a phase I/II clinical trial, performed on 10 disease-free HLA-A2+ melanoma patients, has proven that vaccination with Melan-A and gp100 peptides in combination with DTIC (administered one day before vaccine inoculation) results in an improved cellular immune response as compared with vaccination alone and prevents melanoma relapse [57]. Global transcriptional analysis of peripheral blood mononuclear cells revealed a DTIC-induced activation of genes involved in the immune response and leukocyte stimulation in patients treated with combined chemo-immunotherapy. In particular, it was found a progressive widening of T-cell receptor (TCR) repertoire diversity [58], accompanied by high avidity and high anti-tumor T-cell polyfunctionality [59]. This clinical finding does not appear to be in contrast with the well documented immunosuppressive activity of DTIC previously described, since there is a difference in the dose mainly used in the mouse model (i.e. 100 mg/Kg daily for 5 days) and in the clinical investigation (one single administration of 800 mg/sqm, toxicologically equivalent to 21 mg/Kg in man and 260 mg/kg in mouse [60]).
Adoptive immunotherapy studies in mice showed that transfer of T lymphocytes presensitized against xenogenized leukemia cells can prolong significantly the survival of immunodepressed (i.e. treated with 4 Gy total-body irradiation) recipients bearing the same xenogenized cells [61]. Of particular interest is the finding that a similar effect was attained in mouse brain after intracerebral challenge with xenogenized leukemia followed by intracerebral inoculation with Ag-specific CTL [62].
ICpI and cancer immunotherapy
In the last 5 years we assisted to the steep increase of clinical investigations on cancer immunotherapy [63] - especially with the adoption of ICpI [64] - starting with supposedly immunogenic malignant diseases, such as melanoma and kidney carcinoma. Actually, the first clinical report on the therapeutic and toxic effects of the earliest ICpI utilized in cancer patients, i.e. Ipilimumab, can be traced back in 2003 when Hodi et al. [65] published their studies on this anti-CTLA-4 mAb in a clinical setting. A relatively long period of time was taken in the attempts to establish the best effective dose and schedule, and the safety of ICpI in a limited number of patients, mainly affected by metastatic melanoma. Thereafter, clinical studies involving ICpI-based immunotherapy were extended to a large variety of neoplastic diseases, as summarized in Tables 1 to 5 that illustrate the present state of the art in this area.
Table 1: Approval status and clinical development of immune checkpoint inhibitors.
Agent |
Characteristics |
Clinical studies leading to drug approval and current status of clinical development relative to histological tumor type |
FDA Approval |
|
Anti-CTLA-4 agents |
||||
Ipilimumab |
Fully human anti-CTLA-4 IgG1k mAb |
Melanoma |
In a phase III trial on pretreated pts, median OS was significantly improved in the ipilimumab group (10.1 months vs 6.4 months in the control group) [224]. In another phase III trial, OS was better in the ipilimumab+dacarbazine group than in the dacarbazine group [225]. In phase II-III trials durable responses and prolonged survival have been reported [226]. In a phase III trial, median RFS was 26.1 months (95% CI 19.3-39.3) in the ipilimumab group vs 17.1 months (95% CI 13.4-21.6) in the placebo group (HR 0.75; 95% CI 0.64-0.90; p=0.0013); 3-year RFS was 46.5% (95% CI 41.5-51.3) in the ipilimumab group vs 34.8% (95% CI 30.1-39.5) in the placebo group [227]. |
|
NSCLC |
Under evaluation in phase II/III clinical trials. |
Pending |
||
SCLC, prostate cancer |
Under evaluation in phase II clinical trials. |
Pending |
||
Tremelimumab |
Fully human anti-CTLA-4 IgG2 mAb |
Melanoma |
In a phase III trial, median OS was 12.6 months (95% CI 10.8-14.3) for tremelimumab and 10.7 months (95% CI 9.36-11.96) for chemotherapy (HR, 0.88; p=0.127). ORRs were similar in the two arms, but response duration was significantly longer after tremelimumab (35.8 vs 13.7 months; p=0.0011) [228]. |
Not presented for approval. |
HCC, NSCLC |
Under evaluation in phase II clinical trials |
Pending |
||
Various cancer types |
Under evaluation in phase I/II clinical trials |
Pending |
||
Table 2: Approval status and clinical development of immune checkpoint inhibitors.
Agent |
Characteristics |
Clinical studies leading to drug approval and current status of clinical development relative to histological tumor type |
FDA Approval |
|
Anti-PD-1 agents |
||||
Nivolumab |
Fully human anti-PD-1 IgG4k mAb |
Melanoma |
• In a phase III trial, ORR was 31.7% (95% CI 23.5-40.8) in the nivolumab group vs 10.6% (95% CI 3.5-23.1) in the ICC group [229]. In a phase III trial, 1 year-OS was 72.9% (95% CI 65.5-78.9) in the nivolumab group vs 42.1% (95% CI 33.0-50.9) in the dacarbazine group (HR for death, 0.42; 99.79% CI 0.25-0.73; p < 0.001) [230]. In a phase III trial, median PFS was 11.5 months (95% CI 8.9-16.7) with nivolumab+ipilimumab, vs 2.9 months (95% CI 2.8-3.4) with ipilimumab (HR for death or disease progression, 0.42; 99.5% CI 0.31-0.57; p < 0.001), and 6.9 months (95% CI 4.3-9.5) with nivolumab (HR for the comparison with ipilimumab, 0.57; 99.5% CI 0.43-0.76; p < 0.001) [231]. |
|
NSCLC |
In a phase III trial (on squamous-NSCLC patients), median OS was 9.2 months (95% CI 7.3-13.3) with nivolumab vs 6.0 months (95% CI 5.1-7.3) with docetaxel. The risk of death was 41% lower with nivolumab than with docetaxel (HR 0.59; 95% CI 0.44-0.79; p < 0.001). At 1 year, the OS rate was 42% (95% CI 34-50) with nivolumab vs 24% (95% CI 17-31) with docetaxel [232]. In a phase III trial (on nonsquamous-NSCLC patients), median OS was 12.2 months (95% CI 9.7-15.0) in the nivolumab group and 9.4 months (95% CI 8.1-10.7) in the docetaxel group (HR for death 0.73; 96% CI 0.59-0.89; p = 0.002). The 18-months OS rate was 39% (95% CI 34-45) with nivolumab vs 23% (95% CI 19-28) with docetaxel [233]. |
|
||
RCC |
In a phase III trial, median OS was 25.0 months (95% CI 21.8-not estimable) with nivolumab and 19.6 months (95% CI 17.6-23.1) with everolimus. HR for death with nivolumab vs everolimus was 0.73 (98.5% CI 0.57-0.93; p = 0.002) [234]. |
|
||
Table 3: Approval status and clinical development of immune checkpoint inhibitors.
Agent |
Characteristics |
Clinical studies leading to drug approval and current status of clinical development relative to histological tumor type |
FDA Approval |
|
Anti-PD-1 agents |
||||
Nivolumab |
Fully human anti-PD-1 IgG4k mAb |
HL |
ORR was 87% (with 13% of patients having stable disease). PFS at 24 weeks was 86% [235]. |
|
Gastric cancer, glioblastoma, SCCHN |
Under evaluation in phase III clinical trials |
Pending |
||
Anal cancer, AML, cervical cancer, NHL, nasopharinx carcinoma, pancreatic cancer |
Under evaluation in phase II clinical trials |
Pending |
||
Pembrolizumab |
Humanized anti-PD-1 IgG4k mAb |
Melanoma |
In a phase III trial, estimated 12-month survival rates were 74.1% in the group who received pembrolizumab 10 mg/Kg every 2 weeks, 68.4% in the group who received pembrolizumab 10 mg/Kg every 3 weeks, and 58.2% in the group who received 4 doses of 3 mg/Kg ipilimumab every 3 weeks (HR for death for pembrolizumab every 2 weeks, 0.63; 95% CI 0.47-0.83; p = 0.0005; and for pembrolizumab every 3 weeks, 0.69; 95% CI 0.52-0.90; p = 0.0036) [236]. |
|
NSCLC |
•In a phase III trial (KEYNOTE-010), median OS was 10.4 months with pembrolizumab 2 mg/kg, 12.7 months with pembrolizumab 10 mg/kg, and 8.5 months with docetaxel. OS was significantly longer for pembrolizumab 2 mg/kg vs docetaxel (HR 0.71, 95% CI 0.58-0.88; p = 0.0008) and for pembrolizumab 10 mg/kg vs docetaxel (0.61, 0.49-0.75; p < 0.0001) [237]. |
|
||
Table 4: Approval status and clinical development of immune checkpoint inhibitors.
Agent |
Characteristics |
Clinical studies leading to drug approval and current status of clinical development relative to histological tumor type |
FDA Approval |
|
Anti-PD-1 agents |
||||
Pembrolizumab |
Humanized anti-PD-1 IgG4k mAb |
NSCLC |
In another open label phase III trial (KEYNOTE-024: first line pembrolizumab at a fixed dose of 200 mg every 3 weeks vs CDDP-based CT) median PFS was 10.3 months (95% CI 6.7 to not reached) in the pembrolizumab group vs 6.0 months (95% CI 4.2-6.2) in the CT group (HR for disease progression or death 0.50; 95% CI 0.37-0.68; p < 0.001). The estimated rate of OS at 6 months was 80.2% in the pembrolizumab group vs 72.4% in the CT (HR for death 0.60; 95% CI 0.41-0.89; p = 0.005) [238]. |
|
Gastric/GEJ cancer, SCCHN, urothelial cancer |
Under evaluation in phase III clinical trials |
Pending |
||
CRC, glioblastoma, HL, NHL, Merkel cell carcinoma, 3-BC |
Under evaluation in phase II clinical trials |
Pending |
||
Pidilizumab |
Humanized anti-PD-1 IgG1k mAb |
Multiple myeloma, pancreatic and prostate cancer, RCC, sarcoma, thymic cancer, NHL |
Under evaluation in phase II clinical trials |
Pending |
Table 5: Approval status and clinical development of immune checkpoint inhibitors.
Agent |
Characteristics |
Clinical studies leading to drug approval and current status of clinical development relative to histological tumor type |
FDA Approval |
|
Anti-PD-L1 agents |
||||
BMS-936559 |
A fully human anti-PD-L1 IgG4 mAb |
NSCLC, RCC, CRC, melanoma |
Under evaluation in phase I clinical trials |
Pending |
Atezolizumab (MPDL3280A) |
Human anti-PD-L1 IgG1 mAb |
Bladder cancer, NSCLC |
Under evaluation in phase III clinical trials |
Pending |
RCC |
Under evaluation in phase II clinical trials |
Pending |
||
CRC |
Under evaluation in phase IIb clinical trials |
Pending |
||
Melanoma |
Under evaluation in phase I/II clinical trials |
Pending |
||
Various cancer types |
Under evaluation in phase I clinical trials |
Pending |
||
Durvalumab (MEDI4736) |
Fully human anti-PD-L1 IgG1k mAb |
NSCLC, SCCHN, CRC |
Under evaluation in phase III clinical trials |
Pending |
Various cancer types |
Under evaluation in phase I clinical trials |
Pending |
||
Avelumab (MSB0019718C) |
Fully human anti-PD-L1 IgG1 mAb |
RCC, NSCLC |
Under evaluation in phase III clinical trials |
Pending |
Bladder, gastric and ovarian cancer, HNC, mesothelioma |
Under evaluation in phase II clinical trials |
Pending |
||
3-BC, triple negative breast cancer; CDDP, cisplatin; CI, confidential interval; CRC, colorectal cancer; CT, chemotherapy; HCC, hepatocellular carcinoma; GEJ, gastroesophageal junction; HL, Hodgkin lymphoma; HNC, head and neck cancer; HR, hazard ratio; ICC, investigator's choice of chemotherapy; mAb, monoclonal antibody; NHL, non-Hodgkin lymphoma; NSCLC, non-small cell lung cancer; PD-1, programmed death 1; PD-L1, programmed death-ligand 1; ORR, overall response rate; PFS, progression-free survival; RCC, renal cell carcinoma; SCCHN, squamous cell cancer of head and neck; OS, overall survival; RFS, relapse-free survival; vs, versus.
Why immune checkpoints as target of cancer immunotherapy?
A detailed comprehension of the mechanisms involved in the antitumor immune response is essential for explaining the development of innovative therapeutic strategies based on ICpI. The generation of an effective antitumor T-cell response involves the passage through different steps including Ag-specific T-cell priming, differentiation, trafficking and killing of tumor cells by cytotoxic CD8+ T cells [66]. Furthermore, the amplitude and the quality of Ag-specific T-cell activation is a finely regulated process where the balance between TCR engagement and co-stimulatory as well as inhibitory signals is critical in order to maintain the self-tolerance and prevent T-cell over-activation [67]. Co-stimulatory molecules play a critical role during T-cell activation by inducing cytokine production and promoting T-cell proliferation [68] and are essential for lowering the threshold of TCR activation, supporting the responses against low-affinity Ags [69]. Poorly functional, differentiated CD8+ and CD4+ T cells are characterized by the loss of co-stimulatory molecules such as CD27 and CD28, which have been described also as useful markers for defining T-cell subpopulations at various stages of differentiation [70-73]. However a substantial redundancy in co-stimulatory receptor usage has been demonstrated and co-stimulation through inducible T-cell co-stimulator (a ICOS CD28 family member), CD137 and CD134 (members of the tumor necrosis factor family) contributes to enhance T-cell activation [73-77] in CD28- T cells [73]. Therefore, additional changes to the loss of co-stimulatory receptors are involved in T-cell differentiation-related dysfunction. One such change is a rise in the expression of co-inhibitory receptors, commonly referred to as immune checkpoints, which negatively modulate the response of T cells to self proteins, chronic infection and tumor Ags and have been extensively studied over the last two decades.
The number of immune checkpoint proteins is unexpectedly high
Co-inhibitory receptors including Programmed Death-1 (PD-1), CTLA-4, Lymphocyte-Activation Gene since LAG-3 is acronim of Lymphocyte-Activation Gene 3 (LAG-3), T cell immunoglobulin mucin (TIM-3) and other molecules - some of which are described in the following paragraphs - are gradually up-regulated in T cells as they progress towards the acquisition of an “exhausted” phenotype. This phenotype is characterized by decreased production of effector cytokines and severely impaired anti-tumor and anti-viral functionality [78-80]. Tumors are capable of exploiting the homeostatic mechanisms regulated by these checkpoint molecules, affecting the immune system through several strategies including alterations in Ag expression and interference with T-cell priming and activation. Therefore, in addition to other regulatory effects on the surrounding microenvironment, the tumors escape immune-mediated detection and eradication [81].
Cancer cell defenses against host’s immunity
Although effector T cells are able to enter the tumor micro-environment (TME), once there, they are negatively regulated by the cancer cells themselves and by immunosuppressive factors and infiltrating cells. Among these immunosuppressive elements are transforming growth factor-β, IL-6, vascular endothelial growth factor, Treg, [82] and myeloid-derived suppressor cells (MDSCs, [83-84]). In addition, potential antitumor T cells present in TME are exposed to checkpoint pathways which play a critical role in regulating T-cell phenotype and function [85-89]. Then, the immunosuppressive nature of TME drives the infiltrating tumor-specific T cells towards terminal differentiation and exhaustion [90]. Exhausted T cells lose their functional activity in terms of cytokine production in a hierarchical way: IL-2 production is lost first, tumor necrosis factor-α production at the intermediate phase, while interferon (IFN)-γ and granzyme B are lost at an advanced stage of exhaustion [91]. PD-1 is one of the major regulator of T-cell exhaustion, however immune checkpoint receptors operate by using rather distinctive and non-redundant signaling pathways. Moreover, co-inhibitory receptors have been shown to function synergistically, mostly in concert with PD-1, suggesting that distinct molecules provide individual functions able to impair T-cell responses [92-95].
The CTLA-4 protein
CTLA-4, the first immune checkpoint receptor to be clinically targeted, primarily controls the amplitude of T-cell activation, contributing to the preservation of T-cell homeostasis [5, 67]. CTLA-4 knockout mice die within three weeks from immune destruction of multiple organs [96], which proves a critical role as regulator of T-cell-dependent immune responses. CTLA-4 mainly antagonizes the activity of CD28 by sharing its ligands, CD80 and CD86 - expressed by Antigen Presenting Cells (APCs) - with a much higher overall affinity for both molecules compared to the contender co-stimulatory receptor [97]. Thus CTLA-4 expression reduces T-cell activity by competing with CD28 in the binding with CD80 and CD86 and by delivering inhibitory signals to the T cell [98-100]. Although CTLA-4 is also expressed by activated CD8+ effector T cells, this molecule plays its main physiologic role by down-regulating CD4+ helper T-cell activity. Importantly, CTLA-4 engagement improves the suppressive activity of Tregs [101, 102] which are typically concentrated in tumor tissues where they contribute to the inhibition of anti-tumor effector responses. It follows that CTLA-4 blockade results in a wide enhancement of the immune responses dependent on CD4+ helper T cells. The expression of CTLA-4 is constitutive on Tregs, where it represents a target gene of the forkhead transcription factor Foxp3 [103, 104], a major player in the development and function of Tregs. The critical function played by CTLA-4 in regulating Treg function is confirmed by the observation that mice with CTLA-4-lacking Tregs show systemic lympho-proliferation, fatal T cell-mediated autoimmune diseases and potent anti-tumor immunity.
The engagement of CTLA-4 has been shown to inhibit CD3/CD28-mediated activation of the serine/threonine kinase AKT in T cells mainly through the recruitment and activation of the protein phosphatase 2A [105, 106] and not through phosphatidylinositol 3-kinase (PI3K) inactivation. However, CTLA-4 has been reported to bind also the tyrosine phosphatase SHP-2. SHP-2 activation suppresses the CD3/CD28-induced T-cell transcriptional profile and results in the inactivation of LCK and ZAP-70 kinases as well as the dephosphorylation of the CD3-ζ chains of the TCR complex, thus reducing activation of the TCR signaling [105, 106]. However, the role of SHP-2 in the CTLA-4 negative regulation T-cell function has not been fully elucidated yet.
The intrinsic inhibitory mechanisms mediated by the intracellular domain of CTLA-4 are accompanied by a cell-extrinsic mechanism which involves the capture of CD80 and CD86 from APCs by trans-endocytosis, the degradation of these molecules inside the T cell and the inhibition of T-cell activation through the CD28 engagement [107].
The PD-1 protein and its ligands
The inhibitory mechanisms triggered by PD-1 are distinct from those described for CTLA-4. The relatively milder and more chronic clinical pathological phenotypes which result from PD-1 blockade are probably related to the cell-intrinsic function and to the regulation of PD-1 expression.
PD-1 is a member of the CD28 superfamily, and plays its physiologic inhibitory role by regulating the induction and maintenance of peripheral tolerance, thus protecting tissues from autoimmunity, especially during inflammatory and Ag-specific responses [108-111, 67]. In particular, PD-1 exerts its inhibitory functions only after T-cell activation, following Ag recognition, thus contributing to restrain the effector phase of T-cell mediated tumor rejection [112]. The amount and source of Ag determines the strength and the kinetics of T-cell activation as well as the extent and regulation of PD-1 expression. Moreover, the ligands of PD-1, able to activate the molecule on T cells, are expressed to a different extent depending on the cell type [109].
PD-1 is expressed on activated T cells as well as on B and natural killer (NK) cells, activated monocytes and some subsets of dendritic cells (DCs) [109], implying a broad contribution in the immune regulation. The two ligands for PD-1 are PD-L1 (B7-H1, CD274) and PD-L2 (B7-DC, CD273) [109, 113-115]. Moreover, it has been unexpectedly observed a molecular interaction between PD-L1 and CD80, which indicates that CD80 expressed on T cells and APCs can possibly work as a receptor rather than a ligand, eliciting inhibitory signals when engaged by PD-L1 [116].
PD-L1 is expressed on resting T cells, B cells, DCs, macrophages, vascular endothelial cells, pancreatic islet cells and in various types of cancers, including Non Small Cell Lung Cancer (NSCLC), melanoma, renal cell carcinoma, gastric cancer, hepatocellular carcinoma as well as cutaneous lymphoma, multiple myeloma and various leukemias [117-125]. Elevated expression of PD-L1 has been found to represent an adverse prognostic biomarker in NSCLC [126]. PD-L1 can be up-regulated by IFN-γ produced by tumor-infiltrating T cells thus promoting the progression of cancer [127]. Moreover, in vitro exposure of human lymphoma cells to T cells or monocytes has been shown to up-regulate blast-associated PD-L1 expression [128]. Also radiation or chemotherapy can up-regulate PD-L1 [129, 130]. In particular, radiotherapy may induce direct killing of tumor cells and multiple immune-modulatory changes that can potentially influence the effectiveness of immunotherapy [129, 131]. On the other hand, NSCLC patients are significantly more responsive to anti-PD-1 mAbs when malignant cells over-express PD-L1 [132].
PD-L2, although less expressed by tumors than PD-L1, also binds to PD-1 and regulates T-cell function [133]. PD-L2 is constitutively expressed at low levels but can be induced on DCs, macrophages and mast cells in response to IL-4 and type 1 IFNs [134]. These differences in the pattern of expression suggest different functions played by the engagement of PD-1 in the immune regulation within distinct cell contexts. The limited expression of PD-L2 primarily to APC reflects a function in the control of the T-cell priming, while the wide occurrence of PD-L1 speaks in favor of an overall protective function of peripheral tissues from extreme inflammation.
Viruses and tumors exploit the negative regulatory function of PD-1, causing the inhibition of effector T-cell functionality, which translates into the onset of chronic infections and represents a major mechanism of immune resistance within the TME favoring the tumor progression.
Upon stimulation, PD-1, through its association with the SHP2 phosphatase, inhibits the proximal TCR signaling, leading to a strong reduction of T-cell functionality. PD-1 also prevents AKT phosphorylation by inhibiting CD28-mediated stimulation of PI3K [105, 106]. Therefore, CTLA-4 and PD-1 inhibit AKT by distinctive mechanisms.
Of remarkable importance is the finding that PD-1 blocks T-cell cycle progression through the G1 phase by suppressing the transcription of SKP2, a factor encoding a component of the ubiquitin ligase SCFSkp2 able to degrade p27kip1, an inhibitor of cyclin-dependent kinases [135]. PD-1-mediated decrease of SKP2 transcription is achieved through the inhibition of PI3K/AKT, Ras and extracellular signal-regulated kinase (ERK) signaling. IL-2 partially restores SKP2 expression, probably through the activation of ERK, but not the AKT signaling, demonstrating that PD-1 is able to impair T-cell proliferative potential by affecting multiple regulators of the cell cycle.
T-cell exhaustion is related to physical T-cell depletion especially in cancer. Apoptosis is one of the several potential mechanisms involved in PD-L1-associated T-cell death, which is supported by the inverse correlation existing between PD-L1 expression in tumor tissues and the number of tumor infiltrating lymphocytes (TILs) [136].
Foxp3+ Tregs express PD-1 and PD-L1 [137] and a critical function for the PD-1 engagement in the generation of Tregs has been clearly demonstrated [138]. In particular, the engagement of PD-1 on naive T cells can lead to the development of induced Treg cells partly through the inhibition of AKT/mTOR signaling [139]. Being many tumors highly infiltrated with Tregs which further suppress T-cell effector responses, PD-1 blockade can potentially enhance the antitumor responses also by decreasing the amount and the suppressive action of intra-tumoral Tregs.
The inhibitory function of the PD-1/PD-L1 engagement plays a critical role in reducing the immune-surveillance against tumors by inducing T-cell exhaustion. Therefore, the PD-1/PD-L1 pathway has become an attractive therapeutic target in the setting of cancer. PD-1 is highly expressed on exhausted T cells that develop in the setting of chronic Ag stimulation such as cancer and blockade of PD-L1 or PD-1 can reinvigorate the function of exhausted T cells. Blockade of PD-L1/PD-1 has been extensively shown to enhance T-cell anti-tumor function [140] including immune responses affecting malignant cell growth in the brain [141]. Interestingly, while the PD-L1/PD-1 signaling pathway is abundantly engaged in the TME, expression of PD-1 on peripheral CD4+ and CD8+ T cells has been shown to increase with tumor progression [142].
Several clinical studies have shown that high expression of PD-1 ligands on tumors correlates with poor prognosis [126, 143], which strongly suggests that the engagement of PD-L1/PD-1 pathway supports tumor escape from antitumor T-cell control. However, although co-inhibitory receptors have so far been considered to mark terminally differentiated “exhausted” T cells, they have recently been also associated with the activation status and the differentiation profile of Ag-specific T cells [59, 144] and must be considered instrumental for limiting the self-tissue damage at the tumor site. As stated before, the fate of T cells after the encounter with the specific Ag is determined also by additional inputs through co-receptors which finely regulate strength, duration and properties of the response upon interaction with their ligands. Co-expression of PD-1 with other co-stimulatory or co-inhibitory molecules in a particular Ag-context may then represent a rheostat in the control of highly reactive stimulated T cells [59, 145].
The Killer-cell Lectin like Receptor G1 (KLRG1)
The expression of KLRG1 on Ag-experienced T cells and NK cells increases considerably with age and differentiation [146-148], with the highest expression observed in memory and in end-stage differentiated T cells [149]. An inhibitory role for KLRG1 on the cytolytic activity of polyclonal human NK cells has been suggested [150]. Moreover a critical role has been demonstrated for KLRG1 in the inhibition of AKT phosphorylation at the ser473 site, which results in a compromised proliferation of primary CD8+ T cells [148]. Actually, the loss of the ability to phosphorylate AKT at ser473 is a functional alteration that occurs throughout progressive T-cell differentiation and ageing [73, 151].
The LAG-3 protein
LAG-3 belongs to the immunoglobulin superfamily and is a crucial regulator of T-cell function [92, 152]. It is expressed on activated T cells, B cells, NK cells, DCs and TILs, but not on resting T cells [153, 154].
LAG-3 associates with the CD3-TCR complex after the TCR engagement and negatively regulates the TCR signaling in effector T cells in a pattern similar to that observed for CTLA-4 [155], impairing T-cell proliferation, homeostasis and functionality through the inhibition of calcium fluxes (156]. Similar to CD4 molecule, LAG-3 oligomerizes at the surface of T cells and binds to MHC class II molecules on APCs but with significantly higher affinity as compared with the CD4 molecule [157], thus decreasing the Ag-dependent stimulation of CD4+ T cells. However, a double role in regulating T-cell functionality has been suggested by the observation that expression of LAG-3 on T cells is associated with either the down-regulation of cytokine secretion [158, 159] or the induction of Th1 cytokine production, including IL-2 [160]. LAG-3 plays also a critical role in the control of the functional activity of both natural and induced immunosuppressive Tregs. As described for CTLA-4 and PD-1, LAG-3 is essential for the extrinsic regulation of Treg homeostasis and development [161]. A population of expanded CD4+CD25highFoxp3+ Tregs expressing LAG-3 has been identified in the peripheral blood, in T cells of tumor-invaded lymph nodes and in T cells infiltrating the visceral metastasis of melanoma and sarcoma patients [162]. Of particular interest is the observation that high LAG-3-expression makes effector T cells more susceptible to Treg-mediated suppression [154].
LAG-3 is highly co-expressed with other immune inhibitory molecules, including TIM-3, PD-1, 2B4 and T-cell immunoglobulin and ITIM domain (TIGIT), on tumor-specific CD4+ effector T cells [163]. On Melan-A specific CD8+ T cells isolated from melanoma patients, LAG-3 is highly co-expressed with PD-1 and TIM-3 [59]. LAG-3 itself can directly modulate the activity of PD-1+ T cells [163] and the synergy between LAG-3 and PD-1 can potentiate the tumor-induced tolerance [164].
TIM-3 protein
TIM-3 is a glycoprotein which possesses on its extracellular portion both the immunoglobulin and the mucin domain. TIM-3 has been initially observed on terminally differentiated IFNγ-producing CD4+ Th1 cells and CD8+ cytotoxic T cells [165, 166], as well as on Th17, DCs, monocytes, Tregs, mast cells, NK cells, TILs and tumor cells, including melanoma, squamous cell carcinoma, gastric cancer and NSCLC cells, but not on CD4+ Th2 cells [154].
The most important role of TIM-3 is the negative control of Th1 immunity and the induction of peripheral tolerance [167]. The TIM-3/galectin-9 pathway engagement reduces the proliferative potential and functionality of Th1 and Th17 cells [168] and contributes to the immune-suppressive environment of TME through the promotion of Treg development [169]. TIM-3 also provides a new surface marker able to describe activated tumor infiltrating Tregs, which have been found to co-express higher levels of PD-1, CTLA-4 and the glucocorticoid-induced tumour necrosis factor receptor-related protein [170].
Consistent with the observation that the TIM-3 and PD-1 expressing CD8+ T cells represent the most exhausted TIL population, co-blockade of the TIM-3 and PD-1 signaling pathways has been shown to induce a more vigorous antitumor outcome as compared with PD-1 blockade alone [171].
The TIGIT receptor
The co-inhibitory receptor TIGIT, that contains the immunoreceptor tyrosine-based inhibitory motif (ITIM), has been initially described as a modest inhibitor of CD4+ T cell priming and NK cell killing activity. More recently, it has been shown to be highly expressed by tumor-infiltrating CD8+ T cells in parallel with PD-1 [172], as well as in models of chronic viral infection. In particular, TIGIT has been proven to be a critical and specific regulator of CD8+ T cell-dependent chronic immune responses. Co-blockade of TIGIT and PD-L1 improves synergistically the CD8+ T-cell effector functional activity [173], an effect abolished by blockade of the TIGIT’s corresponding co-stimulatory receptor, CD226. The hypothesis that TIGIT may represent a critical collaborator of the PD-1/PD-L1 engagement in order to limit the activity of chronically stimulated CD8+ T cells is supported by the observation that co-blockade of TIGIT and PD-1 is essential to restore the anti-tumor functional activity of effector CD8+ T cells within the highly immunosuppressive TME.
TIGIT has been shown to identify the most dysfunctional subset of effector CD8+ T cells in tumors, and the tumor-infiltrating Tregs characterized by a highly suppressive phenotype. TIGIT signaling controls Treg phenotype, and Tregs show an increased expression of TIM-3 in the tumor tissue, where TIM-3 and TIGIT synergize in the suppression of the immune responses [174]. TIGIT expressing Foxp3+ Tregs have been recently identified as a subset able to specifically suppress pro-inflammatory Th1 and Th17, but not Th2 cell responses [175].
The NK cell receptor 2B4
2B4 belongs to the CD2 family and is expressed on NK, γ/δ and memory CD8+ T cells [176]. The murine NK receptor 2B4 displays both inhibitory and activating functions, whereas human 2B4 has been reported to be mainly an activating molecule. In what way murine 2B4 can act both as an activating and inhibitory molecule and what distinguishes its function from human 2B4 receptor is still under investigation [177]. The amount of 2B4 expression and the level of 2B4 cross-linking play a significant role in the regulation of 2B4-mediated T-cell signaling pathway. A substantial reduction of T-cell activation has been observed with high levels and a strong engagement of 2B4, in the presence of a weak activation of T-cell signaling-associated molecules. Therefore 2B4 can have opposite effects depending on the degree of receptor expression, the level of its ligation, and the relative abundance of selected adaptor molecules [176].
The B and T lymphocyte attenuator (BTLA)
This molecule has been identified as another co-inhibitory receptor with structural and functional similarities to CTLA-4 and PD-1 [178]. BTLA is induced during T-cell activation and remains expressed on Th1 but not on Th2 CD4+ T cells. The engagement of BTLA induces its tyrosine phosphorylation and association with the tyrosine phosphatases SHP-1 and SHP-2, thus attenuating, through the subsequent inhibition of TCR activation, the production of IL-2. BTLA is also expressed on activated CD8+ T cells where it induces functional inhibition through its ligand herpes virus entry mediator (HVEM). In human virus Ag-specific CD8+ T cells the expression of BTLA is gradually down-regulated as they differentiate into effector cells [179]. In contrast, human melanoma Ag-specific effector CD8+ T cells persistently express high levels of BTLA and remain susceptible to the functional inhibition mediated by its ligand HVEM. Such persistence of BTLA expression has also been found in tumor Ag-specific CD8+ T cells isolated from melanoma patients with spontaneous anti-tumor immune responses and after peptide vaccination. Interestingly, the co-expression of BTLA with PD-1 and TIM-3 has been shown to identify the most dysfunctional NY-ESO-1-specific CD8+ T cell population in melanoma patients [80].
The indoleamine 2,3 dioxygenase (IDO)
The control of the access to nutrients is an important strategy able to regulate cellular responses to proliferative stimuli. The IDO enzyme is responsible for a critical step in the metabolic pathway that converts the essential amino acid L-tryptophan into L-kynurenine, and has been shown to exert a highly suppressive activity on T cells [180]. Both L-tryptophan depletion and L-kynurenine accumulation appear implicated in the immunosuppressive activity of IDO [180].
IDO is expressed in different cell types, including DCs and macrophages and plays a critical role in immunological tolerance. Several cancer types can express themselves IDO or induce its expression in host APCs, either directly or indirectly, thus leading to the impairment of T-cell functionality [181]. Tumors are then able to create an immunosuppressive microenvironment able to impair T-cell mediated antitumor immune response by inducing IDO over-expression [182]. Interestingly, a tumor-mediated IDO-dependent activation of suppressive Tregs has also been documented [183].
In a mouse model, IDO expressed by the host immune cells has been shown to reduce the infiltration of tumor-reactive T cells in B16 tumors, inducing resistance to immunotherapy with mAbs targeting CTLA-4 and PD-1 [184]. Differently, IDO-KO mice have been shown to mount a good anti-tumor response following treatment with anti-CTLA-4 mAbs. This IDO-induced resistance to T-cell-targeting immunotherapies has been associated with a general increase in the recruitment of MDSCs into the TME. A comparable association between IDO expression and MDSC infiltration into the TME has been observed in human melanoma samples and animal tumor models naturally expressing high levels of IDO. These observations prove that IDO represents a key regulator of immunosuppression both at the systemic and TME level, and provide a strong rationale for therapeutic targeting of this pathway.
Cancer immunoediting
The paradigm of cancer immunoediting shows that, during the different phases of cancerogenesis and tumor progression, the immune response shapes the tumor with the selection of tumor variants that escape immune recognition [81, 185, 186].
One of the crucial challenges in immunology is the comprehension of the control of the immune system on cancer development and progression. The immune system plays a dual role in cancer development, being able to contrast tumor growth but also to promote progression either by selecting tumor cells resistant to the immunological control or by modeling conditions within the TME. An essential principle of cancer immunosurveillance is that cancer cells express a number of Ags different from their non-transformed counterparts. These Ags include differentiation, mutated, overexpressed, viral and cancer/testis molecules [186]. Changes capable of conferring resistance to the attack by the immune system include the loss of expression of tumor Ags and an altered expression pattern of class I molecules, as a consequence of critical deficiencies in the Ag processing pathway. This promotes a reduced expression or a total loss of class I peptide presentation, which in turn allows tumor cell escape from Ag-specific effector CD8+ T-cell killing.
In 2002 Dunn et al. [185] showed that tumors developed in the absence of an intact immune system (classified as “unedited” tumors) were more immunogenic than similar tumors derived from immunocompetent mice (classified as “edited” tumors). This observation demonstrated that the immune system controls tumors also in terms of quality and immunogenicity. The concept that the immune system, besides its protective role, also shapes tumor immunogenicity constitutes the starting point of the “cancer immunoediting hypothesis”. Cancer immunoediting consists of three sequential phases: elimination, equilibrium, and escape [186]. In the elimination phase, the immune system works at eliminating developing tumors before they become clinically apparent. Cancer immunoediting enters the equilibrium phase when cancer cells are not totally eliminated but cancer growth is prevented by immune-mediated mechanisms. In particular, the editing of tumor immunogenicity takes place during the equilibrium phase, when some tumor cells become not recognizable by the immune system (due to Ag loss or defects in Ag processing or presentation), or resistant to effector T-cells, or induce an immunosuppressive environment within the TME. These tumor cells may then enter the escape phase, where cancer growth is no longer inhibited by the immune system and the tumor becomes clinically apparent. However, external factors including the immune senescence associated with ageing may influence this directionality, and cancer cells may directly progress toward the escape phase (186].
The role of mutation-induced tumor neoantigens and susceptibility to ICpI.
As tumors grow, they acquire mutations, some of which generate neoantigens potentially able to elicit host’s immune responses [187]. In relatively recent years, a growing number of clinical studies has revealed a direct correlation between mutation load detectable in malignant cells and susceptibility to the therapeutic effectiveness of ICpI. In particular, a direct relationship between the extent of non-synonymous mutation burden and durable clinical benefit was found in NSCLC [188] and in ovarian cancer harboring mutated BRCA1/2 gene [189]. Moreover, Le et al. [190] demonstrated that pembrolizumab, a mAb able to suppress PD-1 function, was significantly more active against metastatic colorectal cancer when malignant cells were deficient for the MMR system. In this case, whole-exome sequencing showed that MMR-deficient tumor cells had a number of somatic mutations approximately 25 times higher than that detectable in MMR-proficient tumors.
Pharmacologically-driven non-synonymous somatic mutations leading to increased immunogenicity of tumors could be induced not only by triazenes but potentially also by other antitumor agents. Actually, cisplatin that was found to be highly mutagenic in a lymphoblastic cell line model [191] merits further investigation for possible DIX effects. In addition, drugs able to influence the epigenome profile of malignant cells can provide novel immunogenic targets through up-regulation of the expression of non-pathogenic retroviruses inserted in human cell genome (“viral mimicry”) as found for colorectal cancer [192] and supposedly applicable to neuroendocrine tumors [193].
The role of somatic mutations in the immunogenicity of neoplastic cells and its relevance not only in the therapeutic response to ICpI but also in vaccine development has been widely confirmed by several authors [194-198]. However, a note of caution stems from a clinical investigation on NSCLC reported by McGranahan [198] who stressed that, in spite of large neoepitope burden, an excess of neoantigen intratumor heterogeneity accompanied by the absence of common antigenic determinants in all malignant cells does not provide therapeutic advantage upon ICpI administration. The authors termed “clonal neoantigens” tumor-specific Ags - generally originated by mutational mechanisms - that are present in the whole tumor cell population. On the contrary, a number of different non cross-reacting tumor-associated neoantigens present in the bulk malignant cells were designated as “subclonal neoantigens”. Therefore, it is reasonable to predict that in case of clonal neoantigens, host’s immune response can be efficiently directed against a common epitope present in all tumor cells. Conversely, in case of subclonal neoantigens, each of the great number of malignant cell-associated neoantigens does not reach the threshold required to elicit an effective cell-mediated response even if assisted by ICpI-based therapy.
Therapeutic potential of DIX in appropriate combination with ICpI
In the last few years we assisted to the enormous expansion of preclinical and clinical studies on cancer immunotherapy based on tumor-associated neoantigens targeted by cytotoxic effector cells generated by immune mechanisms amplified by ICpI. Since, as previously stated, a large number of investigations found that tumor neoantigens are generally the result of somatic mutations, DIX appears to open new and exciting perspectives in the future development of ICpI-dependent cancer immunotherapy. In animal models, DIX was found to be inducible in vivo or in vitro in a number of mouse leukemia syngeneic with strains homozygous for different types of MHC, such as H-2d (e.g. L1210 [1, 12, 15, 18, 19, 36, 199], LSTRA [9], L5178Y [13], P815 [200, 201]) H-2b (RBL-5 [9]) and H-2k (K36 [202]) although in this case the immunogenicity of DTIC-treated cells was found to be modest. This observation implies that DIX is not limited to a specific genetic pattern in mouse models. However, it must be stressed that all these malignant cell types are characterized by complete homozygosity of the entire genome and we do not know the influence that this particular biological situation could have on the mutational profile induced by triazenes.
As previously mentioned in this report, the earliest hint of triazene-induced DIX in human neoplasias was obtained by D’Atri et al. in 1994 [11] who found that selected CD8+ CTL clones directed against in vitro triazene-treated H-125 human lung adenocarcinoma cell line were able to lyse the drug-treated cells but not the untreated parental cells.
Several difficulties must be overcome before formulating an adequate DIX-based design of cancer immunotherapy centered on ICpI administration to amplify host’s cell-mediated responses targeting pharmacologically-induced tumor neoantigens. Firstly, somatic mutations affecting steps of the complex apparatus involved in Ag presentation by malignant cells could compromise its function [203, 204] thus subtracting target cells from lethal attack by effectors of host’s immunity. One of the most utilized devices adopted by tumors to evade T-cell mediated immunity consists in down-regulation of MHC class I molecule expression thereby precluding non-self peptide presentation that can be recognized by cytotoxic T cells [203, 205]. Several approaches have been described to be able to increase MHC expression and to overcome defects in Ag presentation pathways. In particular, upregulation of HLA class I expression in malignant cells has been obtained using pharmacological agents such as DNA hypomethylating compounds (e.g. azacytidyne, [205, 206] or, more recently, SGI-110 [207]) and histone deacetylase inhibitors. Likewise, several studies indicated that histone deacetylase inhibition is able to increase consistently various mechanisms involved in Ag presentation, including, for example, DC function [208].
Unexpectedly, neoantigen pattern of each single patient could also provide additional problems concerning the efficiency of ICpI treatment. Several data from the literature point out that patients with tumors endowed with mutations that are predicted to be immunogenic, show survival benefit from ICpI administration [209]. However, as previously mentioned in this review, drug-induced generation of extremely high numbers of different neoantigens within the tumor cell population (i.e. “subclonal neoantigens” [198]) could be unable to elicit an adequate host’s response. It is reasonable to predict that treatment with high-dose classical cytotoxic or targeted chemotherapy could reduce substantially the polyclonality of malignant cells pre-exposed to drug-induced mutagenization. It follows that a relatively low number of drug-resistant clones could provide an adequate antigenic stimulus to the host’s immune apparatus. The findings described by Nicolin et al. [210], who found that several different L1210 cell lines resistant to a number of antitumor agents are limitedly but significantly immunogenic for the histocompatible host, could be interpreted on this basis.
As an example, a purely speculative therapeutic design stemming from a rational application of triazene-related DIX followed by ICpI should contain at least 3 sequential phases. The first step (i.e. DIX phase) includes temozolomide treatment in two types of patients, i.e. those bearing MGMT-deficient neoplasia, or those affected by MGMT-proficient malignancy. In the latter case, temozolomide-induced xenogenization is prevented by the efficient removal of methyl adducts at O6-guanine [211, 212] and the triazene compound must be associated with an MGMT inhibiting agent [213], including O6-benzylguanine [214, 215], cisplatin [216-218] or Lomeguatrib, that is the best drug available today [3, 47, 214, 219-221]. However, Lomeguatrib downregulates profoundly MGMT expression in both neoplastic and normal cells. Therefore, the dosage of temozolomide must be carefully controlled since this association is characterized by elevated myelotoxicity [221]. Alternatively, if MGMT inhibitors are not available, a sequential administration of high-dose triazenes that deplete target cells of MGMT followed by a DIX generating treatment with the same triazene could be taken into consideration [ 222]. During the DIX phase, it is possible that normal hematopoietic cells undergo malignant transformation. Actually, treatment with temozolomide, especially in brain tumor cases, has been found to be followed by the appearance of leukemia or myelodysplastic syndrome (reviewed in [223]). It can be suggested that rare clones of target hematopoietic cells survive temozolomide-induced apoptosis thank to MMR deficiency [47] or inadequacy of the apoptotic function. On the other hand, it is reasonable to hypothesize that these clones could be also endowed with at least a limited degree of immunogenicity that renders the cells susceptible to host’s immune attack, especially after extreme amplification by ICpI treatment.
The second phase (“clonal simplification”) comes after the completion of DIX, and is directed to reduce the number of xenogenized tumor cell clones through treatment with a standard chemotherapy approach. During this phase it is quite possible that the number of immunogenic xenogenized subclones will be sensibly reduced, since a limited number of drug-resistant clones are selected. Therefore, at the end of phase 2 the host should be able to mount an even minimal immune response, although largely inefficient to control tumor growth.
The third phase (ICpI-immune amplification) should complete the work through treatment with ICpI, leading to immune-mediated suppression of target xenogenized malignant cells and bone-marrow-derived pre-malignant cells.
In conclusion, triazene-induced DIX could open up a new avenue in the area of tumor immunotherapy. Indeed, this treatment modality discloses the invaluable opportunity to design a pharmacological control of neoantigen generation that provides the molecular bases of efficient ICpI-dependent suppression of malignant cell growth.
Abbreviations
Ag(s), antigen(s)
APC, antigen presenting cells
BCNU, bis-chloroethyl-nitrosourea
BTLA, B and T lymphocyte attenuator
CTL, cytotoxic T lymphocytes
CTLA-4, cytotoxic T lymphocyte antigen-4
CX, chemical xenogenization
DC, dendritic cells
DIX, drug-induced xenogenization
DTIC, dimethyltriazene-imidazole-4-carboxamide,
ERK, extracellular signal-regulated kinase
HVEM, herpes virus entry mediator
ICpI, immune checkpoint inhibitors
IDO, indoleamine 2,3 dioxygenase
IFN, interferon
IL, interleukin
ITIM, immunoreceptor tyrosine-based inhibitory motif
KLRG1, killer-cell lectin like receptor G1
LAG-3, lymphocyte activation gene-3
mAb, monoclonal antibody
MDSCs, myeloid-derived suppressor cells
MGMT, O6-methylguanine-DNA methyltransferase
MHC, major histocompatibility complex
MMR, mismatch repair
MNNG, N-methyl-N’-nitro-N-nitrosoguanidine
MST, median survival time
NK, natural killer
NSCLC, non small cell lung cancer
O6-MeG, O6-methylguanine
PD-1/2, programmed death-1/2
PD-L1/2 programmed death ligand 1/2
PI3K, phosphatidylinositol 3-kinase
TCR, T-cell receptor
TIGIT, T-cell immunoglobulin and ITIM domain
TIL, tumor infiltrating lymphocytes
TIM-3, T cell immunoglobulin mucin-3
TME, tumor micro-environment
Treg, regulatory T cells
Acknowledgments
The authors wish to thank Graziano Bonelli (University of Rome Tor Vergata, School of Medicine, Rome, Italy) for the artwork.
Conflicts of interest
The authors declare no conflicts of interest.
GRANT SUPPORT
This work was supported by a grant of the Italian Association for Cancer Research (AIRC) to Stefania D’Atri (Investigator Grant Project 17585).
REFERENCES
1. Bonmassar E, Bonmassar A, Vadlamudi S, Goldin A. Immunological alteration of leukemic cells in vivo after treatment with an antitumor drug. Proc Natl Acad Sci. U S A. 1970; 66: 1089-1095.
2. Kobayashi H, Kodama T, Shirai T, Kaji H, Hosokawa M, Sendo F, Saito H, Takeichi N. Artificial regression of rat tumors infected with Friend virus (xenogenization): an effect produced by acquired antigen. Hokkaido J Med Sci. 1969; 44: 133-134.
3. Bonmassar L, Marchesi F, Pascale E, Franzese O, Margison GP, Bianchi A, D’Atri S, Bernardini S, Lattuada D, Bonmassar E, Aquino A. Triazene compounds in the treatment of acute myeloid leukemia: a short review and a case report. Curr Med Chem. 2013; 20: 2389-2401.
4. Fellner C. Ipilimumab (Yervoy) prolongs survival in advanced melanoma: serious side effects and a hefty price tag may limit its use. P&T. 2012; 37: 503-530.
5. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015; 33: 1974-1982.
6. Agarwala SS. Practical approaches to immunotherapy in the clinic. Semin Oncol. 2015; 42 Suppl 3: S20-7.
7. Nardelli B, Puccetti P, Romani L, Sava G, Bonmassar E, Fioretti MC. Chemical xenogenization of murine lymphoma cells with triazene derivatives: immunotoxicological studies. Cancer Immunol Immunother. 1984; 17: 213-217.
8. Campanile F, Houchens DP, Gaston M, Goldin A, Bonmassar E. Increased immunogenicity of two lymphoma lines after drug treatment of athymic (nude) mice. J Natl Cancer Inst. 1975; 55: 207-209.
9. Houchens DP, Bonmassar E, Gaston MR, Kende M, Goldin A. Drug-mediated immunogenic changes of virus-induced leukemia in vivo. Cancer Res. 1976; 36: 1347-1352.
10. Grohmann U, Puccetti P, Belladonna ML, Fallarino F, Bianchi R, Binaglia L, Sagakuchi K, Mage MG, Appella E, Fioretti MC. Multiple point mutations in an endogenous retroviral gene confer high immunogenicity on a drug-treated murine tumor. J Immunol. 1995; 154: 4630-4641.
11. D’Atri S, Romani L, Bonmassar E, Grohmann U, Tricarico M, Christmas SE, Moore M. Untreated or drug-treated tumor cells are differentially recognized by allogeneic lymphocytes. Int J Immunopharmacol. 1994; 16: 569-579.
12. Bonmassar E, Bonmassar A, Vadlamudi S, Goldin A. Antigenic changes of L1210 leukemia in mice treated with 5-(3,3-dimethy-1-triazeno)imidazole-4-carboxamide. Cancer Res. 1972; 32: 1446-1450.
13. Nicolin A, Spreafico F, Bonmassar E, Goldin A. Antigenic changes of L5178Y lymphoma after treatment with 5-(3,3-dimethyl-1-triazeno) imidazole-4-carboxamide in vivo. J Natl Cancer Inst. 1976; 56: 89-93.
14. Giampietri A, Bonmassar E, Goldin A. Drug induced modulation of immune responses in mice: effects of 5-(3,3-dimethyl-1-triazeno)-imidazole-4-carboxamide (DTIC) and cyclophosphamide (Cy). J Immunopharmacol. 1978-1979; 1: 61-86.
15. Contessa AR, Bonmassar A, Giampietri A, Circolo A, Goldin A, Fioretti MC. In vitro generation of a highly immunogenic subline of L1210 leukemia following exposure to 5-(3,3’-dimethyl-1-triazeno) imidazole-4-carboxamide. Cancer Res. 1981; 41: 2476-2482.
16. Mihich E. Modification of tumor regression by immunologic means. Cancer Res. 1969; 29: 2345-2350.
17. Riccardi C, Bartocci A, Puccetti P, Spreafico F, Bonmassar E, Goldin A. Combined effects of antineoplastic agents and anti-lymphoma allograft reactions. Eur J Cancer. 1980; 16: 23-33.
18. Contessa AR, Giampietri A, Bonmassar A, Goldin A. Increased immunogenicity of L1210 leukemia following short-term exposure to 5(3,3’-dimethyl-1-triazeno)-imidazole-4-carboxamide (DTIC) in vivo or in vitro. Cancer Immunol. Immunother. 1979; 7: 71-76.
19. Marelli O, Canti G, Franco P, Prandoni N, Ricci L, Nicolin A. L1210/DTIC antigenic subline: studies at the clone level. Eur J Cancer Clin Oncol. 1986; 22: 1401-1405.
20. Marelli O, Franco P, Canti G, Ricci L, Prandoni N, Nicolin A, Festenstein H. DTIC xenogenized lines obtained from an L1210 clone: clonal analysis of cytotoxic T lymphocyte reactivity. Br J Cancer. 1988; 58: 171-175.
21. Skibba JL, Ertürk E, Bryan G. Induction of thymic lymphosarcoma and mammary adenocarcinomas in rats by oral administration of the antitumor agent 4(5)-(3,3-dimethyl-1-triazeno)imidazole-5(4)-carboxamide. Cancer. 1970; 26: 1000-1005.
22. Beal DD, Skibba JL, Croft WA, Cohen SM, Bryan GT. Carcinogenicity of the antineoplastic agent, 5-(3,3-dimethyl-1-triazeno)-imidazole-4-carboxamide, and its metabolites in rats. J Natl Cancer Inst. 1975; 54: 951-957.
23. Johnson HG, Bach MK. Apparent antimutagenic activity of quinacrine hydrochloride in Detroit-98 human sternal marrow cells grown in culture. Cancer Res. 1969; 29: 1367-1370.
24. Giampietri A, Fioretti MC, Goldin A, Bonmassar E. Drug-mediated antigenic changes in murine leukemia cells: antagonistic effects of quinacrine, an antimutagenic compound. J Natl Cancer Inst. 1980; 64: 297-301.
25. Grohmann U, Ullrich SJ, Mage MG, Appella E, Fioretti MC, Puccetti P, Romani L. Identification and immunogenic properties of an 80-kDa surface antigen on a drug-treated tumor variant: relationship to MuLV gp70. Eur J Immunol. 1990; 20: 629-636.
26. Grohmann U, Fioretti MC, Binaglia L, Belladonna ML, Bianchi R, Puccetti P. Immunogenic properties of retroviral p15E from drug-treated murine mastocytoma P815. Int J Cancer. 1993; 55: 344-350.
27. Hartmann S, Hasenkamp N, Mayer J, Michaux J, Morand S, Mazzoni CJ, Roca AL, Greenwood AD. Endogenous murine leukemia retroviral variation across wild European and inbred strains of house mouse. BMC Genomics. 2015; 16: 613. doi: 10.1186/s12864-015-1766-z.
28. Boon T, Kellermann O. Rejection by syngeneic mice of cell variants obtained by mutagenesis of a malignant teratocarcinoma cell line. Proc Natl Acad Sci U S A. 1977; 74: 272-275.
29. Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem Res Toxicol. 2006; 19: 1580-1594.
30. Van Pel A, Georlette M, Boon T. Tumor cell variants obtained by mutagenesis of a Lewis lung carcinoma cell line: immune rejection by syngeneic mice. Proc Natl Acad Sci U S A. 1979; 76: 5282-5285.
31. De Plaen E, Lurquin C, Van Pel A, Mariamé B, Szikora JP, Wölfel T, Sibille C, Chomez P, Boon T. Immunogenic (tum-) variants of mouse tumor P815: cloning of the gene of tum- antigen P91A and identification of the tum- mutation. Proc Natl Acad Sci U S A. 1988; 85: 2274-2278.
32. Szikora JP, Van Pel A, Brichard V, André M, Van Baren N, Henry P, De Plaen E, Boon T. Structure of the gene of tum- transplantation antigen P35B: presence of a point mutation in the antigenic allele. EMBO J. 1990; 9: 1041-1050.
33. Altevogt P, Apt D. High-frequency generation of altered Mr 70,000 env glycoproteins in N-methyl-N’-nitro-N-nitrosoguanidine-treated murine tumor cells. Cancer Res. 1988; 48: 1137-1142.
34. Sibille C, Chomez P, Wildmann C, Van Pel A, De Plaen E, Maryanski JL, de Bergeyck V, Boon T. Structure of the gene of tum- transplantation antigen P198: A point mutation generates a new antigenic peptide. J Exp Med. 1990; 172: 35-45.
35. Riccardi C, Fioretti MC, Giampietri A, Puccetti P, Goldin A, Bonmassar E. Growth inhibition of normal or drug-treated lymphoma cells in lethally irradiated mice. J Natl Cancer Inst. 1978; 60: 1083-1090.
36. Marelli O, Franco P, Canti G, Ricci L, Prandoni N, Nicolin A, Festenstein H. DTIC xenogenized lines obtained from an L1210 clone: Clonal analysis of cytotoxic T lymphocyte reactivity. Br J Cancer. 1988; 58: 171-175.
37. Marelli O, Franco P, Canti G, Prandoni N, Ricci L. Induction of new antigenic properties on DTIC-treated L1210 clones. Tumori. 1988; 74: 387-392.
38. Nicolin A, Bini A, Coronetti E, Goldin A. Cellular immune response to a drug-treated L5178Y lymphoma subline. Nature. 1974; 251: 654-655.
39. Nicolin A, Vadlamudi S, Goldin A. Antigenicity of L1210 leukemic sublines induced by drugs. Cancer Res. 1972; 32: 653-657.
40. Testorelli C, Morelli S, Goldin A, Nicolin A. Characterization of a monoclonal antibody to L1210 leukaemia. Br J Cancer. 1982; 45: 395-402.
41. Romani L, Puccetti P, Fioretti MC, Mage MG. Humoral response against murine lymphoma cells xenogenized by drug treatment in vivo. Int J Cancer. 1985; 36: 225-231.
42. Grohmann U, Puccetti P, Fioretti MC, Mage MG, Romani L. Cell-mediated immunity to chemically xenogenized tumors--III. Generation of monoclonal antibodies interfering with reactivity to novel antigens. Int J Immunopharmacol. 1988; 10: 803-809.
43. Grohmann U, Romani L, Principato GB, Puccetti P, Mage MG, Fioretti MC. Generation of monoclonal antibodies to a chemically xenogenized murine lymphoma. Pharmacol Res Commun. 1988; 20: 443-444.
44. Riccardi C, Fioretti MC, Giampietri A, Puccetti P, Goldin A. Growth and rejection patterns of murine lymphoma cells antigenically altered following drug treatment in vivo. Transplantation. 1978; 25: 63-68.
45. Cudkowicz G, Rossi GB. Hybrid resistance to parental DBA-2 grafts: independence from the H-2 locus. I. Studies with normal hematopoietic cells. J Natl Cancer Inst. 1972; 48: 131-139.
46. Cudkowicz G, Nakamura I. Genetics of the murine hemopoietic-histocompatibility system: an overview. Transplant Proc. 1983; 15: 2058-2063.
47. Marchesi F, Turriziani M, Tortorelli G, Avvisati G, Torino F, De Vecchis L. Triazene compounds: Mechanism of action and related DNA repair systems. Pharmacol Res. 2007; 56: 275-287.
48. Drabløs F, Feyzi E, Aas PA, Vaagbø CB, Kavli B, Bratlie MS, Peña-Diaz J, Otterlei M, Slupphaug G, Krokan HE. Alkylation damage in DNA and RNA--repair mechanisms and medical significance. DNA Repair (Amst). 2004; 3: 1389-1407.
49. Catapano CV, Broggini M, Erba E, Ponti M, Mariani L, Citti L, D’Incalci M. In vitro and in vivo methazolastone-induced DNA damage and repair in L-1210 leukemia sensitive and resistant to chloroethylnitrosoureas. Cancer Res. 1987; 47: 4884-4889.
50. Gavande NS, VanderVere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS, Turchi JJ. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol Ther. 2016; 160: 65-83.
51. Fioretti MC, Bianchi R, Romani L, Bonmassar E. Drug-induced immunogenic changes of murine leukemia cells: Dissociation of onset of resistance and emergence of novel immunogenicity. J Natl Cancer Inst. 1983; 71: 1247-1251.
52. Giampietri A, Bonmassar A, Puccetti P, Circolo A, Goldin A, Bonmassar E. Drug-mediated increase of tumor immunogenicity in vivo for a new approach to experimental cancer immunotherapy. Cancer Res. 1981; 41: 681-687.
53. Wada S, Yoshimura K, Hipkiss EL, Harris TJ, Yen HR, Goldberg MV, Grosso JF, Getnet D, Demarzo AM, Netto GJ, Anders R, Pardoll DM, Drake CG. Cyclophosphamide augments antitumor immunity: Studies in an autochthonous prostate cancer model. Cancer Res. 2009; 69: 4309-4318.
54. Lee CH, Chiang YH, Chang SE, Chong CL, Cheng BM, Roffler SR. Tumor-localized ligation of CD3 and CD28 with systemic regulatory T-cell depletion induces potent innate and adaptive antitumor responses. Clin Cancer Res. 2009; 15: 2756-2766.
55. Zheng Y, Dou Y, Duan L, Cong C, Gao A, Lai Q, Sun Y. Using chemo-drugs or irradiation to break immune tolerance and facilitate immunotherapy in solid cancer. Cell Immunol. 2015; 294: 54-59.
56. Bracci L, Moschella F, Sestili P, La Sorsa V, Valentini M, Canini I, Baccarini S, Maccari S, Ramoni C, Belardelli F, Proietti E. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation and specific tumor infiltration. Clin Cancer Res. 2007; 13: 644-653.
57. Nisticò P, Capone I, Palermo B, Del Bello D, Ferraresi V, Moschella F, Aricò E, Valentini M, Bracci L, Cognetti F, Ciccarese M, Vercillo G, Roselli M, et al. Chemotherapy enhances vaccine-induced antitumor immunity in melanoma patients. Int J Cancer. 2009; 124: 130-139.
58. Palermo B, Del Bello D, Sottini A, Serana F, Ghidini C, Gualtieri N, Ferraresi V, Catricalà C, Belardelli F, Proietti E, Natali PG, Imberti L, Nisticò P. Dacarbazine treatment before peptide vaccination enlarges T-cell repertoire diversity of melan-a-specific, tumor-reactive CTL in melanoma patients. Cancer Res. 2010; 70: 7084-7092.
59. Franzese O, Palermo B, Di Donna C, Sperduti I, Ferraresi V, Stabile H, Gismondi A, Santoni A, Nisticò, P. Polyfunctional Melan-A-specific tumor-reactive CD8+ T cells elicited by dacarbazine treatment before peptide-vaccination depends on AKT activation sustained by ICOS. Oncoimmunology. 2016; 5: e1114203. doi: 10.1080/2162402X.2015.1114203. eCollection 2016.
60. Freireich EJ, Gehan EA, Rall DP, Schmidt LH, Skipper HE. Quantitative comparison of toxicity of anticancer agents in mouse, rat, hamster, dog, monkey and man. Cancer Chemother Rep. 1966; 50: 219-244.
61. Canti G , Ricci L, Marelli O, Franco P, Nicolin A. Adoptive immunity in mice challenged with L1210/DTIC clones. Cancer Immunol Immunother. 1987; 24: 64-67.
62. Romani L, Fioretti MC, Bianchi R, Nardelli B, Bonmassar E. Intracerebral adoptive immunotherapy of a murine lymphoma antigenically altered by drug treatment in vivo. J Natl Cancer Inst. 1982; 68: 817-822.
63. Suzuki S, Ishida T, Yoshikawa K, Ueda R. Current status of immunotherapy. Jpn J Clin Oncol. 2016; 46: 191-203.
64. Kreamer KM. Immune checkpoint blockade: A new paradigm in treating advanced cancer. J Adv Pract Oncol. 2014; 5: 418-431.
65. Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, Davis T, Henry-Spires R, MacRae S, Willman A, Padera R, Jaklitsch MT, Shankar S, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci U S A. 2003; 100: 4712-4717.
66. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013; 39: 1-10.
67. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012; 12: 252-264.
68. Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. 2003; 3: 939-951.
69. van Gisbergen KP, Klarenbeek PL, Kragten NA, Unger PP, Nieuwenhuis MB, Wensveen FM, ten Brinke A, Tak PP, Eldering E, Nolte MA, van Lier RA. The costimulatory molecule CD27 maintains clonally diverse CD8+ T cell responses of low antigen affinity to protect against viral variants. Immunity. 2011; 35: 97-108.
70. Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, Ogg GS, King A, Lechner F, Spina CA, Little S, Havlir DV, Richman DD, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002; 8: 379-385.
71. Azuma M, Phillips JH, Lanier LL. CD28- T lymphocytes. Antigenic and functional properties. J Immunol. 1993; 150: 1147-1159.
72. Hamann D, Baars PA, Rep MH, Hooibrink B, Kerkhof-Garde SR, Klein MR, van Lier RA. Phenotypic and functional separation of memory and effector human CD8+ T cells. J Exp Med. 1997; 186: 1407-1418.
73. Plunkett FJ, Franzese O, Finney HM, Fletcher JM, Belaramani LL, Salmon M, Dokal I, Webster D, Lawson AD, Akbar AN. The loss of telomerase activity in highly differentiated CD8+CD28-CD27- T cells is associated with decreased Akt (Ser473) phosphorylation. J Immunol. 2007; 178: 7710-7719.
74. Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, Flavell RA. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature. 2001; 409: 97-101.
75. Bukczynski J, Wen T, Wang C, Christie N, Routy JP, Boulassel MR, Kovacs CM, Macdonald KS, Ostrowski M, Sekaly RP, Bernard NF, Watts TH. Enhancement of HIV-specific CD8 T cell responses by dual costimulation with CD80 and CD137L. J Immunol. 2005; 175: 6378-6389.
76. Serghides L, Bukczynski J, Wen T, Wang C, Routy JP, Boulassel MR, Sekaly RP, Ostrowski M, Bernard NF, Watts TH. Evaluation of OX40 ligand as a costimulator of human antiviral memory CD8 T cell responses: comparison with B7.1 and 4-1BBL. J Immunol. 2005; 175: 6368-6377.
77. Waller EC, McKinney N, Hicks R, Carmichael AJ, Sissons JG, Wills MR. Differential costimulation through CD137 (4-1BB) restores proliferation of human virus-specific “effector memory” (CD28-CD45RAHI) CD8+ T cells. Blood. 2007; 110: 4360-4366.
78. McMahan RH, Golden-Mason L, Nishimura MI, McMahon BJ, Kemper M, Allen TM, Gretch DR, Rosen HR. Tim-3 expression on PD-1+ HCV-specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte-directed in vitro cytotoxicity. J Clin Invest. 2010; 120: 4546-4557.
79. Shin H, Wherry EJ. CD8 T cell dysfunction during chronic viral infection. Curr Opin Immunol. 2007; 19: 408-415.
80. Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015; 6: e1792. doi:10.1038/cddis.2015.162.
81. Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest. 2015; 125: 3338-3346.
82. Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010; 127: 759-767.
83. Allavena P, Mantovani A. Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol. 2012; 167: 195-205.
84. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009; 182: 4499-4506.
85. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-b induction of transcription factor Foxp3. J Exp Med. 2003; 198: 1875-1886.
86. Massague J. TGF beta in cancer. Cell. 2008; 134: 215-230.
87. Trapani JA. The dual adverse effects of TGF-beta secretion on tumor progression. Cancer Cell. 2005; 8: 349-350.
88. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014; 2014: 149185. doi:10.1155/2014/149185.
89. Burkholder B, Huang RY, Burgess R, Luo S, Jones VS, Zhang W, Lv ZQ, Gao CY, Wang BL, Zhang YM, Huang RP. Tumor-induced perturbations of cytokines and immune cell networks. Biochim Biophys Acta. 2014; 1845: 182-201.
90. Wherry EJ. T cell exhaustion. Nat Immunol. 2011; 12: 492-499.
91. Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003; 77: 4911-4927.
92. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013; 13: 227-242.
93. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009; 10: 29-37.
94. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010; 107: 4275-4280.
95. Nakamoto N, Cho H, Shaked A, Olthoff K, Valiga ME, Kaminski M, Gostick E, Price DA, Freeman GJ, Wherry EJ, Chang KM. Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLoS Pathog. 2009; 5: e1000313. doi:10.1371/journal.ppat.1000313.
96. McCoy KD, Le Gros G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol Cell Biol. 1999; 77: 1-10.
97. Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell. 1992; 71: 1065-1068.
98. Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994; 1: 793-801.
99. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, Wei B, Hogg N, Garside P, Rudd CE. Reversal of the TCR stop signal by CTLA-4. Science. 2006; 313: 1972-1975.
100. Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 2002; 16: 23-35.
101. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008; 322: 271-275.
102. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009; 206: 1717-1725.
103. Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, Benoist C. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 2007; 27:786-800.
104. Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007; 445:771-775.
105. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005; 25: 9543-9553.
106. Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signalling pathways and implications for cancer immunotherapy. Clin Cancer Res. 2013; 19: 4917-4924.
107. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, Hou TZ, Futter CE, Anderson G, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011; 332: 600-603.
108. Ishida Y, Agata, Y, Shibahara K & Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992; 11: 3887-3895
109. Keir M, Butte M, Freeman G, Sharpe A. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008; 26: 677-704.
110. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999; 11: 141-151.
111. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001; 291: 319-322.
112. Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004; 64: 1140-1145.
113. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001; 2: 261-268.
114. Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, Shalabi A, Shin T, Pardoll DM, Tsuchiya H. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001; 193: 839-846.
115. Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int. Immunol. 2007; 19: 813-824.
116. Butte MJ , Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007; 27: 111-122.
117. Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother. 2007; 56: 739-745.
118. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002; 99: 12293-12297.
119. Blank C, Gajewski T, Mackensen A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother. 2005; 54: 307-314.
120. Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, Okazaki T, Tokura Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. 2010; 116: 1757-1766.
121. Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, Higuchi T,Yagi H, Takakura K, Minato N, Honjo T, Fujii S. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A. 2007; 104: 3360-3365.
122. Mu CY, Huang JA, Chen Y, Chen C, Zhang XG. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med Oncol. 2011; 28: 682-688.
123. Thompson RH, Kuntz SM, Leibovich BC, Dong H, Lohse CM, Webster WS, Sengupta S, Frank I, Parker AS, Zincke H, Blute ML, Sebo TJ, Cheville JC, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res. 2006; 66: 3381-3385.
124. Kan G, Dong W. The expression of PD-L1 APE1 and P53 in hepatocellular carcinoma and its relationship to clinical pathology. Eur Rev Med Pharmacol Sci. 2015; 19: 3063-3071.
125. Paiva B, Azpilikueta A, Puig N, Ocio EM, Sharma R, Oyajobi BO, Labiano S, San-Segundo L, Rodriguez A, Aires-Mejia I, Rodriguez I, Escalante F, de Coca AG, et al. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia. 2015; 29: 2110-2113.
126. Inoue Y, Yoshimura K, Mori K, Kurabe N, Kahyo T, Mori H, Kawase A, Tanahashi M, Ogawa H, Inui N, Funai K, Shinmura K, Niwa H, et al. Clinical significance of PD L1 and PD L2 copy number gains in non-small-cell lung cancer. Oncotarget. 2016; 7: 32113-32128. doi: 10.18632/oncotarget.8528.
127. Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K, Yoshioka Y, Baba T, Konishi I, Mandai M. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br J Cancer. 2015; 112: 1501-1509.
128. Schuhmacher B, Rengstl B, Döring C, Bein J, Newrzela S, Brunnberg U, Kvasnicka HM, Vornanen M, Küppers R, Hansmann ML, Hartmann S. A strong host response and lack of MYC expression are characteristic for diffuse large B cell lymphoma transformed from nodular lymphocyte predominant Hodgkin lymphoma. Oncotarget. 2016; 7: 72197-72210. doi: 10.18632/oncotarget.12363.
129. Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: A paradigm shift. J Natl Cancer Inst. 2013; 105: 256-265.
130. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. 2013; 39: 74-88.
131. Ma W, Gilligan BM, Yuan J, Li T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J Hematol Oncol. 2016; 9: 47. doi: 10.1186/s13045-016-0277-y.
132. Passiglia F, Bronte G, Bazan V, Natoli C, Rizzo S, Galvano A, Listì A, Cicero G, Rolfo C, Santini D, Russo A. PD-L1 expression as predictive biomarker in patients with NSCLC: A pooled analysis. Oncotarget. 2016; 7: 19738-19747. doi: 10.18632/oncotarget.7582.
133. Zhang Y, Chung Y, Bishop C, Daugherty B, Chute H, Holst P, Kurahara C, Lott F, Sun N, Welcher AA, Dong C. Regulation of T cell activation and tolerance by PDL2. Proc Natl Acad Sci U S A. 2006; 103: 11695-11700.
134. Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y, Zang X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol Med. 2015; 21: 24-33.
135. Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. 2012; 5: ra46. doi:10.1126/scisignal.2002796. 5:ra46.
136. Lu B, Finn OJ. T-cell death and cancer immune tolerance. Cell Death Differ. 2008; 15: 70-79.
137. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010; 236: 219-242.
138. Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci U S A. 2008; 105: 9331-9336.
139. Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med. 2008; 205: 565-574.
140. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366: 2443-2454.
141. Zhang X, Zhu S, Li T, Liu YJ, Chen W, Chen J. Targeting immune checkpoints in malignant glioma. Oncotarget. 2017; 8:7157-7174. doi: 10.18632/oncotarget.12702.
142. MacFarlane AW 4th, Jillab M, Plimack ER, Hudes GR, Uzzo RG, Litwin S, Dulaimi E, Al-Saleem T, Campbell KS. PD-1 expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor resection. Cancer Immunol Res. 2014; 2: 320-331.
143. Zhang Y, Kang S, Shen J, He J, Jiang L, Wang W, Guo Z, Peng G, Chen G, He J, Liang W. Prognostic significance of programmed cell death 1 (PD-1) or PD-1 ligand 1(PD-L1) Expression in epithelial-originated cancer: A meta-analysis. Medicine. 2015; 94: e515. doi: 10.1097/MD.0000000000000515.
144. O’Byrne K. Stimulating immune responses to fight cancer: Basic biology and mechanisms. Asia Pac J Clin Oncol. 2015; 11 Suppl 1: 9-15.
145. Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013; 14: 1212-1218.
146. Ouyang Q, Wagner WM, Voehringer D, Wikby A, Klatt T, Walter S, Müller CA, Pircher H, Pawelec G. Age-associated accumulation of CMV-specific CD8+ T cells expressing the inhibitory killer cell lectin-like receptor G1 (KLRG1). Exp Gerontol. 2003; 38: 911-920.
147. Ito M, Maruyama T, Saito N, Koganei S, Yamamoto K, Matsumoto N. Killer cell lectin-like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J Exp Med. 2006; 203: 289-295.
148. Henson SM, Franzese O, Macaulay R, Libri V, Azevedo RI, Kiani-Alikhan S, Plunkett FJ, Masters JE, Jackson S, Griffiths SJ, Pircher HP, Soares MV, Akbar AN. KLRG1 signaling induces defective Akt (ser473) phosphorylation and proliferative dysfunction of highly differentiated CD8+ T cells. Blood. 2009; 113: 6619-6628.
149. Voehringer D, Koschella M, Pircher H. Lack of proliferative capacity of human effector and memory T cells expressing killer cell lectin-like receptor G1 (KLRG1). Blood. 2002; 100: 3698-3702.
150. Schwartzkopff S, Gründemann C, Schweier O, Rosshart S, Karjalainen KE, Becker KF, Pircher H. Tumor-associated E-cadherin mutations affect binding to the killer cell lectin-like receptor G1 in humans. J Immunol. 2007; 179: 1022-1029.
151. Lanna A, Coutavas E, Levati L, Seidel J, Rustin MH, Henson SM, Akbar AN, Franzese O. IFN-α inhibits telomerase in human CD8+ T cells by both hTERT downregulation and induction of p38 MAPK signaling. J Immunol. 2013; 191: 3744-3752.
152. Triebel F. LAG-3: A regulator of T-cell and DC responses and its use in therapeutic vaccination. Trends Immunol. 2003; 24: 619-622.
153. Shih K, Arkenau HT, Infante JR. Clinical impact of checkpoint inhibitors as novel cancer therapies. Drugs. 2014; 74: 1993-2013.
154. Li X, Hu W, Zheng X, Zhang C, Du P, Zheng Z, Yang Y, Wu J, Ji M, Jiang J, Wu C. Emerging immune checkpoints for cancer therapy. Acta Oncol. 2015; 54: 1706-1713.
155. Workman CJ, Vignali DA. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur J Immunol. 2003; 33: 970-979.
156. Workman CJ, Cauley LS, Kim IJ, Blackman MA, Woodland DL, Vignali DA. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol. 2004; 172: 5450-5455.
157. Hemon P, Jean-Louis F, Ramgolam K, Brignone C, Viguier M, Bachelez H, Triebel F, Charron D, Aoudjit F, Al-Daccak R, Michel L. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J Immunol. 2011; 186: 5173-5183.
158. Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur J Immunol. 1995; 25: 2718-2721.
159. Workman CJ, Dugger KJ, Vignali DA. Cutting edge: Molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J Immunol. 2002; 169: 5392-5395.
160. Macon-Lemaitre L, Triebel F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology. 2005; 115: 170-178.
161. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G. Role of LAG-3 in regulatory T cells. Immunity. 2004; 21: 503-513.
162. Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, Parmiani G, Belli F, Rivoltini L, Castelli C. LAG-3 expression defines a subset of CD4+CD25highFoxp3+ regulatory T cells that are expanded at tumor sites. J Immunol. 2010; 184: 6545-6551.
163. Grosso JF, Goldberg MV, Getnet D, Bruno TC, Yen HR, Pyle KJ. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J Immunol. 2009; 182: 6659-6669.
164. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012; 72: 917-927.
165. Anderson AC. Tim-3, a negative regulator of anti-tumor immunity. Curr Opin Immunol. 2012; 24: 213-216.
166. Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science. 2007; 318: 1141-1143.
167. Sakuishi K, Jayaraman P, Behar SM, Anderson AC, Kuchroo VK. Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol. 2011; 32: 345-349.
168. Sabatos CA, Chakravarti S, Cha E, Schubart A, Sanchez-Fueyo A, Zheng XX. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. 2003; 4: 1102-1110.
169. Yan J, Zhang Y, Zhang JP, Liang J, Li L, Zheng L. Tim-3 expression defines regulatory T cells in human tumors. PLOS One. 2013; 8: e58006.
170. Raimondi G, Shufesky WJ, Tokita D, Morelli AE, Thomson AW. Regulated compartmentalization of programmed cell death-1 discriminates CD4+CD25+ resting regulatory T cells from activated T cells. J Immunol. 2006; 176: 2808-2816.
171. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011; 117: 4501-4510.
172. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, Kirkwood JM, Chen TH, Maurer M, Korman AJ, Zarour HM. TIGIT and PD-1 impair tumor antigen-specific CD8⁺ T cells in melanoma patients. J Clin Invest. 2015; 125: 2046-2058.
173. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, Park S, Javinal V, Chiu H, Irving B, Eaton DL, Grogan JL. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell. 2014; 26: 923-937.
174. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, Smyth MJ, Kuchroo VK, Anderson AC. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. 2015; 125: 4053-4062.
175. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, Xia J, Tan TG, Sefik E, Yajnik V, Sharpe AH, Quintana FJ, Mathis D, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. 2014; 40: 569-581.
176. Chlewicki LK, Velikovsky CA, Balakrishnan V, Mariuzza RA, Kumar V. Molecular basis of the dual functions of 2B4 (CD244). J Immunol. 2008; 180: 8159-8167.
177. Vaidya SV, Mathew PA. Of mice and men: different functions of the murine and human 2B4 (CD244) receptor on NK cells. Immunol Lett. 2006; 105: 180-184.
178. Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, Murphy TL, Russell JH, Allison JP, Murphy KM. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003; 4: 670-679.
179. Derré L, Rivals JP, Jandus C, Pastor S, Rimoldi D, Romero P, Michielin O,Olive D, Speiser DE. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest. 2010; 120: 157-167.
180. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013; 34: 137-143.
181. Munn DH. Blocking IDO activity to enhance anti-tumor immunity. Front Biosci. 2012; 4: 734-745.
182. Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, Muller AJ. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. 2014; 63: 721-735.
183. Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007; 117: 1147-1154.
184. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013; 210: 1389-1402.
185. Dunn GP, Bruce AT, Ikeda H, Old Lj, Schreiber RD. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002; 3: 991-995.
186. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011; 331: 1565-1570.
187. Escors D. Tumour immunogenicity, antigen presentation, and immunological barriers in cancer immunotherapy. New J Sci. 2014; 2014: 734515. doi:10.1155/2014/734515.
188. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015; 348: 124-128.
189. Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, Garber JE, Chowdhury D, Wu CJ, D’Andrea AD, Matulonis UA, Konstantinopoulos PA. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016; 7: 13587-13598. doi: 10.18632/oncotarget.7277.
190. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015; 372: 2509-2520.
191. Szikriszt B, Póti A, Pipek O, Krzystanek M, Kanu N, Molnár J, Ribli D, Szeltner Z, Tusnády GE, Csabai I, Szallasi Z, Swanton C, Szüts D. A comprehensive survey of the mutagenic impact of common cancer cytotoxics. Genome Biology. 2016; 17: 99.
192. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, O’Brien C, De Carvalho DD. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015; 162: 961-973.
193. Cives M, Simone V, Rizzo FM, Silvestris F. NETs: Organ-related epigenetic derangements and potential clinical applications. Oncotarget. 2016; 7: 57414-57429. doi: 10.18632/oncotarget.10598.
194. Van Pel A, Boon T. Immunology protection against a nonimmunogenic mouse leukemia by an immunogenic variant obtained by mutagenesis (tumor immunology) Proc. Natl Acad. Sci. U S A. 1982; 79: 4718-4722.
195. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, Mulder GE, Toebes M, Vesely MD, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014; 515: 577-581.
196. Parmiani G, Maccalli C, Maio M. Integrating immune checkpoint blockade with anti-neo/mutated antigens reactivity to increase the clinical outcome of immunotherapy. Vaccines. 2015; 3: 420-428.
197. Trajanoski Z, Maccalli C, Mennonna D, Casorati G, Parmiani G, Dellabona P. Somatically mutated tumor antigens in the quest for a more efficacious patient-oriented immunotherapy of cancer. Cancer Immunol Immunother. 2015; 64: 99-104.
198. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, Watkins TB, Shafi S, Murugaesu N, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016; 351: 1463-1469
199. Puccetti P , Romani L, Fioretti MC. Chemical xenogenization of experimental tumors. Cancer and Metastasis Rev. 1987; 6: 93-111.
200. Romani L, Puccetti P, Grohmann U, Cenci E, Mage MG, Fioretti MC. Cell-mediated immunity to chemically xenogenized tumors--IV. Production of lymphokine activity by, and in response to, highly immunogenic cells. Int J Immunopharmacol. 1989; 11: 537-542.
201. Belladonna ML, Fioretti MC, Bianchi R, Puccetti P, Grohmann U. A retroviral peptide encoded by mutated env p15E gene is recognized by specific CD8+ T lymphocytes on drug-treated murine mastocytoma P815. Int J Immunopharmacol. 1996; 18: 563-576.
202. Bonmassar A, Frati L, Fioretti MC, Romani L, Giampietri A, Goldin A. Changes of the immunogenic properties of K36 lymphoma treated in vivo with 5(3,3-dimethyl-1-triazeno) imidazole-4-carboxamide (DTIC). Eur J Cancer. 1979; 15: 933-939.
203. Aptsiauri N, Garcia-Lora AM, Cabrera T. MHC class I antigens in malignant cells: immune escape and response to immunotherapy eBook: Briefs in cancer research, 2013. Springer, New York, NY.
204. Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, Stevens J, Lane WJ, Dellagatta JL, Steelman S, Sougnez C, Cibulskis K, Kiezun A, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015; 3: 1152-1158.
205. Haworth KB, Leddon JL, Chen CY, Edwin M, Horwitz EM , Mackall CL, Cripe TP. Going back to class I: MHC and immunotherapies for childhood cancer. Pediatr Blood Cancer. 2015; 62: 571-576.
206. Campoli M, Ferrone S. HLA antigen changes in malignant cells: Epigenetic mechanisms and biologic significance Oncogene. 2008; 27: 5869-5885.
207. Srivastava P, Paluch BE, Junko Matsuzaki J, James SR, Collamat-Lai G, Pietro Taverna P, Karpf AR, Griffiths EA. Immunomodulatory action of the DNA methyltransferase inhibitor SGI-110 in epithelial ovarian cancer cells and xenografts. Epigenetics. 2015; 10: 237-246.
208. Frikeche J, Simon T, Brissot E, Grégoire M, Gaugler B, Mohty M. Impact of valproic acid on dendritic cells function. Immunobiology. 2012; 217: 704-710.
209. Brown SD, Warren RL, Gibb EA, Spencer D, Martin SD, Spinelli JJ, Nelson BH , Holt RA. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014; 24: 743-750.
210. Nicolin A, Vadlamudi S, Goldin A. Antigenicity of L1210 leukemic sublines induced by drugs. Cancer Res. 1972; 32: 653-657.
211. Bianchi R, Citti L, Beghetti R, Romani L, D’Incalci M, Puccetti P, Fioretti MC.O6-methylguanine DNA methyltransferase activity and induction of novel immunogenicity in murine tumor cells treated with methylating agents. Cancer Chemother Pharmacol. 1992; 29: 277-282.
212. Graziani G, Faraoni I, Grohmann U, Bianchi R, Binaglia L, Margison GP, Watson AJ, Orlando L, Bonmassar E, D’Atri S. O6-alkylguanine-DNA alkyltransferase attenuates triazene-induced cytotoxicity and tumor cell immunogenicity in murine L1210 leukemia. Cancer Res. 1995; 55: 6231-6236.
213. Christmann M, Verbeek B, Roos WP, Kaina B. O6-Methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: enzyme activity, promoter methylation and immunohistochemistry. Biochim Biophys Acta. 2011; 1816: 179-190.
214. Kaina B, Margison GP, Christmann M. Targeting O6-methylguanine-DNA methyltransferase with specific inhibitors as a strategy in cancer therapy. Cell Mol Life Sci. 2010; 67: 3663-3681.
215. Dolan ME, Pegg AE, Dumenco LL, Moschel RC, Gerson SL. Comparison of the inactivation of mammalian and bacterial O6-alkylguanine-DNA alkyltransferases by O6-benzylguanine and O6-methylguanine. Carcinogenesis. 1991; 12: 2305-2309.
216. Piccioni D, D’Atri S, Papa G, Caravita T, Franchi A, Bonmassar E, Graziani G. Cisplatin increases sensitivity of human leukemic blasts to triazene compounds. J Chemother. 1995; 7: 224-229.
217. D’Atri S, Graziani G, Lacal PM, Nisticò V, Gilberti S, Faraoni I, Watson AJ, Bonmassar E, Margison GP. Attenuation of O6-methylguanine-DNA methyltransferase activity and mRNA levels by cisplatin and temozolomide in Jurkat cells. J Pharmacol Exp Ther. 2000; 294: 664-671.
218. Seiter K, Katragadda S, Ponce D, Rasul M, Ahmed N. Temozolomide and cisplatin in relapsed/refractory acute leukemia. J Hematol Oncol. 2009; 2: 21. doi: 10.1186/1756-8722-2-21.
219. Khan O, Middleton MR. The therapeutic potential of O6-alkylguanine DNA alkyltransferase inhibitors. Expert Opin Investig Drugs. 2007; 16:1573-1584.
220. Caporaso P, Turriziani M, Venditti A, Marchesi F, Buccisano F, Tirindelli MC, Alvino E, Garbin A, Tortorelli G, Toppo L, Bonmassar E, D’Atri S, Amadori S. Novel role of triazenes in haematological malignancies: Pilot study of Temozolomide, Lomeguatrib and IL-2 in the chemo-immunotherapy of acute leukaemia. DNA Repair 2007; 6: 1179-1186.
221. Ranson M, Middleton MR, Bridgewater J, Lee SM, Dawson M, Jowle D, Halbert G, Waller S, McGrath H, Gumbrell L, McElhinney RS, Donnelly D, McMurry TB, et al. Lomeguatrib, a potent inhibitor of O6-alkylguanine-DNA-alkyltransferase: phase I safety, pharmacodynamic, and pharmacokinetic trial and evaluation in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2006; 12:1577-1584.
222. Middleton MR, Lee SM, Arance A, Wood M, Thatcher N, Margison GP. O6-methylguanine formation, repair protein depletion and clinical outcome with a 4 hr schedule of temozolomide in the treatment of advanced melanoma: results of a phase II study. Int J Cancer. 2000; 88: 469-473.
223. Dixit S, Baker L, Walmsley V, Hingorani M. Temozolomide-related idiosyncratic and other uncommon toxicities: a systematic review. Anticancer Drugs. 2012; 23:1099-1106.
224. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med.2010; 363: 711-723.
225. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Davidson N, Richards J, Maio M, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011; 364: 2517-2526.
226. Specenier P. Ipilimumab in melanoma. Expert Rev Anticancer Ther. 2016; 16: 811-826.
227. Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, Hamid O, Robert C, Ascierto PA, Richards JM, Lebbé C, Ferraresi V, Smylie M, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015; 16: 522-530.
228. Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, Garbe C, Gogas H, Schachter J, Linette G, Lorigan P, Kendra KL, Maio M, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013; 31: 616-622.
229. Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr, Lao CD, Linette GP, Thomas L, Lorigan P, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015; 16: 375-384.
230. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, Savage KJ, Hernberg MM, Lebbé C, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015; 372: 320-330.
231. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015; 373: 23-34.
232. Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015; 373: 123-135.
233. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, Barlesi F, Kohlhäufl M, Arrieta O, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015; 373: 1627-1639.
234. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, Castellano D, Choueiri TK, Gurney H, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015; 373: 1803-1813.
235. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, Rodig SJ, Chapuy B, Ligon AH, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015; 372: 311-319.
236. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015; 372: 2521-2532.
237. Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ, Majem M, Fidler MJ, de Castro G Jr, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016; 387: 1540-1550.
238. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe S, O’Brien M, Rao S, Hotta K, et al. Pembrolizumab versus chemotherapy for PD-L1-positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016 Oct 8. [Epub ahead of print]. doi:10.1056/NEJMoa1606774.