
TNF-induced apoptosis is tightly regulated by the NF-κB pathway. Under physiologic conditions, TNFα stimulation induces NF-κB activation and cell survival, due to the regulation of anti-apoptotic genes, including c-FLIP, a caspase-8 inhibitor, whose expression is sufficient to protect cells against TNF-induced apoptosis. TNF triggers cell death only in circumstances where the NF-κB pathway is defective. Rushworth and collaborators have recently demonstrated, however, that the heme oxygenase-1 (HO-1), also known as Heat shock protein 32 (Hsp32) [1], like c-FLIP, can afford protection against TNF-induced cell death in AML cells, despite NF-κB inactivation [2]. They now provide evidence that TNF mediated HO-1 up-regulation, is negatively regulated by c-FLIP, revealing a novel negative regulatory feedback loop controlling apoptosis induced by TNRI (Figure 1).
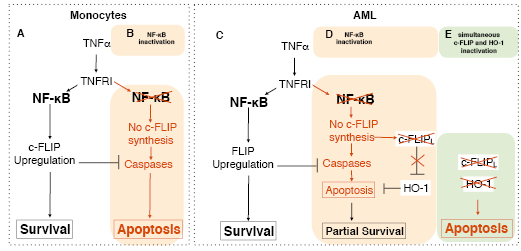
Figure 1: Contribution of HO-1 and c-FLIPL to the regulation of TNF signalling in monocytes and acute myeloid leukemia cells (AML). (A) In monocytes, engagement of TNFR1 by TNFα induces activation of NF-κB, leading to up-regulation of FLIP and inhibition of cell death, however inactivation of NF-κB (B) prevents FLIP neosynthesis, allowing caspase activation and apoptosis. (C) AML cells are resistant to TNFα-induced apoptosis, even upon inactivation of NF- κB (D), due to the up-regulation of HO-1. (E) Simultaneous inactivation of c-FLIP and HO-1 enhances TNF-induced cell death.
In contrast to Fas or TRAIL receptor-mediated cell death, apoptosis induced by TNFRI is a two-step process that requires the formation of two sequential signalling complexes [3]. The plasma membrane-bound complex I, including TNFR1, TRADD, RIP1 and TRAF2, is dedicated to the activation of the survival pathway NF-κB. FADD and caspase-8 are recruited in the “cytosolic” complex, also coined complex II, which is devoid of TNFRI, triggering caspase-8 activation and apoptosis [3]. In the vast majority of cells, however, activation of NF-κB induces protection against TNF-induced cell death [4]. Several anti-apoptotic genes are regulated by NF-κB [5], but so far only c-FLIP has been demonstrated to afford full protection when expressed alone [6,7]. Activation of complex II and thus triggering of the apoptotic program is generally thought to occur in NF-κB defective cells due to the lack of c-FLIP supply [8].
HO-1 is a stress-related anti-apoptotic molecule that has been implicated in enhanced survival of cancer cells and in drug-resistance [1]. Overexpression of HO-1 protects cells from H2O2-, Fas- or TNF-induced apoptosis [9-11]. Unlike HO-2, the second evolutionary conserved heme oxygenase isoenzyme, HO-1 is not expressed constitutively. HO-1 is generally induced under oxidative stress enabling enhanced free heme catabolism and inhibition of programmed cell death[1]. HO-1 mediated cytoprotection has been assigned to the heme catabolism sub-product Fe2+, which triggers reactive oxygen species (ROS) production and NF-κB activation [12]. Induced expression of HO-1 by IL-1 and TNFα was suggested to involve protein kinase c, calcium and phospholipase A2 [13]. Activation of the PkB/Akt pathway and induction of Nrf2 were shown to induce HO-1 up-regulation upon H2O2 stimulation[9]. More recently it was shown that TNF-mediated ROS production, in NF-κB inactivated AML cells, induced the activation of the transcription factor Nrf2 leading to HO-1 up-regulation [2]. The cytoprotective activity of HO-1 in endothelial cells was demonstrated to require NF-κB activation by TNFα [14]. Interestingly, HO-1-mediated inhibition of TNFRI-induced apoptosis, in NF-κB defective cells, can be restored by the ectopic expression of some NF-κB regulated genes such as c-IAP2, A1 or A20 [14]. Furthermore, HO-1-mediated protection against TNF-induced cell death is not restricted to tumour cells, as endothelial cells or human fibroblasts induced to express HO-1 fail to undergo apoptosis [14,15].
Remarkably, and in contrast to most studies demonstrating that inhibition of the NF-κB pathway restores TNF-induced cell death in normal and cancer cells, Rushworth et al. demonstrate in this issue that NF-κB inhibition only affords partial restoration of apoptosis in AML cells, due to the up-regulation of HO-1. Accordingly, inactivation of c-FLIPL expression was sufficient to trigger the accumulation of HO-1 in the absence of TNF, though apoptosis following TNFα stimulation was only partially restored. Accordingly, inactivation of c-FLIPL expression in these cells, albeit partially restoring TNFα-induced apoptosis, in the absence of TNF, triggered the accumulation of HO-1. However, simultaneous inactivation of c-FLIPL and HO-1 significantly enhanced AML cell sensitivity to TNFα. Rushworth et al. make the critical observation that induction of HO-1 expression is negatively regulated at the steady state by c-FLIPL, but not the short forms of c-FLIP, providing a plausible explanation for the resistance of AML cells to TNF-induced apoptosis, despite inactivation of the NF-κB pathway.
These results demonstrate that HO-1 exerts cytoprotection in AML cells, irrespective of NF-κB activation, and suggest in addition that HO-1 and c-FLIPL may negatively regulate TNF-induced cell death in a non-redundant, but exclusive manner. Of particular interest, c-FLIPL down-regulation was unable to promote HO-1 expression in monocytes. Thus the markedly increased expression of c-FLIPL and the constitutive activation of NF-κB in erythroleukemia cells [16] would support the proposal that negative regulation of HO-1 expression by c-FLIPL, at the basal level, might require sustained NF-κB activation. In line with this hypothesis, it has been demonstrated in the past that over-expression of c-FLIP, or at least its amino acid terminal portion, could induce NF-κB activation [17-20]. It is not clear, however, whether NF-κB activation alone is sufficient to repress HO-1. ROS production, through the activation of Nrf2, may also induce the restoration of HO-1 expression in cells in which c-FLIPL has been inactivated, as c-FLIP down-regulation was shown to induce ROS production in some tumour cells [21], while its over-expression produces the opposite effect [22].
While it is clear that the molecular mechanisms underlying c-FLIPL-mediated HO-1 repression at the basal level needs to be explored more precisely, the possibility that HO-1 itself may regulate c-FLIP expression, through its ability to inhibit NF-κB activation, or to induce ROS remains an open question. In line with this hypothesis, it has recently been demonstrated that HO-1 was able to impair NF-κB nuclear translocation in cardiomyocytes [23] and that ROS production can trigger the degradation of c-FLIP in an ubiquitylation-dependent manner [24]. Mutual regulation of these cellular “safeguards” would thus certainly be beneficial for tumour cells to maintain a high level of protection against TNF-induced killing. Altogether these findings uncover a novel cell-decision regulatory mechanism controlling cell death signalling induced by TNFRI, which may extend to other death-inducing ligands of the TNF family.
Acknowledgments
This work is supported by grants of the Conseil Regional de Bourgogne, the INCa (Institut National du Cancer), Cancéropôle Grand-Est, ANR (Agence Nationale de la Recherche, ANR-06-JCJC-0103 and 07-PCV-0031), and the European Community (ApopTrain Marie Curie RTN). Sarah Shirley is supported by a fellowship from the INCa (Polynom174).
References
1. Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol 2010; 50:323-354.
2. Rushworth SA, MacEwan DJ. HO-1 underlies resistance of AML cells to TNF-induced apoptosis. Blood 2008; 111:3793-3801.
3. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114:181-190.
4. Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science 1996; 274:782-784.
5. Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS, Jr. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998; 281:1680-1683.
6. Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol 2001; 21:5299-5305.
7. Kreuz S, Siegmund D, Scheurich P, Wajant H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol 2001; 21:3964-3973.
8. Micheau O. Cellular FLICE-inhibitory protein: an attractive therapeutic target? Expert Opin Ther Targets 2003; 7:559-573.
9. Brunt KR, Fenrich KK, Kiani G, Tse MY, Pang SC, et al. Protection of human vascular smooth muscle cells from H2O2-induced apoptosis through functional codependence between HO-1 and AKT. Arterioscler Thromb Vasc Biol 2006; 26:2027-2034.
10. Kushida T, LiVolti G, Goodman AI, Abraham NG. TNF-alpha-mediated cell death is attenuated by retrovirus delivery of human heme oxygenase-1 gene into human microvessel endothelial cells. Transplant Proc 2002; 34:2973-2978.
11. Pileggi A, Cattan P, Berney T, Molano RD, Vizzardelli C, et al. HO-1 upregulation protects the pancreatic cell line betaTC3 from cytokines and Fas-induced apoptosis. Transplant Proc 2001; 33:266-267.
12. Choi BM, Pae HO, Jeong YR, Oh GS, Jun CD, et al. Overexpression of heme oxygenase (HO)-1 renders Jurkat T cells resistant to fas-mediated apoptosis: involvement of iron released by HO-1. Free Radic Biol Med 2004; 36:858-871.
13. Terry CM, Clikeman JA, Hoidal JR, Callahan KS. TNF-alpha and IL-1alpha induce heme oxygenase-1 via protein kinase C, Ca2+, and phospholipase A2 in endothelial cells. Am J Physiol 1999; 276:H1493-1501.
14. Brouard S, Berberat PO, Tobiasch E, Seldon MP, Bach FH, et al. Heme oxygenase-1-derived carbon monoxide requires the activation of transcription factor NF-kappa B to protect endothelial cells from tumor necrosis factor-alpha-mediated apoptosis. J Biol Chem 2002; 277:17950-17961.
15. Petrache I, Otterbein LE, Alam J, Wiegand GW, Choi AM. Heme oxygenase-1 inhibits TNF-alpha-induced apoptosis in cultured fibroblasts. Am J Physiol Lung Cell Mol Physiol 2000; 278:L312-319.
16. Rae C, Langa S, Tucker SJ, MacEwan DJ. Elevated NF-kappaB responses and FLIP levels in leukemic but not normal lymphocytes: reduction by salicylate allows TNF-induced apoptosis. Proc Natl Acad Sci U S A 2007; 104:12790-12795.
17. Kataoka T, Tschopp J. N-terminal fragment of c-FLIP(L) processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-kappaB signaling pathway. Mol Cell Biol 2004; 24:2627-2636.
18. Kataoka T, Budd RC, Holler N, Thome M, Martinon F, et al. The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr Biol 2000; 10:640-648.
19. Golks A, Brenner D, Krammer PH, Lavrik IN. The c-FLIP-NH2 terminus (p22-FLIP) induces NF-kappaB activation. J Exp Med 2006; 203:1295-1305.
20. Bannerman DD, Eiting KT, Winn RK, Harlan JM. FLICE-like inhibitory protein (FLIP) protects against apoptosis and suppresses NF-kappaB activation induced by bacterial lipopolysaccharide. Am J Pathol 2004; 165:1423-1431.
21. Nakajima A, Kojima Y, Nakayama M, Yagita H, Okumura K, et al. Downregulation of c-FLIP promotes caspase-dependent JNK activation and reactive oxygen species accumulation in tumor cells. Oncogene 2008; 27:76-84.
22. Shim E, Lee YS, Kim HY, Jeoung D. Down-regulation of c-FLIP increases reactive oxygen species, induces phosphorylation of serine/threonine kinase Akt, and impairs motility of cancer cells. Biotechnol Lett 2007; 29:141-147.
23. Yeh CH, Chen TP, Wang YC, Lin YM, Lin PJ. HO-1 activation can attenuate cardiomyocytic apoptosis via inhibition of NF-kappaB and AP-1 translocation following cardiac global ischemia and reperfusion. J Surg Res 2009; 155:147-156.
24. Wang L, Azad N, Kongkaneramit L, Chen F, Lu Y, et al. The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J Immunol 2008; 180:3072-3080.