
INTRODUCTION
Despite the growing appreciation of G protein-coupled receptor (GPCR) signal involvement in cancer pathogenesis, very little is known about the role GPCRs play in tumor etiology. GPCRs are seven transmembrane proteins responsible for transducing signals from a diverse range of ligands that affect numerous physiological processes [1–4]. GPCRs regulate many aspects of tumorigenesis, including proliferation, invasion, and survival at the secondary site, as well as several cancer-associated signaling pathways [5]. Emerging large-scale genomic analyses have recently provided further evidence of frequent GPCR alterations in human tumors, including mutations, copy number, altered expression and promoter methylation [6–8]. Determining the contribution of such alterations to cancer initiation and progression remains a significant challenge, that is critical both for discovery of driver oncogenes and for the development of targeted therapeutics.
Frizzled (Fz) receptors are a subgroup of GPCRs that play a pivotal role in development, tissue homeostasis, and cancer. Activation of the evolutionarily conserved Wnt/β-catenin signaling pathway, also called the canonical Wnt pathway, involves stabilization of β-catenin through binding of Wnt ligands to Fz cell surface receptors and low-density lipoprotein-related protein 5/6 (LRP5/6) coreceptors. In the absence of Wnt, the key effector of this pathway, β-catenin, is continuously degraded by the “degradation complex”. This complex is comprised of Axin, adenomatis polyposis coli (APC), glycogen synthase kinase3β (GSK3β), casein kinase1alpha (CK1α), and the E3 ubiquitin ligase subunit β-TrCP1. Axin provides a scaffolding site for GSK3β to phosphorylate the N-terminus portion of β-catenin (after priming by CK1α), thus generating a phosphorylated form of β-catenin, recognized by the ubiquitin ligase adaptor β-TrCP [9, 10]. Wnt stimulation dismantles the degradation complex, thereby leading to the accumulation of unphosphorylated β-catenin. Once β-catenin is stabilized, it translocates to the cell nucleus. There it alters the activity of members of the lymphoid enhancer factor (Lef)/T-cell factor (Tcf), Lef/Tcf family of HMG-box transcription factors acting as transcriptional switches, recruiting various chromatin modifiers and remodelers to Lef/Tcf target genes inducing expression of an array of genes downstream [10, 11]. A wide range of cancers exhibit hyperactive stabilized β-catenin, either due to oncogenic mutations in its N-terminal phosphorylation site or through mutational inactivation of APC or Axin, its negative regulators [11, 12]. Activated β-catenin can be oncogenic, driving the onset of a wide spectrum of carcinomas [9, 10].
In addition to its pathological role in cell nuclei as a central transcriptional coactivator for Wnt-responsive genes [10, 11], β-catenin is also a membrane-associated protein that constitutes a key component of adherens junctions. Under normal conditions, it has a robust engagement of neighboring adherens junctions and then interacts with the cytoplasmic tail of cadherins [13].
Mammalian protease-activated-receptors PARs, comprise a four-member family subgroup of GPCRs. They are uniquely activated by cleavage of their N-terminal extracellular domain and exposure of internal ligands. PAR1 and PAR2 play central roles in tumor biology [14–19]. Increasing evidence supports the notion that PAR1 and PAR2 exist in close proximity and act as one functional unit, forming PAR1-PAR2 heterodimer [20–23]. We have recently demonstrated that PAR1-induced breast tumor development and the corresponding signaling events are markedly inhibited when PAR2 expression is knocked-down or when it lacks its C-tail portion [24]. This establishes a dominant role for PAR2, which seems essential also for PAR1 function, in a manner that is yet unknown. Previously, we have shown a novel link between human protease-activated receptor-1 (hPar-1) and β-catenin stabilization using a transgenic mouse model whereby hPar1 is overexpressed in the mammary glands and several cancer cell-lines [25–27], demonstrating that PAR1 induces β-catenin stabilization via early formation of the PAR1-Gα13-DVL-DIX axis. Whereas the role of PAR1 in β-catenin stabilization has been addressed, the involvement of PAR2 in this process has not been examined.
In the present study, we demonstrate that PAR2 activation leads to potent β-catenin stabilization accompanied by increased Lef/Tcf transcription activity. The molecular path that links PAR2 with β-catenin stabilization was assessed, identifying LRP6, a known Fz partner, as a novel co-receptor of PAR2 as shown by co-immunoprecipitation, bioluminescence resonance energy transfer (BRET), and confocal microscopy analysis, and supported by bioinformatics using the ZDOCK protein-protein server. Following SLIGKV activation, PAR2 forms a complex with LRP6, which then recruits Axin from the “destruction-complex” pool, leading to β-catenin stabilization and nuclear transcriptional activity. We hereby identify the components that link PAR2 with β-catenin stabilization and characterize PAR induced DVL nuclear localization. Taken together, our data show a novel PAR2-LRP6-Axin axis as key components in PAR2-induced β-catenin stabilization in cancer. This first described axis enhances our understanding in cancer biology, and opens new avenues for future development of an anti-cancer medicament platform.
RESULTS
PAR2 induces β-catenin stabilization and Lef/Tcf transcriptional activity: identification of the minimal PAR2 C-tail region
While previously we have demonstrated a novel link between PAR1 and β-catenin stabilization [25–27], the role of PAR2 in the dynamics of β-catenin stability is unknown. We chose to focus on RKO cells, a colorectal transformed cell line that exhibits intact β-catenin signaling machinery, transformed on a background of microsatellite instability (which leads to hypermethylation of the hMLH1 promoter). RKO cells are of wt APC as well as intact β-catenin and p53 machinery system (frequently used for studies of Wnt/β-catenin pathway). Pretreatment (i.e., 2h) with 40mM LiCl (a known GSK3β inhibitor) followed by activation of PAR2 induced an increase in β-catenin levels (Figure 1A). Moreover, in HEK293T cells transiently transfected with hPar2-wt and flg-β-catenin, the SLIGKV activation of PAR2 leads to nuclear localization and accumulation of β-catenin (Figure 1B). SLIGKV PAR2 activation induces also β-catenin transcriptional activity as shown by the elevated Lef/Tcf luciferase activity (Figure 1D). Therefore, in the presence of a full length hPar2 construct, a significantly marked increase in the level of β-catenin and its transcriptional activity was observed (Figure 1D).
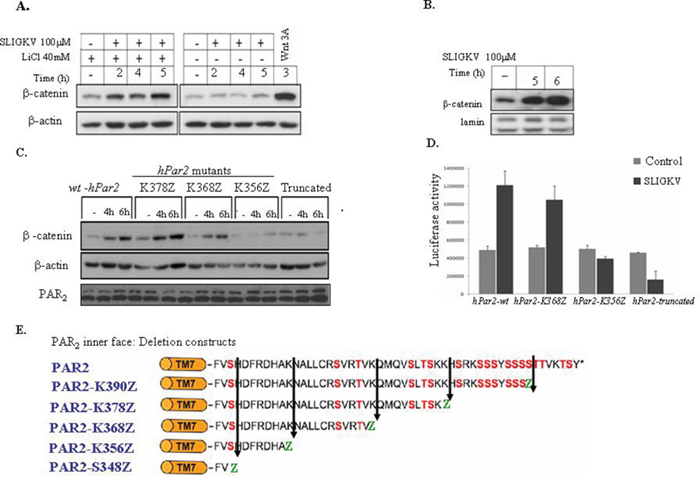
Figure 1: Activation of PAR2 induces β-catenin stabilization and Lef/Tcf transcriptional activity. (A) PAR2 activation induces β-catenin stabilization in RKO cells. RKO cells were pretreated with LiCl (40 mM, 2h) followed by 2, 4, and 5 hours of PAR2 SLIGKV activation (100 μM). Whereas low levels of β-catenin were observed prior to PAR2 activation, a marked increase in β-catenin levels was seen following activation. RKO cells that were not pretreated with LiCl showed basal levels of β-catenin regardless of PAR2 activation. RKO cells pre-treated with Wnt3A conditioned medium for 3 hours were used as a positive control. (B) PAR2 activation induces nuclear β-catenin in HEK-293T cells. HEK-293T cells were transiently transfected with both flg-β-catenin and hPar2-wt plasmids. After PAR2 activation, the nuclear fraction was extracted and immunoblots were analyzed using anti-flg (for flg-β-catenin) and anti-lamin (used as a nuclearprotein loading control) antibodies. The amount of nuclear β-catenin was markedly increased in response to PAR2 activation. (C) PAR2-induced β-catenin stabilization depends on an intact K356 - K368 region. HEK-293T cells transiently transfected with hPar2 deletion constructs lacking the K356-K368 (i.e., hPar2-K356Z or hPar2-truncated) displayed a markedly reduced β-catenin stabilization level. In contrast, cells transfected with hPar2 constructs that possess the above-mentioned region (e.g., hPar2-K368Z/K378Z or wt) displayed increased β-catenin accumulation. (D) An intact K356 - K368 region is essential for PAR2-induced Lef/Tcf transcriptional activity. TOPflash luciferase transcription activity was analyzed in RKO cells following PAR2 activation in the presence of hPar2 wt or deletion constructs. While in hPar2-wt and hPar2-K368Z transfected cells, PAR2 activation elicits markedly elevated luciferase Lef/Tcf activity, in hPar2-K356Z and hPar2-truncated cells PAR2 activation failed to increase luciferase Lef/Tcf activity above the basal level. The results were evaluated using GraphPad InStat software and found to be statistically significant (p<0.01). (E) Scheme of PAR2 C-tail. Schematic representation of PAR2 C-tail and its various deleted constructs.
We next set out to determine the minimal PAR2 C-tail region that is required for β-catenin stabilization. For this purpose, several randomly generated deleted hPar2 C-tail plasmids (e.g. hPar2 K390Z, hPar2 K378Z, hPar2 K368Z, hPar2 K356Z and truncated hPar2 that was devoid the entire cytoplasmic tail) were prepared and analyzed for their ability to elicit β-catenin stabilization. Deletion constructs were obtained through substituting amino acids at various sites along the cytoplasmic tail with a premature stop codon. When HEK293T cells were transiently transfected with each of the hPar2 constructs (including wt), as well as with a flg-β-catenin plasmid, the following outcome was obtained. After PAR2 SLIGKV activation, cell lysates were prepared and Western blot detection was carried out using anti-flg antibodies. In contrast to PAR2-wt, the truncated plasmid as also the shortest C-tail, hPar2 K356Z, both were unable to induce β-catenin stabilization (Figure 1C), whereas the other various deleted hPar2 constructs effectively elicited marked levels of β-catenin. Since deletion of the K356-K368 region potently abrogates PAR2-induced β-catenin stabilization, we conclude that this region, e.g. the sequence corresponding to K356-K368 NH2-NALLCRSVRTV-COOH, is critical for the function of the PAR2-enhanced β-catenin signaling pathway. Residues AKNALLCRSVTV were previously proposed as the Ca++signaling site (38). We did not address here whether Ca++ signaling is a prerequisite for PAR2 induced β-catenin stabilization, and presently it is an open question. However, when we applied the established Ca++ blocker; BAPTA to our PAR2 induced β-catenin stabilization system, a marked inhibition was seen (Supplementary Figure 1). When we compared the potency of SLIGKV versus trypsin in PAR2 induced β-catenin stabilization, the following outcome was observed. While a small increase in β-catenin level is seen following SLIGKV application as soon as 30 min, a marked increase was observed by 4 and 5 hr activation (Supplementary Figure 2). Trypsin showed an increase only after 4 and 5 hr activation (Supplementary Figure 2).
We next assessed PAR2-induced β-catenin transcription activities, and determined the minimal PAR2 C-tail region that is required for this activity. Co-transfection of RKO cells with the TOPflash-luciferase construct (TOP, TCF optimal promoter containing three copies of LEF-1 binding sites) and with hPar2-wt, hPar2-K368Z, hPar2-K356Z or hPar2-truncated constructs, was carried out. As shown in Figure 1D, elicited Lef/Tcf promoter activity was seen following PAR2 activation when either hPar2-wt or hPar2-K368Z were present. In contrast, in the presence of either hPar2-truncated or the shortest C-tail, hPar2-K356Z plasmids, low levels of luciferase promoter activity were observed, similar to control, nonactivated cells. We thus confirm that the sequence NH2-NALLCRSVRTV-COOH of PAR2K356-K368 C-tail is required for β-catenin transcriptional activity, in agreement with the above described β-catenin stabilization data. Hence, the assigned PAR2 C-tail; K356-K368 was identified as necessary for PAR2 enhanced β-catenin stabilization and transcriptional activity.
LRP6 acts as a coreceptor with PAR2
Both LDL-receptor-related proteins 5 and 6; LRP5 and LRP6 are key components for the activation of β-catenin signaling in the canonical Wnt signaling pathway. It should be noted that while LRP5 and LRP6 exhibit high homology, they may not be equivalent in their ability to transduce Wnt signals. LRP6 independently induces axis duplication in Xenopus embryos, whereas LRP5 does not [28]. LRP6 knockout in mice is embryonically lethal, whereas LRP5-deficient mice are viable and fertile [29]. We then analyzed the possible involvement of LRP6 in PAR2-induced β-catenin signaling. We first asked whether LRP6 is recruited to PAR2 following SLIGKV activation. Application of anti-PAR2 antibodies to HEK293T cell lysates that were transfected with lrp6 and hPar2 plasmids showed unequivocally the presence of lrp6 within the same immune-complex of PAR2 (Figure 2Aii). When immunoprecipitation was carried out using YFP-hPar2 plasmid cotransfected with lrp6 and detected by application of anti GFP antibodies, a similar result was obtained (Figure 2Aiii). LRP6 is distinctly seen within PAR2 immunocomplex following SLIGKV activation mainly after 10 min. When the phosphorylation status of the recruited LRP6 was assessed, it showed the presence of phosphorylated LRP6 (Figure 2Aii&2Bii). Hence, LRP6 is a coreceptor of PAR2 present functionally active within one immunocomplex following PAR2 activation. Next, the possibility that Axin is recruited to PAR2 immunocomplex following SLIGKV PAR2 activation was addressed. HEK293T cells were transiently transfected with hPar2, lrp6 and flg-Axin plasmids followed by co-immunoprecipitation analyses. Indeed, a significant level of Axin was found within PAR2 immunocomplexes following SLIGKV activation (Figure 2Bi&ii). Hence, Axin is actively engaged to the cell membrane following PAR2 activation (bound to pLRP6), forming PAR2-LRP6-Axin axis. It appears that the association between PAR2 and LRP6 takes place using the extracellular portion of PAR2, since also in the presence of a truncated form PAR2 lacking its entire cytoplasmic tail, the PAR2-LRP6 complex is formed (Figure 2Ci&ii).
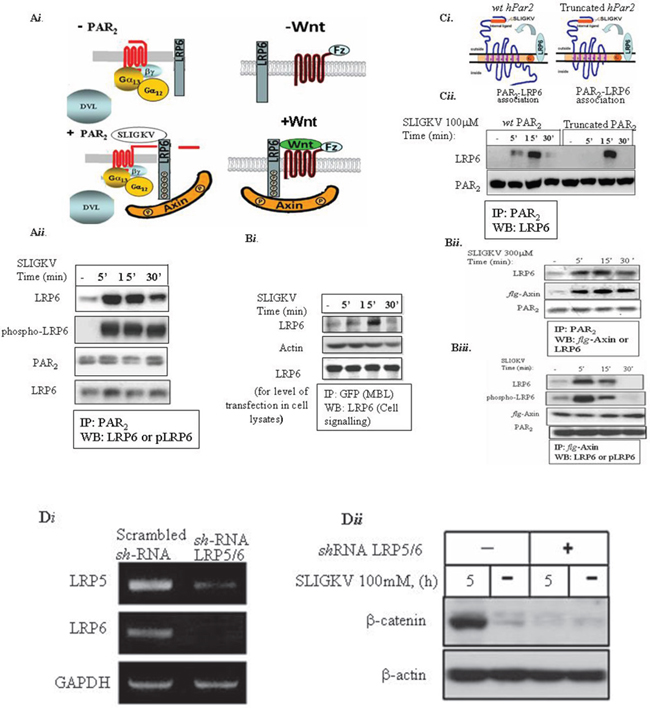
Figure 2: LRP6 is a coreceptor of PAR2. (Ai) A scheme of PAR2 and co-receptor LRP6 (Aii) HEK293T cells were transiently transfected with HA-Lrp6, wt hPar2 and flg-Axin. Following SLIGKV PAR2 activation, co-immunoprecipitation assay was carried out using anti-PAR2 abs, and LRP6 was detected following application of anti-HA (LRP6) antibodies. As early as 5 min following PAR2 activation and for up to 30 min, PAR2 and LRP6 were shown to be within the same immune complex. LRP6 phosphorylation was detected by anti-phospho-LRP6 abs. (Aiii) HEK293T cells were transiently transfected with HA-Lrp6 and YFP- hPar2. Following SLIGKV PAR2 activation, co-immunoprecipitation assay was carried out using anti-GFP abs (MBL), and LRP6 was detected following application of anti-HA (LRP6) antibodies. (Bi) Axin is present within the immune-complex of PAR2 and LRP6. Immunoprecipitation using anti-PAR2 abs shows distinct co-association with Axin as detected by anti-flg antibody application. (Bii) Axin is present in a complex with LRP6 following SLIGKV PAR2 activation. Immunoprecipitation analysis of HEK293T cells following PAR2 SLIGKV activation was carried out using anti-flg-Axin. Detection by either anti-phospho-LRP6 abs or anti-LRP6 showed a profound presence within the same immune complex of LRP6 and Axin. (Ci) A truncated form of PAR2 associates with LRP6. A scheme representing PAR2 and the coreceptor LRP6 association. (Cii) HEK293T cells were transiently transfected with Lrp6, wt hPar2, and truncated hPar2. Following SLIGKV PAR2 activation, co-immunoprecipitation was carried out using anti-PAR2 abs and LRP6 was detected by application of anti-LRP6. For 5 min following PAR2 activation and for up to 30 min, PAR2 and LRP6 were found within the same immune complex. This binding association also occurred when a truncated hPar2 was used. (Di) shRNA-LRP5/6 inhibits effectively LRP5&6 levels. Total cell RNA isolated from HEK-293T cells that were infected either with shRNA-LRP5/6 or nonspecific scrambled shRNA was isolated and RT-PCR analysis was performed using primers for LRP5, LRP6, and GAPDH (used as the internal control). shRNA-LRP5/6 effectively inhibited LRP5/6 levels. (Dii) shRNA-LRP5/6 inhibits PAR2-induced β-catenin stabilization. HEK-293T cells were transfected with flg-β-catenin and hPar2-wt following infection with shRNA-LRP5/6 viral vectors. Next, the cells were activated with SLIGKV (100μM, 5hr). Lysates were prepared and immunoblots were detected using both anti-flg (for flg-β-catenin) and anti-β-actin as a control for protein loading. The shRNA constructs significantly inhibited PAR2-induced β-catenin stabilization.
We next examined whether silencing LRP5/6 would affect PAR2-induced β-catenin stabilization. This was carried out by introducing a mixture of shRNA pool of silenced lrp5 as also silenced lrp6 (e.g., shLRP5/6) that effectively silenced both lrp5 and 6 (see Figure 2Di). Knocked-down lrp5/6 markedly attenuated PAR2-induced β-catenin stabilization (Figure 2Dii), indicating that LRP6 is a critical mediator of PAR2-induced β-catenin stabilization.
We have further confirmed the co-association between PAR2 and LRP6 using BRET2. BRET has been successfully applied to study receptor-receptor interactions, serving as a powerful tool to assess the nature of these interactions at the molecular level. BRET2 assays use the combination of luciferase from Renilla reniformis (Rluc) and GFP variants from Aequorea victoria, which do not interact without a specific trigger, thus limiting non-specific and random associations. A resonance energy transfer and signal emission from the acceptor partner take place if the distance between the two interacting components is less than approximately10 nm [30, 31]. Not only the distance between these components is a limiting factor, but also the extent of the overlap between the emission spectrum of the donor and excitation of the acceptor [30]. When we transfected HU cells with Rluc-hPar2 and lrp6-pEGFP-C1 constructs following SLIGKV PAR2 activation, the BRET ratio revealed that in comparison with the inactive status of PAR2, activated PAR2 is found in a close proximity with the fluorescent-tagged LRP6. This receptor-receptor interaction further induces a measurable energy transfer in the form of a BRET signal. As shown in Figure 3, in a PAR2-LRP6 co-transfected cells, SLIGKV peptide activationfor 5 min induces a highly significant (p=0.002) maximal increment in BRET ratio. This signal declines in a time dependent manner, showing the lowest difference at 30 min of SLIGKV activation (Figure 3).
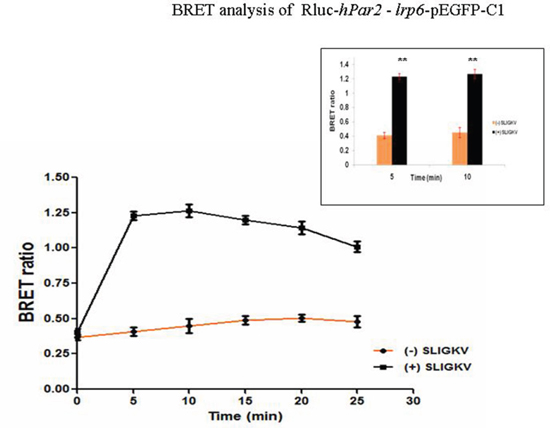
Figure 3: BRET assay shows specific PAR2-LRP6 interaction in fibrocystic HU cells. The existence of PAR2-LRP6 receptor-receptor interaction and SLIGKV agonist regulation is shown by quantitative BRET2 assay in fibrocystic HU cells. HU cells were co-transfected with Rluc-hPar2 and lrp6-pEGFP-C1. Specific maximal signal emission is detected beginning 5 min following SLIGKV activation (P=0.002) for up to 30 min, as compared with the non-activated status. This is a representative curve of three independent experiments performed.
ZDOCK protein-protein docking server information also showed the association between PAR2 and LRP6. The protein domains are predicted by Bioinformatics Tool SMART (Simple Modular Architecture Research Tool), an online resource (http://smart.embl.de/) that allows identification and annotation of mobile domains and analysis of the domain architecture (Figure 4).
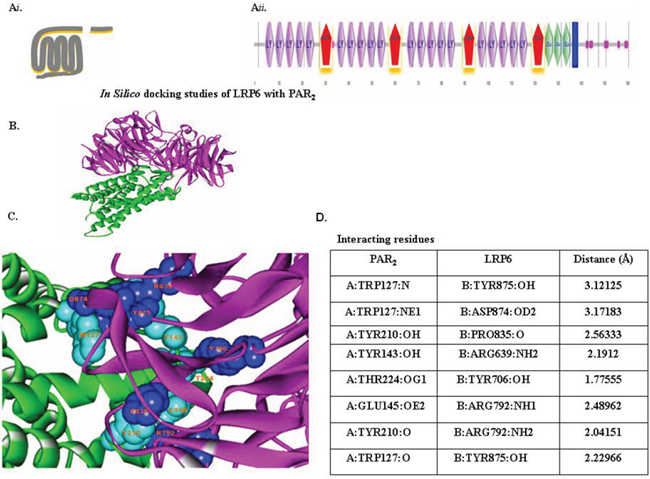
Figure 4: In-silico docking studies of LRP6 with PAR2. (Ai) Schematic presentation of PAR2. (Aii) Schematic presentation of LRP6. The domains present in LRP6 were predicted by Bioinformatics Tool SMART (Simple Modular Architecture Research Tool), an online resource (http://smart.embl.de/) that allows identification and annotation of genetically mobile domains and analysis of domain architecture. LRP6, a member of LDLR family, consists of four EGF domains and three LDL repeats (LDLR). (B) Protein-protein docking study. Interaction between LRP6 (magenta) with PAR2 (green) were determined with the ZDOCK protein–protein docking server, based on a Fourier transform method to search all possible binding modes for the applied proteins. Finally, most probable predictions were ranked on the basis on geometry, hydrophobicity, and electrostatic complementarity of the molecular surface using ZDOCK. (C) Determination of interacting residues (e.g., LRP6 with PAR2). The interacting residues are shown in CPK (Corey-Pauling-Koltun), a ball-shaped interacting amino acid residue, with cyan CPK for PAR2 and blue CPK for LRP6. (D) List of PAR2 and LRP6 interacting residues.
The association between PAR2 and LRP6 was also assessed by in situ confocal microscopy. For this purpose, HEK293T cells were transiently transfected with both HA-lrp6 and YFP-hPar2. After 24 hrs, the cells were serum deprived for additional 24 hrs then activated with SLIGKV for 15 min. PAR2 was visualized by fluorescence (e.g., green) level and LRP6 by anti-HA antibodies followed by Cy3-conjugated IgG secondary antibodies (e.g., red; Figure 5A). SLIGKV activation resulted in a merged yellow staining, indicative of co-localization between PAR2 and LRP6 (Figure 5A). This result is obtained under conditions whereby equal levels of either YFP-PAR2 or HA-lrp6 in the HEK293T cells were observed (Figure 5B).
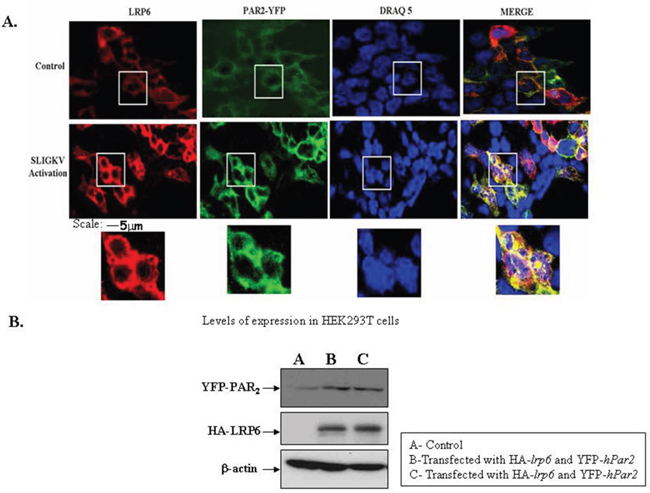
Figure 5: SLIGKV PAR2 activation induces colocalization between LRP6 and PAR2: confocal imaging. (A) Confocal immunofluorescence analysis. HEK293T cells were transiently transfected with HA-lrp6, YFP- hPar2 and activated with SLIGKV. PAR2 was visualized by direct fluorescence (green) and LRP6 by anti-HA antibodies followed by Cy3-conjugated IgG secondary antibodies. For reference, staining of cell nuclei with DRAQ5 (blue) is shown. Merge staining of both LRP6 and PAR2 co-localization is shown (yellow), following SLIGKV PAR2 activation. (B) Western blot analysis indicates levels of YFP-PAR2 and HA-LRP6 following transfection. β-actin served as a reference for loading. Data shown are representative of four independent experiments.
PAR2 induces mammary gland tumors
Stable cell clones (e.g., MCF-7 cells) overexpressing PAR2 were assessed for their capabilities to generate orthotopic mammary fad pat tumors. Following subcutaneous implantation of slow release pellets of β-estradiol (Innovative Research of America, Sarasota, FL, USA), MCF-7 clones were inoculated in the mammary fat pad with MCF7/hPar2 stable clones. After appropriate periods of time the PAR2 overexpressing clones elicited large mammary gland tumors, as compared with control mock-transfected mice, which generated very small tumors (Figure 6A&6B). Tumor volume growth of the PAR2 instigated tumors as compared with control mock transfected cells was evaluated (Figure 6C). RT-PCR analysis confirmed that the MCF-7 clones expressed PAR2, in contrast with the mock transfected cells, which were lacking PAR2 (Figure 6B).
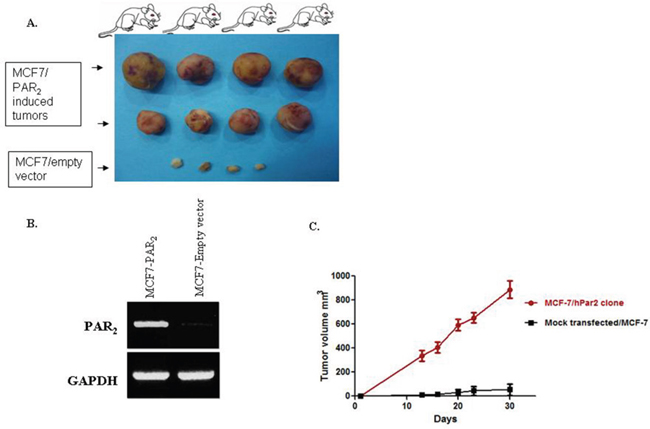
Figure 6: PAR2 elicits mammary gland tumors in a xenograft mouse model. (A) Morphological appearance of PAR2-induced mammary gland tumors shows large and vascularized tumors obtained by orthotopic inoculation of MCF-7 clones stably overexpressing hPar2 (MCF7/hPar2), as compared with mock-transfected clones (MCF7/empty vector). (B) RT-PCR analyses demonstrating the levels of hPar2 in the stable clones, as compared with a house-keeping gene GAPDH. (C) Tumor volume of the two groups is shown. Error bars show s.d.; *P < 0.006. Data shown are representative of three independent experiments.
In order to substantiate in vivo the PAR2 and LRP6 co-association that was observed in vitro, we have analyzed PAR2-driven mammary gland tumor tissue sections by confocal staining for the colocalization between PAR2 and LRP6. Toward this goal, tissues were double-stained for LRP6 (A; red) and PAR2 (C; green). Abundant distribution of either LRP6 or PAR2 was seen in the large mammary gland tumor sections (Figure 7A&7C). Merge analysis indicated colocalization of PAR2 with LRP6, as shown by the yellow staining (Figure 7D). Inset is a magnified view of the boxed area. It is postulated that the tumors exhibit plentiful protease in the vicinity of the tumor microenvironment, sufficient to continuously activate PAR2. As a result, colocalization with LRP6 is observed during PAR2 induction and initiation of large tumor formation, in vivo.
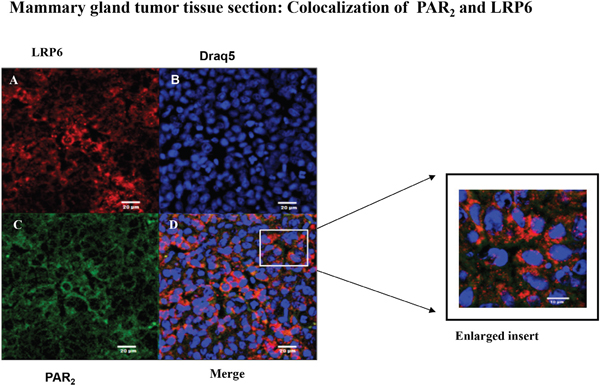
Figure 7: Colocalization between LRP6 and PAR2 in breast mouse cancer tumor biopsies: confocal analysis. Confocal immunohistostaining was carried out on PAR2-induced mouse orthotopic mammary gland tumors. Application of anti-LRP6 (A; red) and anti-PAR2 (C; green) Abs was followed by appropriate Cy2- and Cy3-conjugated IgG secondary antibodies. The cell nuclei were visualized by DRAQ5 (B; blue). High expression levels were observed in both LRP6 and PAR2. Merge analyses indicate co-localization between LRP6 and PAR2 (D; orange). Data shown are representative of three independent experiment. Magnification x40.
Both Gα13 and Gα12 are involved in PAR2-mediated β-catenin stabilization
Since Gα12/13 are the only G-proteins known to be involved in cell transformation [32–34], we sought to analyze the relationship between these G-proteins and PAR2-induced β-catenin stabilization. In order to dissect their relative involvement, dominant negative (DN) forms of Gα12 and Gα13 [26, 34] were utilized. When we transiently transfected HEK293T with flg-β-catenin, wt hPar2, and either a dominant negative Gα12-GA or Gα13-GA mutants, the following data were obtained. After activation of PAR2, cell lysates were prepared and immunoblots were further evaluated. Application of anti-flg (to detect levels of β-catenin) or anti-Gα12 and Gα13 antibodies was carried out. We have found that increased expression of the dominant negative forms of either Gα13 or Gα12 (i.e., Gα13-GA or Gα12-GA) both markedly inhibited PAR2-induced β-catenin stabilization in a dose-dependent manner (Figure 8A&8B). In contrast, PAR1 acts specifically via Gα13 [26]. The presence of increasing concentrations of Gα12-GA did not show any effect on PAR1-induced β-catenin levels, whereas the DN Gα13-GA construct effectively inhibited β-catenin levels [26]. Taken together, these findings suggest that there is, most likely, an overlap in PAR2-induced β-catenin stabilization with regard to the role played by Gα12 and Gα13, and both Gα12 and Gα13 are required to induce PAR2-mediated β-catenin stabilization.
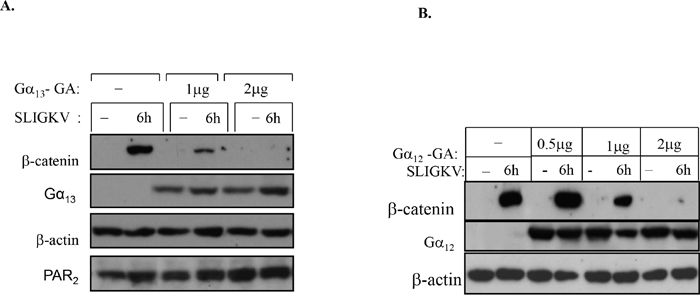
Figure 8: Both Gα13 and Gα12 are involved in PAR2-induced β-catenin stabilization. (A) A dominant-negative (DN) form of Gα13 inhibits β-catenin stabilization. HEK-293T cells were transiently transfected with hPar2-wt, flg-β-catenin, empty vector, or dominant-negative Gα13-GA plasmid. Following PAR2 activation, immunoblots were analyzed using anti-flg (for flg-β-catenin), anti-Gα13, anti-PAR2, and anti-β-actin antibodies. Gα13 potently inhibited PAR2-induced β-catenin stabilization in a dose-dependent manner. (B) A DN form of Gα12 inhibits β-catenin stabilization. HEK-293T cells were transiently transfected with hPar2-wt, flg-β-catenin, empty vector, or dominant-negative Gα12-GA plasmid. Following PAR2 activation, immunoblots were analyzed using anti-flg (for flg-β-catenin), anti- Gα12, and anti-β-actin antibodies. Gα12 as well as Gα13 potently inhibited PAR2-induced β-catenin stabilization in a dose-dependent manner. Since both Gα13-GA and Gα12-GA independently abrogate PAR2-induced β-catenin accumulation, there is most likely an overlap between Gα12 and Gα13 in mediating the induction of β-catenin stabilization.
Translocation of DVL to the cell nuclei by PARs
While DVL acts as an upstream cytoplasmic link initiating the process of β-catenin stabilization, it is now well recognized that DVL also exists in the cell nucleus, where it is required for the Wnt-β-catenin transcriptional activity eliciting downstream targeted gene expression. This renders DVL a more complex role than initially thought. In addition to serving as a scaffolding protein bridging seven transmembrane receptors and signaling components [35–37], it also acts as part of a nuclear transcription complex. At least in the case of PAR1 [25, 26], as also the parathyroid receptor1 (PTH1R), DVL acts as a scaffold bridge, linking to GPCRs and consequently enabling the early-on event in β-catenin stabilization [39]. We demonstrate now that following SLIGKV PAR2 activation DVL is ultimately found within the cell nucleus as soon as 30 min after activation and distinctly by 2hr (Figure 9A). Furthermore, we show here that DVL and c-Jun form a complex within the cell nuclei. This takes place following transient transfection of flg-dvl, HA-c-Jun and hPar2 plasmids in HEK293T cells. Immunoprecipitation using anti-HA antibodies and flg, Western blot detection showed a noticeable complex formation with DVL and c-Jun, in the cell nuclei, by SLIGKV PAR2 activation that became prominent after 5h PAR2 activation (Figure 9B). Indeed, it was previously reported that at a later time period and upon Wnt signaling, DVL translocates to the cell nucleus where it forms a transcription complex among others, with nuclear β-catenin and c-Jun [39]. Similarly, nuclear translocation of DVL was found also following activation of PAR1 (see Supplementary Figures 3&4).
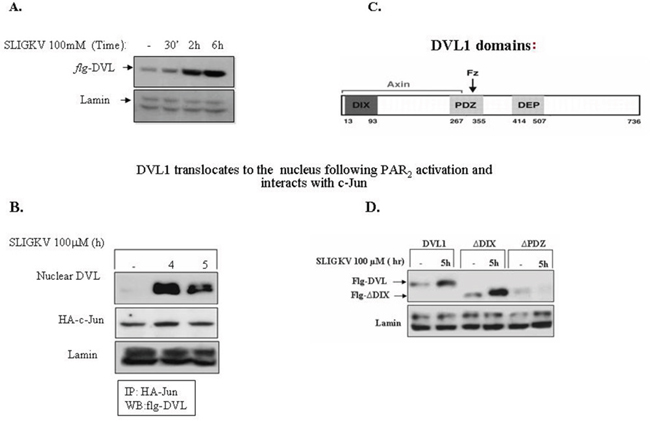
Figure 9: DVL1 translocates to the cell nucleus following PAR2 activation. (A) DVL1 translocates to the cell nucleus following PAR2 activation and PAR2 activation. HEK-293T cells were transiently transfected with flg-dvl1 and hPar2. The nuclear fraction was isolated and Western blots were analyzed using anti-flg for the detection of DVL1 and anti-lamin antibodies as loading control. DVL accumulated in the nucleus in response to PAR2 activation. (B) DVL interacts with c-jun in the nucleus following PAR2 activation. PAR2 induces the interaction between DVL1 and c-Jun. HEK293T cells transfected with both flg-Dvl1 and c-Jun showed nuclear localization of DVL1 as judged by immunoprecipitation analyses. PAR2-induced association of DVL with c-Jun occurred in a manner similar to Wnt signaling. (C) Scheme of DVL1 domains. (D) DVL-PDZ domain is required for PAR2-induced DVL1 nuclear localization. HEK-293T cells were transiently transfected with the following constructs: flg-wt dvl1, DVL1 deletion constructs (e.g., flg-ΔDIX-dvl or flg-ΔPDZ-dvl), and hPar2. The nuclear fraction was isolated after PAR2 activation and immunoblots were analyzed using anti-flg and anti-lamin antibodies. The deleted DVL-PDZ domain construct, but not the DIX domain-deleted construct, failed to translocate to the nucleus, as compared with wt DVL nuclear localization. It is concluded that the PDZ, but not the DIX domain, is necessary for PAR2-induced nuclear localization of DVL1.
DVL protein comprises an N-terminal DIX domain, a central PDZ domain, and a C-terminal DEP domain (see scheme, Figure 9C). Wewished to identify which of these regions are essential for DVL nuclear localization. Toward this purpose, we performed Western blot analyses from nuclear extracts of cells expressing dvl constructs of either wt full length or the deleted domains of DIX or PDZ-(deleted for DIX or for PDZ). Transfections with flg-wt-dvl, flg-dvl-ΔDIX, or flg-dvl-ΔPDZ showed that efficient translocation of DVL is found after 5 hr of PAR2 activation using the DIX deleted construct, similar to the wt DVL (Figure 9D). In contrast, nuclear localization of DVL was completely abrogated in the presence of the PDZ deleted form (Figure 9D). This suggests that PDZ domain is required for DVL nuclear localization. A similar outcome is demonstrated for PAR1 DVL nuclear localization (see Supplementary Figure 5). This stands in contrast to the early-on DVL binding to membrane-anchored PAR1, whereby the DIX domain was found to be essential for the PAR1-Gα13 axis formation [26]. In accordance with the involvement of both Gα12 and Gα13 in PAR2-induced β-catenin stabilization, this also takes place with regard to PAR2-induced DVL nuclear localization (Supplementary Figure 6). DVL is necessary also for PAR1-induced β-catenin stabilization (Supplementary Figure 7).
DISCUSSION
Here we show that PAR2 is a significant inducer of the β-catenin stabilization path in cancer. We demonstrate the novel recruitment of LRP6 following SLIGKV-PAR2 activation, and show for the first time that PAR2 associates with LRP6, a known coreceptor of Frizzled (Fz) proteins in the Wnt/β-catenin route, following SLIGKV activation. The association is demonstrated using various methodologies, namely co-immunoprecipitation, BRET, and also confocal microscopy image analysis. These assays are supported by the ZDOCK protein-protein server, which predicts the tight association between PAR2 and LRP6.
PAR2 association with LRP6 takes place via the interaction of the PAR2 extracellular portion on the cell surface. As a result, LRP6 is phosphorylated and further binds Axin, which is being dislodged from the “degradation complex”, ultimately leading to β-catenin stabilization. Taken together, our data suggest a novel path of PAR2-induced β-catenin stabilization. We show that the shortest PAR2 C-tail capable of eliciting β-catenin stabilization and transcriptional activity is PAR2-K368Z, which includes the amino acid sequence “AKNALLCRSVTV,” a Ca++ binding domain [38]. It is possible that a PAR2-induced Ca++ signal is required for β-catenin stabilization, however this remains to be studied. This possibility is raised by our preliminary observations indicating that in the presence of BAPTA (30μM), a known Ca++ inhibitor, PAR2-induced β-catenin stabilization and transcriptional activity are inhibited (Supplementary Figure 1). Previously, we have demonstrated a direct link between PAR1 and β-catenin stabilization both in a transgenic (tg) mouse model, overexpressing hPar1 in the mammary glands and in a spectrum of transformed epithelial cell lines [25–27]. This was shown to be mediated selectively via the association of Gα13 and the DIX-DVL axis, formed early on to facilitate β-catenin stabilization. PAR2-mediated dynamics of β-catenin hyperactivity takes place in a different manner. We now show the equal involvement of both Gα12 and Gα13 G-proteins in PAR2-induced β-catenin stabilization, and the recruitment of Axin to the PAR2-LRP6 axis. It is demonstrated that shRNA silencing of LRP6 effectively attenuates PAR2-induced β-catenin stabilization; thus, SLIGKV/PAR2-induced recruitment of LRP6 is essential for the PAR induced β-catenin signaling. As a result, DVL is translocated to the cell nucleus where it initiates a transcriptional switch, recruiting various chromatin modifiers to β-catenin, among of which are; c-Jun, and Lef/Tcf family members, consequently leading to the transcription of downstream target genes. While DVL-DIX domain is essential for the formation of PAR1-Gα13-DVL complex, the PDZ domain of DVL is involved in PAR1- and PAR2-induced DVL nuclear translocation.
LRP6 is a well-recognized coreceptor for Wnt signaling in cancer [40–47]. Whereas Wnt is known to function through the classical Fz-LRP6 shared interaction for β-catenin dynamics, our data introduce a new partner for LRP6 association that potently instigates β-catenin stabilization. Similarly, LRP6 was previously shown to act as a coreceptor with another GPCR, the parathyroid receptor1, PTH1R [39]. The PTH hormone activates β-catenin signaling in osteoclasts by the direct recruitment of LRP6 to the PTH/PTH1R complex [36]. This takes place via the extracellular portion of LRP6, that acts with PTH1R, leading to the phosphorylation of the PPPSP motif of the LRP6 cytoplasmic tail [48]. Interestingly, PTH1R was found also capable of recruiting DVL to the PTHR1 C-tail following PTH activation [39], as we previously demonstrated for PAR1 [26].
LRP5 and LRP6 are type I, single-span transmembrane receptors with a large extracellular domain, that were shown to bind several Wnt ligand species shown in vitro experiments [28, 45–47]. In addition to Wnt proteins, the extracellular portions of LRP5 and LRP6 also bind other agonists and antagonists of the Wnt pathway, such as DKK1, Sclerostin, and Wise, members of the Dkk family [46, 48–51]. LRP5 and LRP6 exhibit a high degree of sequence homology, sharing 73% and 64% sequence identity in their extracellular and intracellular domains, respectively [35]. The ectodomain of LRP5/6 is composed of four propeller/epidermal growth factor (EGF) repeats (E1-4), three LDL repeats (LDLR) and five intracellular PPP(S/T)P domains that mediate downstream signaling events [47]. E1-4 but not LDLR is the binding domain of canonical Wnt ligands and the canonical pathway inhibitor Dkk [45, 46, 48, 51]. Until now, the LDLR-binding proteins remain unexplored. The current view is that the close proximity of LRP5/6 and Fz coupled by canonical Wnt ligand binding to E1-4 of LRP5/6 and amino-terminal cysteine-rich domain (CRD) of Fz is needed for canonical pathway activation [45, 49]. In contrast, Dkk1 promotes the internalization of LRP5/6 via binding with Dkk1 receptor Kremen, making LRP5/6 unavailable for Wnt reception and inhibiting the canonical pathway [50].
This, coupled with extensive similarities in structural and biochemical properties, has led to the assumption of functional redundancy between the two receptors; however, in vivo studies show that they mediate unique functions. In addition, homozygous deletion of LRP6 in transgenic mice leads to perinatal lethality, while LRP5 knock-out mice are viable and fertile [29, 52, 53]. Recently however, it was found that LRP5-deficient mice develop low bone mass postnatally [28, 54–56], which was attributed to a direct effect of LRP5 on osteoblast function. Nevertheless, the role of LRP5 in Wnt signaling remains unknown, since the absence of LRP5 has no effect on Wnt3a-mediated transactivation of the canonical Wnt pathway in lrp5_/_ mammary epithelial cells (MECs) [57].
Recently, it was proposed that while LRP5/6 and Fz are oncogenic in nature, through direct binding (Fz and LRP5/6) they are able to prevent Fz-regulated non-canonical pathway activation and the non-canonical mediated tumor metastasis. Knocking down endogenous LRP5/6 promoted otherwise nonmetastatic tumor cells to disseminate throughout the body. Along this line of evidence, the application of soluble recombinant LRP6 extracellular domain effectively inhibited the appearance of metastatic foci from otherwise aggressive metastatic tumor cells. Hence, according to these findings, therapeutic medicaments based on anti-LRP6 should be posibly reconsidered since they may induce metastatic spread [58].
DIX domain is in fact found in three proteins, Axin, DVL, and Ccd1. A previous study suggested that DVL may behave as a dominant-negative protein of Axin, regulating its function via the heterotypic interaction between DVL-DIX and Axin-DAX (disheveled and Axin domains) [59]. The Axin DAX domain mediates homo- and heteropolymerization, which may be important for its function [59–61]. Previous studies have suggested that the Axin N-terminus portion, including the RGS domain and the linker region between the RGS domain and the GSK-binding domain, has an inhibitory role on Axin’s binding with its partners [63, 64]; however, the mechanism underlying this inhibition remains elusive [65]. Two recent publications propose a model in which Axin is regulated via conformational change of an “open” or activated conformation as well as a “closed” or auto-inhibited structure. Axin is a phosphoprotein that is central to assemblies of both destruction [66–69] and signaling complexes [28, 51, 62, 70, 71], and it becomes dephosphorylated upon Wnt stimulation [72]. Kim et al., [65] proposed that without Wnt, Axin is associated with and phosphorylated by GSK3β, which is present in an activated (“open”) conformation for β-catenin binding and is poised for engagement of LRP6. In the presence of Wnt, LRP6 undergoes Fz/DVL-dependent phosphorylation and recruits active Axin from the “destruction complex” to form the signaling queue, in which GSK3β bound to Axin is inhibited by the phospho-LRP6 [73–76], consequently leading to inhibition of β-catenin phosphorylation which tips the balance toward Axin dephosphorylation by PP1 phosphatase. The dephosphorylated Axin form adopts an inactivated (“closed”) conformation through intramolecular auto-inhibition and becomes incompetent for association with β-catenin or phospho-LRP6, leading to the disassembly of destruction and signaling complexes. Similarly, it was suggested by the group of Lin Li [77] that auto-inhibitory conformation of Axin is mediated by interactions between its N- and C-terminal domains. It was shown that conformational change is most likely regulated by the Wnt signaling pathway and is further facilitated by HLY78, a small molecule that binds Axin and activates Wnt signaling. It has been postulated that the direct binding of HLY78 to the DAX domain of Axin triggers the conformational change of Axin from a ‘closed’ auto-inhibitory state to an ‘open’ active state, leading to enhancement of the Axin-LRP6 association and the subsequent phosphorylation and activation of LRP6. Hence, these two independent publications strongly support and strengthen the conformation change scenario in Axin as a key regulatory change controlling Wnt signaling. We find that SLIGKV activation of PAR2 leads to LRP6 recruitment followed by the binding association of the Axin, most likely similar to Wnt activation, yet to be fully described.
Formation of the β-catenin-TCFs complex has been well established as a prerequisite for c-Jun but not of endogenous c-Fos, indicating that c-Fos did not participate in the canonical Wnt signaling as did c-Jun, and knocking down endogenous c-Jun with shRNA markedly suppressed Wnt-3a-induced transcriptional activity in HEK293T cells [75]. In-parallel, the expression of c-myc target gene failed to respond to canonical Wnt stimulation in c-Jun -/-cells [77].
In zebra fish, loss of c-Jun function inhibited the induction of ventral mesoderm and reduced the expression of ventral marker genes in a manner that was very similar to the phenotype obtained by the loss of function of Wnt-8. Two previous studies have demonstrated that DVL, a pivotal regulator of the canonical Wnt pathway, is also localized in the nucleus [35, 37], and that its nuclear localization is required for the canonical Wnt signaling [35]. It is well known that β-catenin directly interacts with TCFs in vitro and in vivo; however, insight as to how these interactions take place has been only recently gained. It was demonstrated [78] that DVL can be recruited in the nucleus to the promoter of Wnt target genes interacting with c-Jun and β-catenin, respectively, further enhancing its association with β-catenin-TCFs transcriptional complex.
Nuclear DVL is suggested to be crucial for the formation of a stable complex between β-catenin and TCFs in mammalian cells and zebrafish [78]. We hereby demonstrate that, similar to Wnt signaling, PAR1 (or PAR2) activation leads to β-catenin-DVL complex formation in the nucleus but not in the cytoplasmic compartment (Supplementary Figure 4). Nuclear DVL is essential for Lef/Tcf transcriptional activity and downstream target gene expression, since shRNA silencing of dvl potently inhibited PAR1- (or PAR2) induced transcriptional activity (Supplementary Figure 4).
The current model thus suggests that DVL may have multiple roles in the canonical Wnt signaling pathway. Initially, cytoplasmic DVL receives a signal from the plasma membrane, resulting in accumulation of β-catenin in the nucleus. Next, DVL nuclear accumulation is promoted [35, 37], involving DVL activation of JNK in a yet unknown manner to promote c-Jun phosphorylation [77, 78]. In the nucleus, DVL binds to phospho-c-Jun and β-catenin, and promotes the formation of a quaternary functional complex consisting of β-catenin, Lef-1/Tcfs, c-Jun, and DVL. We provide evidence that both PAR1 and PAR2 act to instigate nuclear localization of DVL and its interaction with both c-Jun and β-catenin, corresponding with the proposed path of the canonical Wnt signaling.
MATERIALS AND METHODS
Plasmids and reagents
cDNA encoding mouse flg-DVL1, flg-ΔDIX-DVL1, flg- ΔPDX-DVL1, and myc and flg- β-catenin were kindly provided by Dr. Ben-Neria (Hebrew University, Jerusalem). cDNA encoding mouse flg-DVL1-DIX and flg-DVL1-PDZ were kindly provided by Lin Li (State Key Laboratory of Molecular Biology, Shanghai Institute for Biological Science, Chinese Academy of Sciences, Shanghai 200031, China). The cDNA encoding Gα12, Gα13, and Gαq constructs, wt, was constitutively active and dominant negative as previously described [26]. The PAR2 and PAR1 agonists TFLLRN and SLIGKVwere obtained from GeneScript (Piscataway, NJ, USA 08854). Thrombin was obtained from OMRIX Bio Pharmaceutical (Ramat Gan, Israel).
Cells
HCT116, HT29, HEK293T, HU and RKO cells were grown in 10% FCS-DMEM/RPMI supplemented with 50 U/ml penicillin and streptomycin (GIBCO-BRL, Gaithersburg, MD, USA) and maintained in a humidified incubator with 8% CO2 at 37°C.
Mutation generation
PAR2 mutants were generated by site-directed mutagenesis using QuikChange (Stratagene, La Jolla, CA) as previously described [17], Briefly, the procedure utilizes a pcDNA3.1(+) vector containing an insert of hPar2 coding sequence and two synthetic oligonucleotide primers containing the desired mutations. The oligonucleotide primers, each complementary to opposite strands of the vector, are extended during temperature cycling by PfuTurbo DNA polymerase (2.5U per reaction). Incorporation of the oligonucleotide primers generates a mutated plasmid containing staggered nicks. Following temperature cycling, the product is treated with DpnI (10U, 37°C, 1 hr). DNA containing the desired mutations is then transformed into XL1-blue super competent bacteria cellsfollowing plasmid isolation using the PureLink™ Maxiprep Kit (Invitrogen Corporation, Carlsbad, CA, USA). Dideoxy sequencing is performed to confirm insertion of the appropriate mutation. The primers are as follows: S390Z- GCTCTTACTCTTCAAGTTGAACCACTGTTAAGACCTCC, K378Z-CCCTCACCTCAAAGTAACACTCCAGGAAATCCAGC, K368Z- GCCGAAGTGTCCGCACTGTATAGCAGATGCAAGTATCCC, K356Z-GGGATCATGCATAGAACGCTCTCCTTTGCCGAAGTGTCCGC, S348Z-CGACCCCTTTGTCTATTACTTTGTTTCACATGATTTCAGGG.
Immunohistological staining
Tissue samples derived from PAR2 induced mammary glands were fixed with 4% formaldehyde in PBS, embedded in paraffin, and sectioned (5-μm sections). After deparaffinization and rehydration, the sections were stained with H&E or subjected to immunohistochemistry. For this, the slides were incubated 3% H2O2 prior to antigen retrieval. Antigen unmasking was carried out heating (20 min) in a microwave oven in 10mM Tris buffer containing 1mM EDTA. After blocking slides were incubated with the following primary antibodies: anti β-catenin (C-2206, Sigma-Aldrich St Louis MO, USA), anti PCNA (sc-56, Santa Cruz Biotechnology, USA dilution 1:200) anti DVL1 (sc7397, Santa Cruz Biotechnology, Dallas Texas, USA; goat polyclonal IgG) or anti CD31 (Dako, Clone JC70A, Carpinteria, CA). Color was developed using the 3,3’-diaminobenzidine (DAB) (Thermo Scientific, Walham, MA, USA) or the Zymed AEC substrate kit (Zymed Laboratories So, San-Francisco, CA, USA), followed by counter staining with Mayer’s haematoxylin. Controls without addition of primary antibodies showed low or no background staining in all cases.
Cell transfections
Cells grown to 80% confluency were transfected with 0.5-2 μg/mL of plasmid DNA in Fugene 6 transfection reagent (Boehringer-Mannheim, Mannheim, Germany) according to the manufacturer’s instructions.
RNA isolation and RT-PCR
RNA was isolated with Tri-Reagent (MRC, Cincinnati, OH, USA) according to the manufacturer's instructions. After reverse transcription of 1 μg total RNA by oligo (dT) priming, cDNA was amplified using Taq DNA polymerase (Promega, Madison, WI, USA). Comparative semi quantitative PCR was performed as follows: GAPDH mRNA was first amplified at a low cycle number. If needed, cDNAs were adjusted to obtain similar intensities for GAPDH signals with all the samples. The adjusted amounts of cDNA were subjected to PCR. The PCR conditions were an initial denaturation at 94°C for 2 min, denaturation at 94°C for 15 sec, annealing for 45 sec at the appropriate temperature and extension for 1 min at 72°C (24–33 cycles of amplification). Aliquots (15 μl) of the amplified cDNA were separated by 1.5% agarose gel electrophoresis. VEGF PCR product was separated on a 2% Nusieve (FMC Rockland, ME, USA, 3:1 agarose gel) and visualized by ethidium bromide staining under ultraviolet light.
shRNA constructs and lentiviral vector production
To prepare SiRNA constructs we used U6 promoter driver and a lentivirus-mediated delivery cassette of SiRNA (pLentilox 3.7) specific for the target genes: DVL1, LRP5, and LRP6. For this, a sequence of 19 nucleotides of the target gene coding region was selected for stem-and-loop oligonucleotide shRNA. Appropriate DNA oligonucleotides were synthesized to generate the hairpin structure stem-and-loop shRNA expression cassette. The oligos comprised the following: 19 bases of the target gene coding sequence, the loop sequence linker (9 bases), a reverse complement of 19 bases of the target gene coding region, and a terminator sequence poly T. The sticky end of the XhoI site was added to the antisense strand oligos. Both sense and antisense sequences were phosphorylated at the 5' ends. The sense sequence oligos were annealed to their respective antisense oligos. SiRNA cassette sequences were then ligated into pLentilox 3.7 vector (Van Parij Laboratory, Massachusetts Institute of Technology, Cambridge, MA, USA). The sequence of sh RNA for the target genes was as follows: AACAAG ATC ACCTTCTCCGAG (DVL1), CATGATCGAGTCGTCCAAC (LRP5), CCGCATG GTG ATT GA TGA (LRP6).
Western blot analysis
Cells were solubilized in lysis buffer containing 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% triton X-100, and protease inhibitor cocktail including 5 mg/ml aprotinin, 1 mM phenylmethylsulfonylfluoride, PMSF, and 1 mM Na orthovanadate (Sigma, St. Louis, MO, USA), for 20 min at 4°C. After centrifugation at 10,000 g for 20 min at 4°C, the supernatants were transferred and the protein content was measured. Lysates (50 μg) were loaded on a 10% SDS-PAGE followed by transfer to Immobilon-P membrane (Millipore, Bedford, MA, USA). Membranes were blocked and probed with the appropriate antibodies at a concentration of 1 μg/ml. Anti-β-catenin (Cell signaling 9562), anti PAR2 (SAM11 Santa Cruz, Dallas Texas, USA), LRP6 antibodies (Cell signaling (C5C7) rabbit mAb Danver, MA USA), anti DVL1 (mAb; sc 8025, Santa Cruz, Dallas Texas, USA), anti Gα12, anti Gα13 and anti β-actin (Santa Cruz, CA, USA), or anti flg antibodies (mAbF316, Sigma clone M2) and for IP OctA -Probe(D-8) sc807 polyclonal Ab (Santa Cruz Biotechnology Inc. Dallas, TX, USA) were suspended in 3% BSA in 10 mM Tris-HCl pH 7.5, 100 mM NaCl and 0.1% Tween-20. After washes, blots were incubated with secondary antibodies conjugated to horseradish-peroxidase. Immunoreactive bands were detected by the enhanced chemiluminescence (ECL) reagent (Pierce, Rockford, IL, USA).
Nuclear extract
Cells were solubilized in lysis buffer A (10mM HEPES, pH 7.9, 10 mM KCl, 0.1mM EDTA and 1mM DTT), a protease inhibitor cocktail (1:100), 1 mM phenylmethylsulfonylfluoride, PMSF, and 1 mM Na orthovanadate (Sigma) for 15 min at 4°C. After adding 10% NP-40 solution, centrifugation at 10,000 g was performed. The pellet was incubated on ice with buffer C (20 mM HEPES, pH 7.9, 420 mM KCl, 1mM EDTA, and 1mM DTT), a protease inhibitor cocktail, 1 mM PMSF, and 1 mM Na orthovanadate. After centrifugation at 12,000 g for 15 min at 4°C, the supernatants were collected and the protein content was evaluated.
Immunoprecipitation
Protein cell lysates (400 μg) were used for immunoprecipitation analysis. Anti- PAR2, or anti-flg antibodies were added to the cell lysates. After overnight incubation, protein A-Sepharose beads were added to the suspension, which was subsequently rotated at 4°C for 1h. Elution of the reactive proteins was performed by resuspending the beads in protein sample buffer followed by boiling for 5 min. The supernatant was then resolved on a 10% SDS-polyacrylamide gel and treated as indicated above for Western blotting.
BRET assay
Interaction of PAR2 with LRP6 was further validated using bioluminescence resonance energy transfer assay (BRET2). In our BRET2 experiment, we applied Rluc-hPar2 (a kind gift of the Trejo J-A Department of Pharmacology, School of Medicine, University of California San Diego, La Jolla, CA, USA) as a donor and lrp6- pEGFP-C1 as an acceptor. Toward this purpose, we cloned LRP6 downstream of pEGFP. To prepare the lrp6-pEGFP-C1 construct, lrp6 insert was taken from from lrp6-pCS2 plasmid (addgene) using Xba-1 and BamH1 restriction enzymes. Next, the lrp6 insert was subcloned into pEGFP-C1 vector. The cloned construct was confirmed by sequencing and LRP6 expression was also confimed by Western blot using anti-LRP6 antibody (Cell Signaling).
The entire BRET2 experiment was performed using well-defined protocols, with some modifications [30, 31]. Briefly, to perform BRET2, after concentration optimization, 0.5 μg of Rluc-hPar2 alone or together with 1 μg of lrp6-pEGFP-C1 was transfected into HU cells. The day following transfection, cells were detached and re-seeded (50,000 cells/well) into white opaque 96-well microplates (Thermo Fisher, Waltham, MA, USA). The next day, experimental cells were activated with the PAR2 ligand, SLIGKV (200 μM), washed (PBS containing 0.1% D-glucose and 0.5mM MgCl2), and immediately after addition of the cell-permeable substrate DeepBlueCTM (10 μM), luminescence was measured using the Tecan SparkTM10M multimode microplate detection system. The detection system is equipped with two distinct filters. Here, filter 1 (410 ± 70nm) was used to determine luminescence of Rluc-hPar2 donor, and filter 2 (515 ± 30nm) was used to measure lrp6-pEGFP-C1 (acceptor) luminescence, simultaneously. The data obtained are represented as the BRET2 ratio, with the BRET ratio for co-expressed Rluc-hPar2 and lrp6-pEGFP-C1 constructs normalized against the BRET ratio found for the Rluc-hPar2 expression construct alone in the same experiment [30].
BRET2 ratio =[(GFP acceptor emission at 515 ± 30nm)/(Rluc donor emission at 410 ± 70nm)] −cf.
The correction factor (cf), corresponds to (emission at 515 ± 30nm)/(emission at 410 ± 70nm), which is found when the Rluc-hPar2 construct is expressed alone in the same experiment.
The BRET ratio was measured as mean ± standard deviation (SD) (mean ± SD), derived from three independent experiments using T Test statistical analysis. Data have been expressed as scattered and bar diagrams in excel drawing.
Lef/Tcf luciferase reporter assay
RKO cells (0.5×105) were plated in 12-well dishes and incubated overnight at 37°C. The cells were transfected with hPar2-wt or hPar2 mutants (i.e. hPar2-truncated, hPar2-356 or hPar2-368) plasmids, human Lef-1 TOPflash (Tcf Optimal Promoter + luciferase, T cell factor (Tcf) reporter plasmid containing two sets (the second set in reverse orientation) of three copies of the Tcf binding site upstream of the thymidine kinase (TK) minimal promoter and luciferase open reading frame using Fugene 6 transfection reagent (Boehringer-Mannheim). CMV/β-gal plasmid was cotransfected as an internal control for transfection efficiency. After 48 h transfection, the cells were washed in PBS and luciferase assay performed with the Luciferase Reporter System (Promega, Heidelberg, Germany) according to the manufacturer's instructions, and detected on a luminometer Mithras LB940, Berthold Technologies GmbH & Co. KG, Bad Wildbad, Germany).
Immunofluorescence: cells
Either HCT-116 colon cancer or HEK-293 cells were plated on fibronectin (5μg/ml) coated coverslipsand transfected with YFP-hPar2 and Lrp6. After overnight serum deprivation (0.2% BSA), the cells were activated by SLIGKV for various periods of time. Samples were then fixed with 4% paraformaldehyde in PBS containing 5% sucrose for 10 min and permeabilized with 0.5% triton X-100 in PBS for 3 min. Samples were fixed and stained with anti-LRP6 (H-300 rabbit polyclonal sc15399, Santa Cruz Biotechnology, Inc., Dallas Texas, USA) and anti GFP (for the detection of PAR2; SIGMA St Louis MO, USA). Cy2- and Cy3-conjugated anti-mouse IgG (Jackson Laboratories, West Grove, PA, USA) was used as asecond antibody. Cells were examined using laser confocal microscopy (Model 410, Zeiss, Oberkochen, Germany).
Immunofluorescence: tumor tissue biopsy specimens
Paraffin embedded slides derived from PAR2 induced mammary gland tumors were deparaffinized and incubated in 3% H2O2. Antigen retrieval was carried out by heating (20 min) in a microwave oven in 10 mM Tris buffer containing 1 mM EDTA. After blocking the slides were incubated with the following primary antibodies: anti LRP6 (H-300 rabbit polyclonal sc15399, Santa Cruz Biotechnology, Inc. Dallas Texas, USA), anti-PAR2 (4μg/ml [SAM11] Santa Cruz), Cy2 conjugated anti-rabbit IgG, and Cy3-conjugated anti-mouse IgG antibodies (4μg/ml, Jackson Laboratories) were used as secondary antibodies. Nuclear staining was performed using DRAQ5 (4μM, Cell Signaling). Images were obtained using a Zeiss LSM 5 confocal microscope and analyzed with Zen software (Zeiss).
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest
GRANT SUPPORT
This work was supported by grants from the Israel Science Foundation (grant 1337/12 and 1420/16) and Monsa fellowship (to RB-S).
REFERENCES
1. Feigin ME. Harnessing the genome for characterization of G-protein coupled receptors in cancer pathogenesis. The FEBS journal. 2013; 280:4729-4738.
2. Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nature reviews Drug discovery. 2011; 10:47-60.
3. Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999; 283:655-661.
4. Wisler JW, Xiao K, Thomsen AR, Lefkowitz RJ. Recent developments in biased agonism. Current opinion in cell biology. 2014; 27:18-24.
5. Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nature reviews Cancer. 2007; 7:79-94.
6. Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, Moorhead M, Chaudhuri S, Tomsho LP, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010; 466:869-873.
7. O'Hayre M, Vazquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, Gutkind JS. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nature reviews Cancer. 2013; 13:412-424.
8. The Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012; 489:519-525.
9. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006; 127:469-480.
10. Nusse R. Wnt signaling in disease and in development. Cell research. 2005; 15:28-32.
11. Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997; 275:1784-1787.
12. Valenta T, Hausmann G, Basler K. The many faces and functions of beta-catenin. The EMBO journal. 2012; 31:2714-2736.
13. Grimson MJ, Coates JC, Reynolds JP, Shipman M, Blanton RL, Harwood AJ. Adherens junctions and beta-catenin-mediated cell signalling in a non-metazoan organism. Nature. 2000; 408:727-731.
14. Bar-Shavit R, Turm H, Salah Z, Maoz M, Cohen I, Weiss E, Uziely B, Grisaru-Granovsky S. PAR1 plays a role in epithelial malignancies: transcriptional regulation and novel signaling pathway. IUBMB life. 2011; 63:397-402.
15. Booden MA, Eckert LB, Der CJ, Trejo J. Persistent signaling by dysregulated thrombin receptor trafficking promotes breast carcinoma cell invasion. Molecular and cellular biology. 2004; 24:1990-1999.
16. Even-Ram S, Uziely B, Cohen P, Grisaru-Granovsky S, Maoz M, Ginzburg Y, Reich R, Vlodavsky I, Bar-Shavit R. Thrombin receptor overexpression in malignant and physiological invasion processes. Nature medicine. 1998; 4:909-914.
17. Kancharla A, Maoz M, Jaber M, Agranovich D, Peretz T, Grisaru-Granovsky S, Uziely B, Bar-Shavit R. PH motifs in PAR1&2 endow breast cancer growth. Nature communications. 2015; 6:8853.
18. Nierodzik ML, Chen K, Takeshita K, Li JJ, Huang YQ, Feng XS, D'Andrea MR, Andrade-Gordon P, Karpatkin S. Protease-activated receptor 1 (PAR-1) is required and rate-limiting for thrombin-enhanced experimental pulmonary metastasis. Blood. 1998; 92:3694-3700.
19. Versteeg HH, Schaffner F, Kerver M, Ellies LG, Andrade-Gordon P, Mueller BM, Ruf W. Protease-activated receptor (PAR) 2, but not PAR1, signaling promotes the development of mammary adenocarcinoma in polyoma middle T mice. Cancer research. 2008; 68:7219-7227.
20. Blackhart BD, Emilsson K, Nguyen D, Teng W, Martelli AJ, Nystedt S, Sundelin J, Scarborough RM. Ligand cross-reactivity within the protease-activated receptor family. The Journal of biological chemistry. 1996; 271:16466-16471.
21. Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, Covic L, Kuliopulos A. 'Role reversal' for the receptor PAR1 in sepsis-induced vascular damage. Nature immunology. 2007; 8:1303-1312.
22. O'Brien PJ, Prevost N, Molino M, Hollinger MK, Woolkalis MJ, Woulfe DS, Brass LF. Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1. The Journal of biological chemistry. 2000; 275:13502-13509.
23. Sevigny LM, Austin KM, Zhang P, Kasuda S, Koukos G, Sharifi S, Covic L, Kuliopulos A. Protease-activated receptor-2 modulates protease-activated receptor-1-driven neointimal hyperplasia. Arteriosclerosis, thrombosis, and vascular biology. 2011; 31:e100-106.
24. Jaber M, Maoz M, Kancharla A, Agranovich D, Peretz T, Grisaru-Granovsky S, Uziely B, Bar-Shavit R. Protease-activated-receptor-2 affects protease-activated-receptor-1-driven breast cancer. Cellular and molecular life sciences: CMLS. 2014; 71:2517-2533.
25. Turm H, Grisaru-Granvosky S, Maoz M, Offermanns S, Bar-Shavit R. DVL as a scaffold protein capturing classical GPCRs. Communicative & integrative biology. 2010; 3:495-498.
26. Turm H, Maoz M, Katz V, Yin YJ, Offermanns S, Bar-Shavit R. Protease-activated receptor-1 (PAR1) acts via a novel Galpha13-dishevelled axis to stabilize beta-catenin levels. The Journal of biological chemistry. 2010; 285:15137-15148.
27. Yin YJ, Katz V, Salah Z, Maoz M, Cohen I, Uziely B, Turm H, Grisaru-Granovsky S, Suzuki H, Bar-Shavit R. Mammary gland tissue targeted overexpression of human protease-activated receptor 1 reveals a novel link to beta-catenin stabilization. Cancer research. 2006; 66:5224-5233.
28. Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y, Hess F, Saint-Jeannet JP, He X. LDL-receptor-related proteins in Wnt signal transduction. Nature. 2000; 407:530-535.
29. Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 2000; 407:535-538.
30. Borroto-Escuela DO, Flajolet M, Agnati LF, Greengard P, Fuxe K. Bioluminescence resonance energy transfer methods to study G protein-coupled receptor-receptor tyrosine kinase heteroreceptor complexes. Methods Cell Biol. 2013;117:141-64.
31. Xie Q1, Soutto M, Xu X, Zhang Y, Johnson CH. Bioluminescence resonance energy transfer (BRET) imaging in plant seedlings and mammalian cells. Methods Mol Biol. 2011;680:3-28.
32. Chan AM, Fleming TP, McGovern ES, Chedid M, Miki T, Aaronson SA. Expression cDNA cloning of a transforming gene encoding the wild-type G alpha 12 gene product. Molecular and cellular biology. 1993; 13:762-768.
33. Marinissen MJ, Servitja JM, Offermanns S, Simon MI, Gutkind JS. Thrombin protease-activated receptor-1 signals through Gq- and G13-initiated MAPK cascades regulating c-Jun expression to induce cell transformation. The Journal of biological chemistry. 2003; 278:46814-46825.
34. Xu N, Bradley L, Ambdukar I, Gutkind JS. A mutant alpha subunit of G12 potentiates the eicosanoid pathway and is highly oncogenic in NIH 3T3 cells. Proceedings of the National Academy of Sciences of the United States of America. 1993; 90:6741-6745.
35. Itoh K, Brott BK, Bae GU, Ratcliffe MJ, Sokol SY. Nuclear localization is required for Dishevelled function in Wnt/beta-catenin signaling. Journal of biology. 2005; 4:3.
36. Romero G, Sneddon WB, Yang Y, Wheeler D, Blair HC, Friedman PA. Parathyroid hormone receptor directly interacts with dishevelled to regulate beta-Catenin signaling and osteoclastogenesis. The Journal of biological chemistry. 2010; 285:14756-14763.
37. Torres MA, Nelson WJ. Colocalization and redistribution of dishevelled and actin during Wnt-induced mesenchymal morphogenesis. The Journal of cell biology. 2000; 149:1433-1442.
38. Seatter MJ, Drummond R, Kanke T, Macfarlane SR, Hollenberg MD, Plevin R. The role of the C-terminal tail in protease-activated receptor-2-mediated Ca2+ signalling, proline-rich tyrosine kinase-2 activation, and mitogen-activated protein kinase activity. Cell Signal. 2004 Jan;16:21-9.
39. Wan M, Yang C, Li J, Wu X, Yuan H, Ma H, He X, Nie S, Chang C, Cao X. Parathyroid hormone signaling through low-density lipoprotein-related protein 6 Genes Dev. 2008; 22:2968-79.
40. Gan XQ, Wang JY, Xi Y, Wu ZL, Li YP, Li L. Nuclear Dvl, c-Jun, beta-catenin, and TCF form a complex leading to stabilization of beta-catenin-TCF interaction. The Journal of cell biology. 2008; 180:1087-1100.
41. Khan Z, Vijayakumar S, de la Torre TV, Rotolo S, Bafico A. Analysis of endogenous LRP6 function reveals a novel feedback mechanism by which Wnt negatively regulates its receptor. Molecular and cellular biology. 2007; 27:7291-7301.
42. Li L, Mao J, Sun L, Liu W, Wu D. Second cysteine-rich domain of Dickkopf-2 activates canonical Wnt signaling pathway via LRP-6 independently of dishevelled. The Journal of biological chemistry. 2002; 277:5977-5981.
43. Liu G, Bafico A, Harris VK, Aaronson SA. A novel mechanism for Wnt activation of canonical signaling through the LRP6 receptor. Molecular and cellular biology. 2003; 23:5825-5835.
44. Zeng X, Huang H, Tamai K, Zhang X, Harada Y, Yokota C, Almeida K, Wang J, Doble B, Woodgett J, Wynshaw-Boris A, Hsieh JC, He X. Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development. 2008; 135:367-375.
45. Angers S, Moon RT. Proximal events in Wnt signal transduction. Nature reviews Molecular cell biology. 2009; 10:468-477.
46. Chen S, Bubeck D, MacDonald BT, Liang WX, Mao JH, Malinauskas T, Llorca O, Aricescu AR, Siebold C, He X, Jones EY. Structural and functional studies of LRP6 ectodomain reveal a platform for Wnt signaling. Developmental cell. 2011; 21:848-861.
47. van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009; 136:3205-3214.
48. Bhanot P, Brink M, Samos CH, Hsieh JC, Wang Y, Macke JP, Andrew D, Nathans J, Nusse R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996; 382:225-230.
49. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Developmental cell. 2009; 17:9-26.
50. Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, Glinka A, Niehrs C. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature. 2002; 417:664-667.
51. Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, Niehrs C. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature. 2001; 411:321-325.
52. Kato M, Patel MS, Levasseur R, Lobov I, Chang BH, Glass DA 2nd, Hartmann C, Li L, Hwang TH, Brayton CF, Lang RA, Karsenty G, Chan L. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. The Journal of cell biology. 2002; 157:303-314.
53. Lindvall C, Evans NC, Zylstra CR, Li Y, Alexander CM, Williams BO. The Wnt signaling receptor Lrp5 is required for mammary ductal stem cell activity and Wnt1-induced tumorigenesis. The Journal of biological chemistry. 2006; 281:35081-35087.
54. Hendrickx M, Leyns L. Non-conventional Frizzled ligands and Wnt receptors. Development, growth & differentiation. 2008; 50:229-243.
55. Itasaki N, Jones CM, Mercurio S, Rowe A, Domingos PM, Smith JC, Krumlauf R. Wise, a context-dependent activator and inhibitor of Wnt signalling. Development. 2003; 130:4295-4305.
56. Mason JJ, Williams BO. SOST and DKK: Antagonists of LRP Family Signaling as Targets for Treating Bone Disease. Journal of osteoporosis. 2010; 2010.
57. Badders NM, Goel S, Clark RJ, Klos KS, Kim S, Bafico A, Lindvall C, Williams BO, Alexander CM. The Wnt receptor, Lrp5, is expressed by mouse mammary stem cells and is required to maintain the basal lineage. PloS one. 2009; 4:e6594.
58. Ren DN, Chen J, Li Z, Yan H, Yin Y, Wo D, Zhang J, Ao L, Chen B, Ito TK, Chen Y, Liu Z, Li Y, et al. LRP5/6 directly bind to Frizzled and prevent Frizzled-regulated tumour metastasis. Nature communications. 2015; 6:6906.
59. Fiedler M, Mendoza-Topaz C, Rutherford TJ, Mieszczanek J, Bienz M. Dishevelled interacts with the DIX domain polymerization interface of Axin to interfere with its function in down-regulating beta-catenin. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:1937-1942.
60. Hsu W, Zeng L, Costantini F. Identification of a domain of Axin that binds to the serine/threonine protein phosphatase 2A and a self-binding domain. The Journal of biological chemistry. 1999; 274:3439-3445.
61. Sakanaka C, Williams LT. Functional domains of axin. Importance of the C terminus as an oligomerization domain. The Journal of biological chemistry. 1999; 274:14090-14093.
62. Schwarz-Romond T, Fiedler M, Shibata N, Butler PJ, Kikuchi A, Higuchi Y, Bienz M. The DIX domain of Dishevelled confers Wnt signaling by dynamic polymerization. Nature structural & molecular biology. 2007; 14:484-492.
63. Chen T, Li M, Ding Y, Zhang LS, Xi Y, Pan WJ, Tao DL, Wang JY, Li L. Identification of zinc-finger BED domain-containing 3 (Zbed3) as a novel Axin-interacting protein that activates Wnt/beta-catenin signaling. The Journal of biological chemistry. 2009; 284:6683-6689.
64. Erter CE, Wilm TP, Basler N, Wright CV, Solnica-Krezel L. Wnt8 is required in lateral mesendodermal precursors for neural posteriorization in vivo. Development. 2001; 128:3571-3583.
65. Kim SE, Huang H, Zhao M, Zhang X, Zhang A, Semonov MV, MacDonald BT, Zhang X, Garcia Abreu J, Peng L, He X. Wnt stabilization of beta-catenin reveals principles for morphogen receptor-scaffold assemblies. Science. 2013; 340:867-870.
66. Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998; 280:596-599.
67. Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. The EMBO journal. 1998; 17:1371-1384.
68. Kimelman D, Xu W. beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene. 2006; 25:7482-7491.
69. Stamos JL, Weis WI. The beta-catenin destruction complex. Cold Spring Harbor perspectives in biology. 2013; 5:a007898.
70. Bilic J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M, Niehrs C. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science. 2007; 316:1619-1622.
71. Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, Okamura H, Woodgett J, He X. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature. 2005; 438:873-877.
72. Willert K, Shibamoto S, Nusse R. Wnt-induced dephosphorylation of axin releases beta-catenin from the axin complex. Genes & development. 1999; 13:1768-1773.
73. Cselenyi CS, Jernigan KK, Tahinci E, Thorne CA, Lee LA, Lee E. LRP6 transduces a canonical Wnt signal independently of Axin degradation by inhibiting GSK3's phosphorylation of beta-catenin. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105:8032-8037.
74. Piao S, Lee SH, Kim H, Yum S, Stamos JL, Xu Y, Lee SJ, Lee J, Oh S, Han JK, Park BJ, Weis WI, Ha NC. Direct inhibition of GSK3beta by the phosphorylated cytoplasmic domain of LRP6 in Wnt/beta-catenin signaling. PloS one. 2008; 3:e4046.
75. Wu G, Huang H, Garcia Abreu J, He X. Inhibition of GSK3 phosphorylation of beta-catenin via phosphorylated PPPSPXS motifs of Wnt coreceptor LRP6. PloS one. 2009; 4:e4926.
76. Wang S, Yin J, Chen D, Nie F, Song X, Fei C, Miao H, Jing C, Ma W, Wang L, Xie S, Li C, Zeng R, et al. Small-molecule modulation of Wnt signaling via modulating the Axin-LRP5/6 interaction. Nature chemical biology. 2013; 9:579-585.
77. Li L, Yuan H, Xie W, Mao J, Caruso AM, McMahon A, Sussman DJ, Wu D. Dishevelled proteins lead to two signaling pathways. Regulation of LEF-1 and c-Jun N-terminal kinase in mammalian cells. The Journal of biological chemistry. 1999; 274:129-134.
78. Habas R, Dawid IB, He X. Coactivation of Rac and Rho by Wnt/Frizzled signaling is required for vertebrate gastrulation. Genes & development. 2003; 17:295-309.