
INTRODUCTION
As a synthetic glutamic acid derivative, thalidomide was originally prescribed as a sedative and antinausea medicine in pregnant women, but was withdrawn from the market as severe birth defects were observed in patients receiving thalidomide [1, 2]. However, thalidomide has returned to clinical use in recent years for the treatment of several proinflammatory or autoimmune diseases since it was discovered to modulate immune and inflammatory responses [3, 4]. In addition, thalidomide has been demonstrated to be effective in the treatment of multiple myeloma (MM) presumably due to antiangiogenic activity or immune modulation [5]. However, its efficacy is largely limited by the increased incidence of thrombosis, especially when used in combination with other drugs, such as dexamethasone or doxorubicin [6, 7]. Meanwhile, addition of thalidomide has been demonstrated to increase the incidence of 2-year thrombosis from 1.5% to 18.5% in patients administered melphalan or prednisone [8].
Given the association of thalidomide with increased thrombotic risk [9], the precise mechanism by which thalidomide induces thrombogenesis in vivo remains poorly understood. Thrombotic manifestations observed in patients treated with thalidomide are usually venous; however arterial events have also been reported in some individuals [10]. Under physiological conditions, the vascular endothelium mediates vascular dilatation, prevents platelet adhesion and activation, blocks thrombin formation and mitigates fibrin deposition to maintain an anti-thrombotic state [11]. When the endothelium is activated or injured, disrupting these processes, this may lead to a prothrombotic state. A previous study reported that increased endothelial activation was observed in MM patients treated with thalidomide as demonstrated by elevated levels of von Willebrand factor [12], a surrogate marker of endothelial cell injury [13]. Furthermore, increased expression of phosphatidylserine, tissue factor and enhanced thrombin generation were observed in cultured human umbilical vein endothelial cells treated with thalidomide and plasma from MM patients [14], suggesting thalidomide potentially induces thrombogenesis by disrupting the balance between pro-coagulant and anticoagulant proteins on the surface of endothelial cells. Apart from the endothelium, aberrant activation of platelets was also reported in MM patients receiving thalidomide as shown by increased platelet surface P-selectin expression [15], PAC-1 binding [16] and platelet aggregation [17]. However, the exact role of thalidomide on platelet activation has not been thoroughly investigated, and whether thalidomide induces platelet activation directly or through other factors remains poorly understood. In this study, we investigate the potential role of thalidomide on platelet activation in vitro and in vivo, by directly treating isolated human or mouse platelets with thalidomide, and by analyzing platelets and coagulation markers following injection of thalidomide into mice. Together, our findings support a mechanism involving a predominantly procoagulant effect rather than a direct effect on platelets, with implications for the future clinical use of thalidomide.
RESULTS AND DISCUSSION
Several studies have demonstrated increased platelet activation in patients receiving thalidomide therapy [15, 16]. To investigate whether thalidomide was directly activating platelets, we treated washed platelets from healthy individuals with different concentrations of thalidomide (10, 50 and 100 μg/ml) in vitro and evaluated the extent of platelet activation. As shown in Figure 1A, there was no platelet activation observed following thalidomide treatment as compared with vehicle controls, demonstrated by no significant increase of expression of P-selectin on the platelet surface. In addition, platelet integrin αIIbβ3 activation (essential for platelet aggregation) was also measured and showed no increased PAC-1 binding to platelets treated with thalidomide (Figure 1B), indicating thalidomide did not lead to activation of platelet αIIbβ3 in vitro.
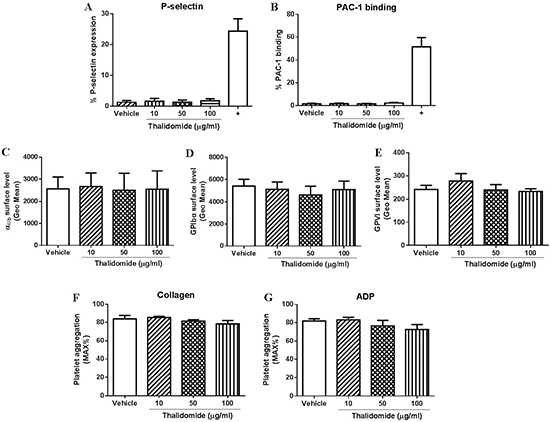
Figure 1: The effect of thalidomide on human platelets in vitro. Isolated human platelets (2.5 × 108/ml) were treated with 10, 50 or 100 μg/ml thalidomide for 1 h at 37°C followed by measuring (A) expression of P-selectin, (B) PAC-1 binding, or expression of (C) αIIb, (D) GPIbα or (E) GPVI by flow cytometry. In addition, platelet aggregation in citrated PRP after thalidomide treatment in response to threshold concentration of (F) collagen (2.5 μg/ml) or (G) ADP (5 μM) was also performed. Collagen (10 μg/ml) was used as a positive control (“+”) for platelet activation. All the data were acquired from at least three independent experiments using different donors.
Platelet receptors, such as GPIbα, GPVI or integrin αIIbβ3 play an important role in regulating platelet function and aberrant expression of these receptors has been shown to affect platelet thrombus formation [18, 19]. As seen in Figure 1C–1E, there was no significant changes in expression of αIIb, GPIbα or GPVI on platelets treated with thalidomide compared with vehicle controls when measured by flow cytometry, revealing thalidomide also does not alter the expression profiles of these important platelet receptors.
Consistent with the lack of effect of thalidomide on platelet activation or receptor expression, pre-treatment of platelets with different concentrations of thalidomide also showed no effect on collagen- or ADP-induced platelet aggregation at threshold concentrations in human citrated PRP (Figure 1F–1G). Further, consistent with these findings in human platelets, thalidomide-treated mouse platelets also displayed no sign of activation as evaluated by P-selectin expression or fibrinogen binding (Figure 2A–2B), while αIIb surface expression and collagen- or ADP-induced platelet aggregation at threshold concentrations were similarly unaffected (Figure 2C–2E). Taken together, these data demonstrate that thalidomide does not induce platelet activation in vitro under experimental conditions described.
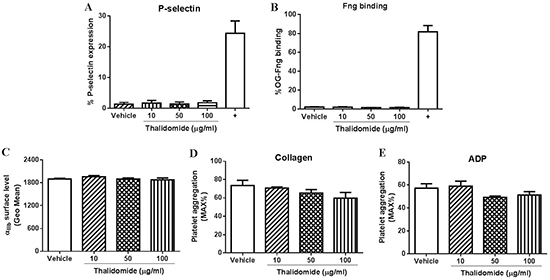
Figure 2: The effect of thalidomide on mouse platelets in vitro. Isolated mouse platelets (2.5 × 108/ml) were treated with indicated concentrations of thalidomide followed by measuring (A) expression of P-selectin, (B) fibrinogen (Fng) binding, or (C) expression of αIIb by flow cytometry. Platelet aggregation in mouse washed platelets after thalidomide treatment in response to threshold concentration of (D) collagen (1 μg/ml) or (E) ADP (10 μM) was performed in the presence of fibrinogen (0.5 mg/ml). CRP (10 μg/ml) was used as a positive control (“+”) for mouse platelet activation. All the data were acquired from at least three independent experiments.
In order to investigate whether thalidomide affects platelet activation in vivo, thalidomide was intraperitoneally injected into mice followed by collection of peripheral blood at 6 h after thalidomide administration. We found no significant changes of platelet count after thalidomide injection (Figure 3A), and no platelet activation or changes of αIIb surface levels (Figure 3B–3C), consistent with the in vitro findings. Interestingly, however, mice injected with thalidomide displayed a significantly shortened tail bleeding time compared to mice receiving vehicle alone (Figure 3D), suggesting thalidomide might affect coagulation in vivo. To further investigate this possibility, plasma was isolated from mice injected with thalidomide or vehicle and coagulation-related parameters were analysed. As shown in Figure 4, significantly increased levels of factor IX (FIX) (122.30 ± 10.76%) (P < 0.05) and fibrinogen (2.81 ± 0.30 g/l) (P < 0.01) were observed in mice after thalidomide administration compared with vehicle (90.06 ± 2.46% for FIX and 1.65 ± 0.10 g/l for fibrinogen). In addition, thrombin time (TT) (12.50 ± 0.60 s) (P < 0.05), prothrombin time (PT) (8.46 ± 0.09) (P < 0.05), PT-Ratio (0.74 ± 0.01) (P < 0.05) and normalized prothrombin time (PT-INR) (0.73 ± 0.01) (P < 0.05) of mice injected with thalidomide were significantly shortened compared with mice receiving vehicle (14.03 ± 0.32 s for TT, 8.83 ± 0.12 for PT, 0.77 ± 0.01 for PT-Ratio, and 0.77 ± 0.01 for PT-INR). Interestingly, thalidomide treatment did not alter the levels of coagulation factor VIII (FVIII), anti-thrombin III (ATIII) as well as the activated partial thrombin time (APTT) (Figure 4). Taken together, our study demonstrates that thalidomide induces increased pro-coagulant activity in mice, which was consistent with a previous study demonstrating increased expression of tissue factor and phosphatidylserine exposure on the surface of endothelial cells and monocytes by a thalidomide-based regimen [20].
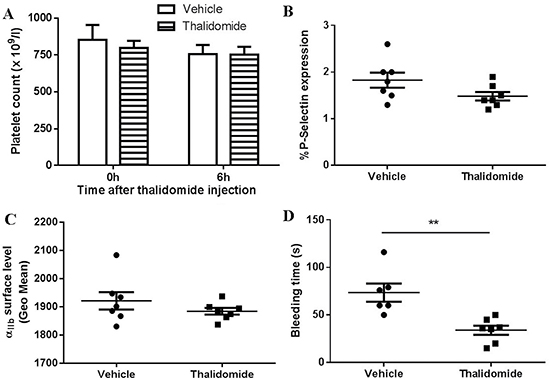
Figure 3: The effect of thalidomide on platelets in vivo. Thalidomide (200 mg/kg) was intraperitoneally injected into mice followed by collection of peripheral blood at 6 h after administration for analysis of (A) platelet count, (B) platelet surface P-selectin expression and (C) αIIb expression. (D) Tail bleeding time was also measured. **P < 0.01 (Unpaired student t-test). All the data were acquired from at least three independent experiments.
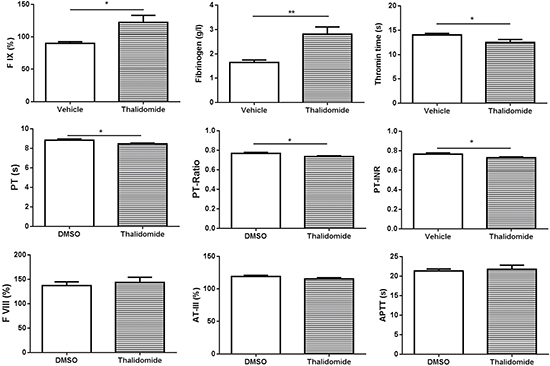
Figure 4: Analysis of coagulation parameters following thalidomide administration into mice. Plasma was isolated from mice after thalidomide administration for 6 h followed by analysis of the level of factor IX (FIX), fibrinogen, thrombin time (TT), prothrombin time (PT), PT-Ratio, PT-INR, FVIII, AT-III, and APTT using an automated coagulation analyzer. *P < 0.05 (Unpaired student t-test). **P < 0.01 (Unpaired student t-test). PT-INR: Prothrombin Time-International Normalized Ratio; APTT: Activated Partial Thrombin Time; AT: Anti-thrombin.
In conclusion, this study demonstrates that thalidomide dose not trigger platelet activation in vitro or in vivo. An important limitation of our study is that it used platelet samples from healthy humans and wildtype mice not afflicted with disease. Thus, increased platelet activation in patients receiving thalidomide therapy might be due to the underlying disease, for example, making platelets more susceptible to activation, or an indirect effect of coagulopathy. Further studies using platelets isolated from patients with active myeloma disease are required to confirm that thalidomide treatment does not directly affect platelet function. A further possibility not excluded by our results is that thalidomide treatment indirectly leads to platelet hyperreactivity by a mechanism involving activation of endothelial cells. However, further studies are required to confirm these hypotheses.
Whilst the thalidomide/thrombosis mechanistic link has not been fully elucidated here, the data obtained in our study highlight that thalidomide does not trigger or enhance platelet reactivity. That is, our data indicate that the effect is not via direct influence on platelet reactivity or disruption of normal platelet responsiveness. Further, our data suggest that thalidomide treatment is able to modulate the procoagulant state. This is potentially significant for the clinical use of thalidomide as an anti-inflammatory therapeutic and in the setting of neoplasia, particularly when anti-coagulant/antiplatelet therapies form part of the clinical management of patients with complex disease aetiologies. Our data, for example, may be relevant for management of prophylactic anticoagulation in patients with myeloma receiving thalidomide as part of their treatment regime.
MATERIALS AND METHODS
Reagents
Thalidomide was purchased from Changzhou Pharmaceutical Factory Co., Ltd (Changzhou, Jiangsu, China). Collagen and ADP were from Helena laboratories (Beaumont, Texas, USA). Collagen-related peptide (CRP) was prepared as previously described [21]. FITC-conjugated anti-human CD41a and PAC-1 antibody were from BD Biosciences (San Jose, CA) and Becton Dickinson (San Jose, CA) respectively. PE-conjugated anti-human/mouse CD62p (P-Selectin) and purified anti-human Glycoprotein (GP) VI antibody were purchased from eBioscience (San Diego, CA). FITC-conjugated anti-CD42b antibody was from Abcam (Cambridge, MA). FITC-conjugated goat anti-mouse IgG was from ZSGB-BIO (Beijing, China). Oregon Green-labelled fibrinogen (OG-Fng) was from Thermo Fisher Scientific (Waltham, MA).
Platelet preparation
All procedures involving collection of human and mouse blood were approved by the Medical Ethics Committee of Xuzhou Medical University. Informed consent has been obtained and all clinical investigations have been conducted in accordance with the ethical standards and according to the principles expressed in the Declaration of Helsinki. Human or mouse platelets were isolated as previously described [22, 23]. Briefly, venous human blood was collected into trisodium citrate and centrifuged to obtain platelet-rich plasma (PRP). For washed platelet studies, blood was collected into acid-citrate-dextrose (ACD) and centrifuged to get PRP. Platelet were washed three times in citrate-glucose-saline (CGS) and resuspended in Tyrode’s buffer. Mouse platelets were isolated from ACD anti-coagulated blood, washed using CGS buffer and resuspended in Tyrode’s buffer.
Platelet activation
Platelet activation was evaluated by measuring the surface expression of the α-granule glycoprotein, P-selectin, and by the activation-dependent binding of either PAC-1 (human) or fibrinogen (mouse) to platelet αIIbβ3. Isolated human or mouse platelets (2.5 × 108/ml) were incubated with different concentrations of thalidomide (10, 50 or 100 μg/ml) according to published studies [24, 25] for 60 min at 37°C followed by addition of PE-conjugated anti-P-selectin antibody (2.5 μg/ml), FITC-conjugated PAC-1 antibody (1 μg/ml), or Oregon Green-labelled fibrinogen and analysed by flow cytometry. Collagen (10 μg/ml) and CRP (10 μg/ml) were used as a positive control for human and mouse platelet activation, respectively.
Surface expression of platelet receptors
Washed platelets were untreated or treated with thalidomide for 60 min at 37°C, then FITC-conjugated anti-CD42b antibody (GPIbα) (1 μg/ml), FITC-conjugated mouse anti-human CD41a antibody (αIIb) (10 μl/test) or anti-human GPVI antibody (2.5 μg/ml) (detected by FITC-conjugated goat anti-mouse IgG) were added and incubated for 30 min at room temperature and analysed by flow cytometry. FITC-conjugated anti-mouse CD41a antibody (2 μg/ml) was used for analysing αIIb surface expression on mouse platelets.
Platelet aggregation
Platelet aggregation was performed using human citrated PRP or mouse washed platelets. After thalidomide treatment as described above, human or mouse platelet aggregation (250 × 106/ml) in response to collagen or adenosine diphosphate (ADP) at threshold concentrations (defined as the lowest dose that induced more that 50% of aggregation ) was determined by light transmittance aggregometry (Helena Aggram, Helena Laboratories, Beaumont, USA) as previously described [22]. The extent of platelet aggregation was defined as the percentage of maximum platelet aggregation (monitored for 5 min) determined in the absence of drug. Mouse washed platelet aggregation was performed in the presence of fibrinogen (0.5 mg/ml).
In vivo studies in mice
BALB/c mice, aged 10–12 weeks and weighted 20–25 g, were purchased from SLAC Laboratory Animal, Shanghai, China. The mice were housed in sterilized cages at the Experimental Animal Center of Xuzhou Medical University. The experiments on animals were performed in accordance with the Xuzhou Medical University concerned with the ethics of experimentation. Thalidomide (200 mg/kg) was intraperitoneally injected into mice followed by collecting peripheral blood after 6 h for analysis of platelet count by an automatic blood cell analyzer (BC5300, Mindray, Shenzhen, Guangdong, China), and platelet surface expression of P-selectin, platelet receptor αIIb expression by flow cytometry as described above. An equal volume of dimethyl sulfoxide (DMSO) was used as vehicle only control.
Tail bleeding time
At 6 h following thalidomide or DMSO administration, a 10-mm segment of tail tip of each mouse was cut off and the tail was immersed in pre-warmed sterile saline solution (37°C). Tail bleeding time was defined as the time taken for bleeding to stop.
Coagulation analysis
Plasma was isolated from mice treated with thalidomide or DMSO for analysis of the level of factor IX factor VIII, anti-thrombin III, and fibrinogen, and for determining the thrombin time, activated partial thrombin time and prothrombin time using an automated coagulation analyzer (Sysmex CS-5100).
Statistical analysis
Data were represented as mean ± standard error (SE). At least three independent experiments were performed for each assay. For comparison between vehicle and different concentrations of thalidomide, data were assessed by one-way ANOVA with Tukey multiple comparison post-hoc analysis using GraphPad Prism software. Two tailed student t-test was used to evaluate the difference between vehicle and thalidomide-injected mouse. P < 0.05 indicates statistically significance.
Abbreviations
MM: multiple myeloma; CRP: collagen-related peptide; ADP: adenosine diphosphate; OG-Fng: Oregon Green-labelled fibrinogen; PRP: platelet-rich plasma.
Authors′ contributions
J. Qiao designed the study, performed the experiments, analyzed and interpreted data and wrote the manuscript. Y. Wu, X. Wu, Y. Liu, X. Li, W. Ju, K. Qi and D. Li performed the experiments and analyzed data. E. E. Gardiner and R. K. Andrews provided intellectual input. J. Qiao, L. Zeng and K. Xu conceived, designed the study and co-wrote the manuscript.
CONFLICTS OF INTEREST
All authors have no conflicts of interest to declare.
FUNDING
This research was supported by National Natural Science Foundation of China (grant no. 81400082 and 81370602), the Natural Science Foundation of Jiangsu Province (grant no. BK20140219), China Postdoctoral Science Foundation funded project (project no. 2015M570479), the funding for the Distinguished Professorship Program of Jiangsu Province, the Six Talent Peaks Project of Jiangsu Province (project no. WSN-133), the Shuangchuang Project of Jiangsu Province, the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, and the Science and Technology Foundation for the Selected Overseas Chinese Scholars, State Ministry of Human Resources and Social Security.
REFERENCES
1. Lenz W, Knapp K. Thalidomide embryopathy. Arch Environ Health. 1962; 5:100–105.
2. Vargesson N. Thalidomide-induced teratogenesis: history and mechanisms. Birth Defects Res C Embryo Today. 2015; 105:140–156.
3. Zhu YX, Kortuem KM, Stewart AK. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk Lymphoma. 2013; 54:683–687.
4. Matthews SJ, McCoy C. Thalidomide: a review of approved and investigational uses. Clin Ther. 2003; 25:342–395.
5. Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, Munshi N, Anaissie E, Wilson C, Dhodapkar M, Zeddis J, Barlogie B. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999; 341:1565–1571.
6. Rajkumar SV. Thalidomide therapy and deep venous thrombosis in multiple myeloma. Mayo Clin Proc. 2005; 80:1549–1551.
7. Barbui T, Falanga A. Thalidomide and thrombosis in multiple myeloma. J Thromb Haemost. 2003; 1:421–422.
8. Palumbo A, Rus C, Zeldis JB, Rodeghiero F, Boccadoro M. Enoxaparin or aspirin for the prevention of recurrent thromboembolism in newly diagnosed myeloma patients treated with melphalan and prednisone plus thalidomide or lenalidomide. J Thromb Haemost. 2006; 4:1842–1845.
9. El Accaoui RN, Shamseddeen WA, Taher AT. Thalidomide and thrombosis. A meta-analysis. Thromb Haemost. 2007; 97:1031–1036.
10. Scarpace SL, Hahn T, Roy H, Brown K, Paplham P, Chanan-Khan A, van Besien K, McCarthy PL Jr. Arterial thrombosis in four patients treated with thalidomide. Leuk Lymphoma. 2005; 46:239–242.
11. Becker BF, Heindl B, Kupatt C, Zahler S. Endothelial function and hemostasis. Z Kardiol. 2000; 89:160–167.
12. Minnema MC, Fijnheer R, De Groot PG, Lokhorst HM. Extremely high levels of von Willebrand factor antigen and of procoagulant factor VIII found in multiple myeloma patients are associated with activity status but not with thalidomide treatment. J Thromb Haemost. 2003; 1:445–449.
13. Vischer UM. von Willebrand factor, endothelial dysfunction, and cardiovascular disease. J Thromb Haemost. 2006; 4:1186–1193.
14. Gao Y, Ma G, Liu S, Teng Y, Wang Y, Su Y. Thalidomide and multiple myeloma serum synergistically induce a hemostatic imbalance in endothelial cells in vitro. Thromb Res. 2015; 135:1154–1159.
15. Dunkley S, Gaudry L. Thalidomide causes platelet activation, which can be abrogated by aspirin. J Thromb Haemost. 2007; 5:1323–1325.
16. Abdullah WZ, Roshan TM, Hussin A, Zain WS, Abdullah D. Increased PAC-1 expression among patients with multiple myeloma on concurrent thalidomide and warfarin. Blood Coagul Fibrinolysis. 2013; 24:893–895.
17. Baz R, Li L, Kottke-Marchant K, Srkalovic G, McGowan B, Yiannaki E, Karam MA, Faiman B, Jawde RA, Andresen S, Zeldis J, Hussein MA. The role of aspirin in the prevention of thrombotic complications of thalidomide and anthracycline-based chemotherapy for multiple myeloma. Mayo Clin Proc. 2005; 80:1568–1574.
18. Rivera J, Lozano ML, Navarro-Nunez L, Vicente V. Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica. 2009; 94:700–711.
19. Swieringa F, Kuijpers MJ, Heemskerk JW, van der Meijden PE. Targeting platelet receptor function in thrombus formation: the risk of bleeding. Blood Rev. 2014; 28:9–21.
20. Hoshi A, Matsumoto A, Chung J, Isozumi Y, Koyama T. Activation of coagulation by a thalidomide-based regimen. Blood Coagul Fibrinolysis. 2011; 22:532–540.
21. Arthur JF, Qiao J, Shen Y, Davis AK, Dunne E, Berndt MC, Gardiner EE, Andrews RK. ITAM receptor-mediated generation of reactive oxygen species in human platelets occurs via Syk-dependent and Syk-independent pathways. J Thromb Haemost. 2012; 10:1133–1141.
22. Qiao J, Wu Y, Liu Y, Li X, Wu X, Liu N, Zhu F, Qi K, Cheng H, Li D, Li H, Li Z, Zeng L, et al. Busulfan Triggers Intrinsic Mitochondrial-Dependent Platelet Apoptosis Independent of Platelet Activation. Biol Blood Marrow Transplant. 2016; 22:1565–1572.
23. Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ, Josefsson EC, Alwis I, Ono A, Willcox A, Andrews RK, Mason KD, Salem HH, et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood. 2011; 118:1663–1674.
24. DeCicco KL, Tanaka T, Andreola F, De Luca LM. The effect of thalidomide on non-small cell lung cancer (NSCLC) cell lines: possible involvement in the PPARgamma pathway. Carcinogenesis. 2004; 25:1805–1812.
25. Li Y, Jiang Z, Xiao Y, Li L, Gao Y. Metabolism of thalidomide by human liver microsome cytochrome CYP2C19 is required for its antimyeloma and antiangiogenic activities in vitro. Hematol Oncol. 2012; 30:13–21.