
INTRODUCTION
Prostate cancer is the most common cancer in men and targeting androgen receptor (AR) is an effective treatment. In the clinical setting, inhibition of AR activity frequently leads to the development of castration-resistant prostate cancer, a condition with no curative treatment options [1, 2]. AR signaling remains active in castration-resistant disease, and a recent study used chromatin immunoprecipitation (ChIP) coupled to sequencing (seq), to identify a gene signature of putative AR-target genes in this lethal form of cancer [3]. Among this signature was a poorly characterized enzyme, Enoyl-CoA delta isomerase 2 (ECI2). ECI2 has been purified, and using enzymatic assays, was shown to catalyze isomerization of lipids [4]. Lipid metabolism is of high interest in prostate cancer, since it offers a route to intra-tumoral steroid-hormone synthesis after or during androgen deprivation therapy [5]. The importance of steroidogenesis is further underlined by the anti-androgen abiraterone, which targets steroid metabolism and offers clinical benefit for castration-resistant prostate cancer patients [6].
The metabolic status of the normal prostate is different from the rest of the body due to the incomplete TCA cycle and secretion of citrate, a TCA metabolite, important for the viability of sperm [7]. In the early stages of prostate cancer, citrate secretion decreases, accompanied by a decrease in aerobic glycolysis, which ‘masks’ tumors from the conventional tracers of proliferating cells, such as glucose-PET [8, 9]. Lipid metabolizing enzymes are up-regulated in the early phase of prostate cancer and remain elevated throughout the disease progression, which suggests an active role in the disease [9]. In agreement with this, statin intake to lower blood cholesterol levels is associated with prostate cancer prevention [10]. The importance of lipid metabolism is exemplified further by the lipid producing enzyme, fatty acid synthase, which is over-expressed during prostate cancer progression and can be targeted with Orlistat, a drug approved for the treatment of obesity [11, 12].
Since AR activity and lipid metabolism appear important in prostate cancer, here we investigate ECI2, an AR-target gene. We show that ECI2 is over-expressed in prostate cancer patient samples in mRNA and protein levels, and that increased expression predicts poor outcome. Knockdown of ECI2 had minimal effect on gene expression in a cell line representing normal prostate epithelium but resulted in down-regulation of cell cycle-associated genes in prostate cancer cells. The difference in gene expression translated to growth inhibition of prostate cancer cells, but had only modest effect on a cell line representing normal prostate epithelium. Integration of the RNA-seq data with metabolomics data revealed that inhibition of ECI2 expression led to acute metabolic stress in prostate cancer cells.
RESULTS
ECI2 is over-expressed in clinical prostate cancer
ECI2 was identified as a putative AR target gene in castration-resistant prostate cancer tissue samples using ChIP-seq technology [3]. As the first step, we evaluated ECI2 expression in matched benign and prostate cancer patient tissue samples, and observed a 2-fold increased expression of ECI2 mRNA (p = 0.024, Figure 1A). Encouraged by this result, we evaluated ECI2 protein level expression using immunohistochemistry, and found out that elevated ECI2 protein predicted mortality (p = 0.0086, Figure 1B, see also Supplementary Figure 1).
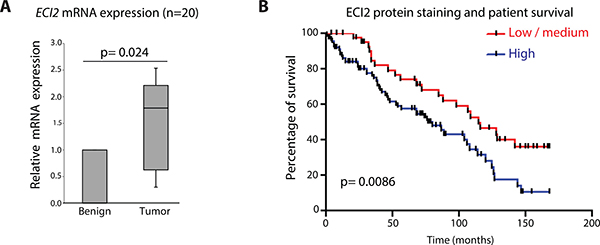
Figure 1: Enoyl-CoA delta isomerase 2 (ECI2) is over-expressed in prostate cancer. (A) ECI2 expression was evaluated in prostate cancer tissue samples. The data shown represents matched normal epithelium and adenocarcinoma from 20 radical prostatectomy specimens. Relative expression of the different transcripts were calculated using the comparative CT method, where the matched benign tissue of the same patient were set to 1 and normalized to the geometric mean CT value of GAPDH, TBP and 18s. Wilcoxon matched-pairs signed rank test was used to test for significance in the differential expression of ECI2 between the matched benign and cancer tissue. (B) Kaplan Meier curves for the low/medium group versus the high ECI2 expressing group. We evaluated whether ECI2 expression levels are associated with survival in prostate cancer patients. The difference in overall survival between the low/medium expressing group and high expressing group was 77 months vs 115 months, p = 0.0086. Here stating that an overview of the clinical cohorts use in Figures 1A and 1B and the statistical analysis are to be found in Supplementary Tables 2, 4 and 5.
Since ECI2 was over-expressed in prostate cancer patient samples, we moved on to assess AR-dependent regulation of this gene. We re-analyzed AR ChIP-seq data from human tissue samples [3], and putative AR-binding site in castration-resistant prostate cancer patient samples was found inside the ECI2 gene body (chromosome coordinates in Human Genome 18: chr6:4,075,826-4,076,114). In order to confirm these data, we designed primers against this site, and assessed potential AR binding using ChIP-qPCR. Androgen-stimulation resulted in 6-fold increased AR binding to this site, once compared to vehicle and an IgG antibody control (Figure 2A). We next confirmed that androgen stimulation increases ECI2 expression at the mRNA and protein levels in LNCaP and VCaP cells (Figure 2B and 2C). Information on the primers and probes used in this study for ChIP-qPCR and RT-PCR are to be found in Supplementary Table 3 and more detailed methodology is provided in Supplementary Materials.
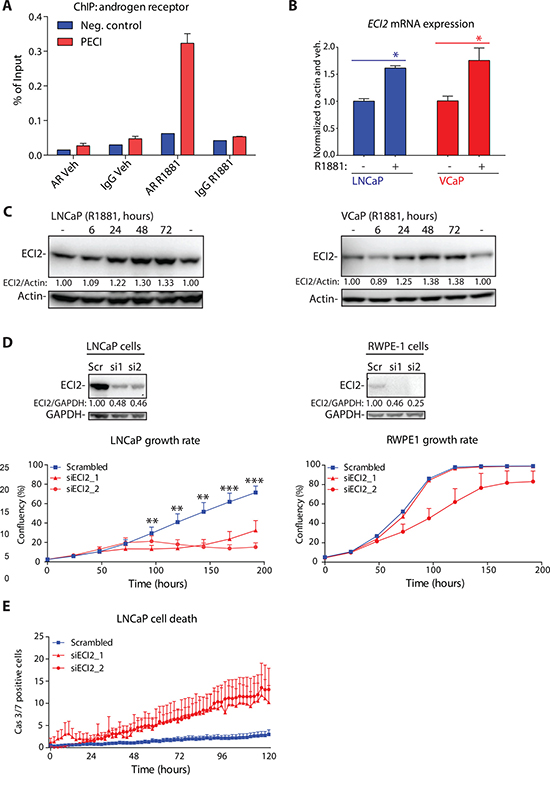
Figure 2: Androgen receptor (AR) regulates Enoyl-CoA delta isomerase 2 (ECI2) expression. (A) Chromatin immunoprecipitation (ChIP) of androgen receptor (AR) in VCaP cells. Cells were deprived of androgens for 3 days and treated either with 1nM R1881 or vehicle, as indicated. The putative AR binding site for ECI2 was identified from a published AR ChIP-seq data set [3]. The data shown is representative of two biological replicates. (B) LNCaP and VCaP cells were treated as in A. Total mRNA was isolated at 12 hours and the expression of ECI2 and actin was evaluated using RT-qPCR. The data shown are an average of three independent experiments with SEM, and significance was evaluated using paired samples Student’s t-test, *< 0.05. (C) LNCaP and VCaP cells were deprived of androgens for 3 days, treated either with 1nM R1881 or vehicle (-) and protein lysates were collected. Densitometry was used to evaluate ECI2 levels. (D) Western blot was used to confirm ECI2 knockdown in LNCaP and RWPE-1 cells after 96 hours. Densitometry was used to evaluate ECI2 levels. The growth rate of cells was evaluated with life-cell imaging. The data shown are an average of three independent biological replicates with SEM. The significance was evaluated using paired samples Student’s t-test, **< 0.01, ***< 0.001. (E) Cell death activation after ECI2 knockdown. LNCaP cells were reverse-transfected and allowed to attach for 24 hours. At this point, a dye detecting caspase 3/7 activation was added and cumulative activation of caspase 3/7 was followed in real-time using life-cell imaging. Data shown is an average of three biological replicates with SEM.
Since ECI2 was over-expressed in prostate cancer patient samples, we tested whether the enzyme is important for prostate cancer cell growth by following cell proliferation using life-cell imaging. ECI2 knockdown strongly inhibited proliferation of LNCaP prostate cancer cell line but had only modest effects on RWPE-1, a cell line derived from normal prostate epithelium (Figure 2D). In addition, knockdown of ECI2 activated cell death response in LNCaP cells (Figure 2E).
These data suggest that ECI2 has a role in promoting the proliferation of prostate cancer cells.
Metabolomic profiling after ECI2 knockdown revealed profound changes in lipid composition
ECI2 has been reported to function in lipid processing and the enzyme has been shown to isomerize 3-cis-octenoyl-CoA to 2-trans-octenoyl-CoA [4] (Figure 3A). This isomerization reaction is important for the subsequent degradation of unsaturated lipids [4]. Degradation maintains correct lipid homeostasis, supports citric acid cycle by production of acetyl-CoA and replenishes NADH and FADH pools.
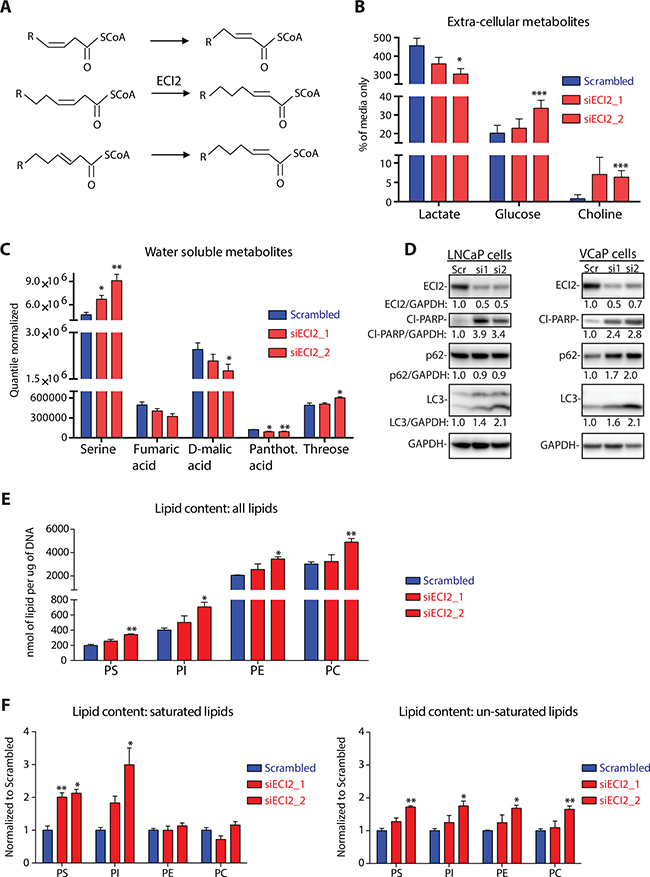
Figure 3: Metabolomic profiling after ECI2 knockdown in LNCaP cells. (A) Reaction catalyzed by Enoyl-CoA Delta Isomerase 2 (ECI2). (B) The level of extra-cellular lactate, glucose and choline were determined using nuclear magnetic resonance after 72 hours of ECI2 knockdown. The data are presented as % of complete media without cells and are an average (with SEM) of five biological replicates. The significance was evaluated using paired samples Student’s t-test, *< 0.05, ***< 0.001. (C) The levels of water-soluble intra-cellular metabolites were determined using mass-spectrometry after 72 hours of ECI2 knockdown. Only metabolites that were affected significantly by at least one siRNA are shown. Data shown are an average (with SEM) of five biological replicates. The significance was evaluated using paired samples Student’s t-test, *< 0.05, **< 0.01. (D) ECI2 knockdown in LNCaP (for 72 hours) and VCaP cells (for 96 hours). The data shown are representative of at least three biological replicates for LNCaP cells and two replicates for VCaP cells. Densitometry was used to evaluate signal intensity. (E) The levels of intra-cellular phosphatidylserine (PS), phosphatidylcholine (PC), phosphatidylethanolamine (PE) and phosphatidylinositol (PI) were determined using mass-spectrometry after 72 hours of ECI2 knockdown. Identification of lipid composition is provided in Supplementary Figure 2. Data shown are an average (with SEM) of three biological replicates. The significance was evaluated using paired samples Student’s t-test, *< 0.05, **< 0.01. (F) The levels of saturated and un-saturated lipids were determined using mass-spectrometry after 72 hours of ECI2 knockdown (same data-set as presented in Figure 3E). Scrambled samples were set to 1 for all four lipid classes, and siECI2 samples were normalized to this. Data shown are an average (with SEM) of three biological replicates. The significance was evaluated using paired samples Student’s t-test, *< 0.05, **< 0.01.
ECI2 knockdown decreased glucose consumption and lactate production, as measured from cell culture media (Figure 3B). In order to get a clearer picture of the prostate cancer cell metabolome after ECI2 knockdown, we used mass spectrometry-based untargeted metabolite profiling of intra-cellular metabolites. This approach revealed decrease in TCA cycle metabolites fumarate and malate, while serine accumulated (Figure 3C).
These changes suggest that ECI2 knockdown imposes an acute metabolic stress on cells. Therefore we would expect stress-resistance promoting pathways, such as autophagy, to be up-regulated [13]. ECI2 knockdown induced prominent accumulation of the canonical autophagy marker LC3 [14], in two prostate cancer cell lines, LNCaP and VCaP (Figure 3D). However, p62, an adaptor protein for autophagy [14], was not degraded, which suggests incomplete autophagy and sustained stress. Indeed, prostate cancer cells were unable to respond to ECI2 knockdown induced metabolic stress, which resulted in cell death activation (PARP cleavage, Figure 3D).
The major task for ECI2 enzyme is believed to be lipid processing [4], and we observed choline accumulation in culture media after ECI2 knockdown (Figure 3B). Based on these facts, we performed targeted phospholipidomic profiling of LNCaP prostate cancer cells after ECI2 knockdown, which revealed accumulation of all major classes of phospholipids, phosphatidylserine (PS), phosphatidylcholine (PC), phosphatidylethanolamine (PE) and phosphatidylinositol (PI) (Figure 3E and Supplementary Figure 2). ECI2 knockdown had prominent effect on the saturated lipids of PS and PI (Figure 3F). However, this lipid pool represents a minor fraction of total cellular lipids, and in this context it is important to point out that the most abundant lipids in scrambled condition were profoundly increased upon ECI2 knockdown (Supplementary Figure 2). For example lipids with single double-bond, especially 36:1, were prominently increased in all four lipid classes.
We next used Oil Red O staining to confirm a statistically significant increase of lipids (Supplementary Figure 3A). Based on Oil Red O staining, we noted possible accumulation of lipid droplets, and this was confirmed with AdipoRed stain (Supplementary Figure 3B).
We have so far shown that ECI2 knockdown inhibits cancer cell proliferation, which is associated with decreased glucose consumption and lipid accumulation. This caused acute stress and cell death activation, but exact mechanism(s) remain unclear.
ECI2 knockdown represses cell cycle associated genes
In order to better understand metabolomics data, we performed RNA-sequencing after ECI2 knockdown in the LNCaP prostate cancer cell line and RWPE-1 cell line, which represents normal prostate epithelial cells. In order to set stringent criteria to establish ECI2-dependent transcripts, we performed RNA-seq on two biological replicates and selected only the transcripts that were affected by both of the two siRNAs targeting ECI2. A summary of the differentially expressed genes in each cell-line and in each biological replicate is provided in Supplementary Table 6. Based on this approach, the expression of 4 genes was altered in RWPE-1 cells and the expression of 102 genes was altered in LNCaP cells after ECI2 knockdown at the selected 48 hour time point (Figure 4A). We first inspected the individual genes differently regulated in RWPE-1 and LNCaP cells. Interestingly, RWPE-1 but not LNCaP cells, increased the expression of stress-resistance factor HSPB1 (heat shock protein family B (small) member 1) (Supplementary Table 1), which is known to protect cells from stress-induced apoptosis [15]. On the other hand, we found a number of genes known to be over-expressed in prostate cancer and promote prostate cancer cell survival, which were profoundly down-regulated only in LNCaP cells (Supplementary Table 1). Two examples are MELK (Maternal Embryonic Leucine Zipper Kinase) [16] and WASF3 (WAS Protein Family Member 3) [17, 18]. These data support the hypothesis that ECI2 has more important function(s) in prostate cancer cells.
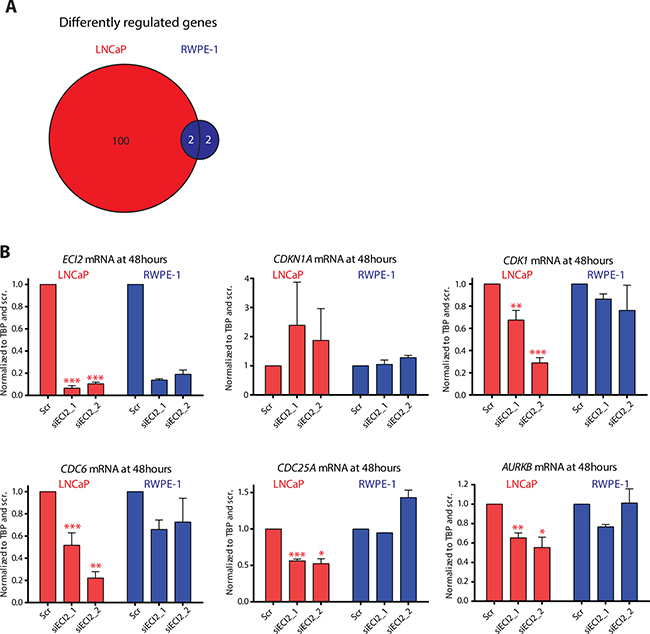
Figure 4: RNA-seq after ECI2 knockdown in LNCaP and RWPE-1 cells. The expression of ECI2 was reduced by treating LNCaP and RWPE-1 cells for 48 hours with siRNA and RNA was collected and used for RNA-seq. (A) Venn diagram shows the number of genes that were differentially regulated by both siRNAs in either LNCaP or RWPE-1 cells, and regulated differentially between the two cell lines. (B) Validation of the RNA-seq data using RT-qPCR. The data shown are an average of at least two biological replicates for both RNA-seq and validation, and the significance was evaluated using paired samples Student’s t-test, *< 0.05, **< 0.01, ***< 0.001.
In order to understand potential transcriptional programs of how prostate cancer cells respond to knockdown of ECI2, we next focused on the genes that were up-regulated after ECI2 knockdown only in LNCaP cells and performed pathway enrichment analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID) [19, 20]. This analysis highlighted two pathways, ‘p53 signaling pathway’ and ‘Chronic myeloid leukemia’, with one common gene, CDKN1A (Supplementary Table 1). CDKN1A functions as a cyclin-dependent kinase inhibitor [21]. We next performed the pathway enrichment analysis with the genes that were down-regulated in LNCaP cells after ECI2 knockdown. Four pathways were highlighted, and the most significant one was ‘Cell cycle’, through decreased expression of CDK1, CDC6, MAD2L1, CCND3, CDKN2C, CDC25A, WEE1 and CDC25B (Supplementary Table 1). We used RT-qPCR to confirm the statistically significant differential expression of CDKN1A, CDK1, CDC6, CDC25A and AURKB in LNCaP and RWPE-1 cells after ECI2 knockdown (Figure 4B). ECI2 knockdown also statistically significantly decreased the expression of CDC6 in VCaP cells (Supplementary Figure 4).
RNA-seq and subsequent validation revealed that CDC6 is one of the most prominently down-regulated genes in both prostate cancer cell lines. Potential correlation between CDC6 expression and prostate cancer aggressiveness has not been previously evaluated, but this protein is normally found in proliferating cells [22]. We therefore evaluated the correlation between elevated CDC6 expression and metastasis in a cohort of high-risk prostate cancer patients [23]. In this cohort, increased CDC6 expression predicted metastasis (p = 7.41e-04), and was also prominently associated with prostate cancer-specific mortality (p = 1.09e-02, Supplementary Figure 5).
So far, we have identified a gene, ECI2, for which knockdown inhibited prostate cancer cell proliferation and in addition suppressed expression of CDC6. Elevated expression of CDC6 is associated with poor patient outcome. Based on these data, ECI2, or more generally, lipid degradation, might represent a novel drug target to limit prostate cancer cell proliferation.
Targeting lipid degradation inhibits prostate cancer cell proliferation
There are no drugs directly inhibiting ECI2 activity but small molecule inhibitors targeting lipid degradation are available. Some of these compounds, including perhexiline, are available clinically [24]. Perhexiline is used to inhibit degradation of lipids for energy production, and thereby to promote more oxygen-efficient utilization of glucose as an energy source in chronic ischemic cardiomyopathy [25]. Based on our metabolomics and RNA-seq data, prostate cancer cells are unable to increase glucose utilization once lipid degradation is inhibited. Instead, an acute metabolic stress is induced, which culminates in cell death response activation (Figure 3D). We hypothesized that perhexiline might be able to cause similar response in prostate cancer cells.
We first confirmed that perhexiline treatment leads to dose-dependent lipid accumulation, similar to that observed upon ECI2 knockdown (Figure 5A). This lipid accumulation was mirrored by concomitant decrease in prostate cancer cell proliferation (Figure 5B). Similar to ECI2 knockdown, prostate cancer cells responded by activation of incomplete autophagy, as measured by prominent accumulation of autophagy markers LC3 and adaptor protein p62 [14] (Figure 5C). Prostate cancer cells were unable to respond to perhexiline-induced metabolic stress, and higher doses induced cell-death, as measured by PARP cleavage and activation of caspases 3/7 (Figure 5C and 5D). In addition, this compound let to prominent down-regulation of CDC6 (Supplementary Figure 6).
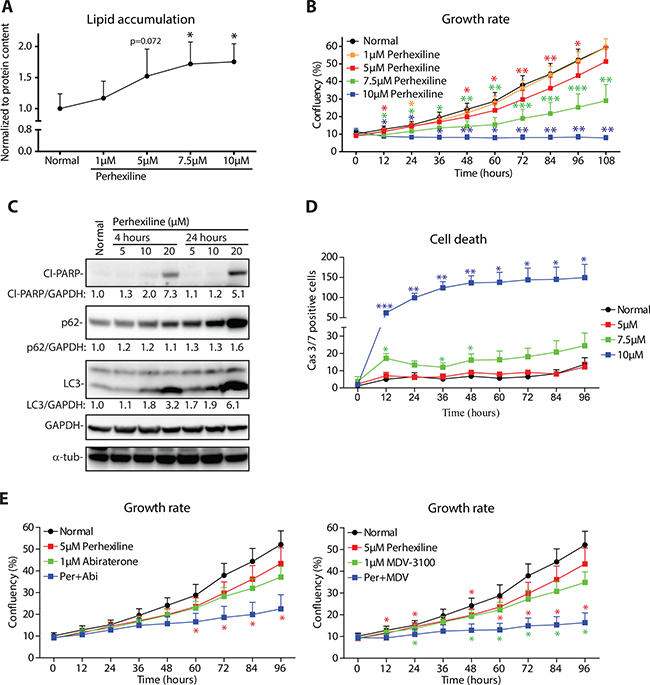
Figure 5: Lipid degradation inhibitor perhexiline activates cell death in prostate cancer cells. (A) Oil red O staining of LNCaP cells after 24 hours of perhexiline treatment. Data shown are an average (with SEM) of five biological replicates. The significance was evaluated using paired samples Student’s t-test, *< 0.05. (B) LNCaP cells were treated with increasing doses of perhexiline and growth rate was followed using life-cell imaging. Data shown is an average of four biological replicates with SEM. Significance was evaluated using paired samples Student’s t-test, *< 0.05, **< 0.01, ***< 0.001. (C) LNCaP cells were treated with perhexiline for 4 hours or 24 hours, and protein lysates were collected for western blotting. The data shown are representative of three biological replicates. Densitometry was used to evaluate signal intensity. (D) LNCaP cells were treated with perhexiline as indicated and cell death activation was evaluated using life-cell imaging detecting activation of caspases 3 and 7. Data shown is an average of four biological replicates with SEM. Significance was evaluated with paired samples Student’s t-test, *< 0.05, **< 0.01. (E) LNCaP cells were treated with perhexiline alone or in combination with androgen deprivation therapy (either Abiraterone or MDV-3100) and growth rate was followed using life-cell imaging. Data shown are an average of four biological replicates with SEM. Significance was evaluated using paired samples Student’s t-test, *< 0.05. Red stars indicate comparison between perhexiline and combinatorial treatment, while green stars indicate comparison between androgen deprivation and combinatorial treatment.
These data suggest that perhexiline could have potential in limiting the proliferation of prostate cancer cells, but one of the challenges in utilizing perhexiline in clinical setting are ‘poor-metabolizers’, patients who have altered activity of CYP2D6 (cytochrome P450 family 2 subfamily D member 6) [26]. This problem can be overcome by dose-reduction and/or genetic testing [27]. The dose-limitation challenge motivated us to evaluate whether combinatorial treatment with perhexiline and anti-androgens would allow dose reduction. For these experiments, we selected a low dose of perhexiline (5 μM), which on its own led to prominent increase in intracellular lipid content but only modestly inhibited proliferation (Figure 5A and 5B). Interestingly, combinatorial treatment of LNCaP cells with perhexiline and either Abiraterone or Enzalutamide (MDV-3100) almost completely blocked proliferation (Figure 5E).
Taken together, these data suggest that a novel AR target gene, ECI2, supports aberrant metabolic homeostasis of prostate cancer cells. Inhibition of lipid degradation either by knocking down this enzyme or by a small molecule inhibitor leads to metabolic stress and activation of the cell death response.
DISCUSSION
Prostate cancer is the most common cancer in men, and increased AR activity is associated with prostate cancer development and progression. AR-driven complex transcriptional programs have previously been used as a starting point to identify prostate cancer-specific metabolic vulnerabilities [28, 29]. In this study, we showed that ECI2, an enzyme involved in degradation of unsaturated lipids, is a direct AR-target and over-expressed in clinical prostate cancer (Figures 1 and 2). Both ECI2 and CDC6 can be induced by AR (this paper and Mallik & al. 2008 [30], respectively). Inhibition of ECI2 expression decreased the growth rate and activated cell death response in prostate cancer cells (Figures 2D and 3D).
Proliferating cancer cells have increased need for lipids [31]. Interestingly, here we observed that inhibition of ECI2 expression and perhexiline treatment led to prominent accumulation of lipids into cells, which did not promote cancer cell proliferation. Normal prostate cells have an incomplete TCA cycle and therefore cannot rely on typical energy producing pathways [9, 11]. This metabolic adaptation has been shown to sensitize prostate cancer cells to compounds inhibiting mitochondrial activity, and specifically sensitize the cancer cells to other treatments [32]. Here we propose that this metabolic adaptation forces prostate cancer cells to derive a significant fraction of their energy through alternative pathways, such as lipid degradation. In agreement with this model, inhibition of lipid degradation decreased glucose utilization and led to accumulation of starvation /autophagy markers (Figure 3D and Figure 5C) [14]. Instead of successful, cell-survival promoting autophagy, inhibition of lipid degradation led to the activation of the cell death response in prostate cancer cells. Similar dependency on fatty acid oxidation was recently reported in MYC-overexpressing triple-negative breast cancer cells [33]. In breast cancer cells, perhexiline has been shown to inhibit HER3-signaling [34] and induce autophagy [35]. It is not known whether these effects are caused directly by the compound or are down-stream effects from inhibition of lipid metabolism, as it is well established that perhexiline affects lipid metabolism of cancer cells [36, 37]. Interestingly, a number of studies have reported that perhexiline is able to sensitize, and in some cases, reverse resistance, to clinically relevant drugs, including doxorubicin in breast cancer cells [38] and murine leukemia cell line [39] and cisplatin in neuroblastoma cells [40]. We are the first to show that perhexiline has prominent anti-tumor activity once combined with second-generation anti-androgens, Abiraterone and Enzalutamide (Figure 5E). However, because cancer metabolism is influenced by the tumour microenvironment and can be compartmentalized between different cell populations [41–43], it will be important in the future to extend studies of ECI2 function and perhexiline response to more patient-derived pre-clinical models (eg. Explants or PDX) as a stepping stone to clinical trials.
Perhexiline can cause severe toxic side-effects, including neuro- and hepatotoxicity, in a sub-set of patients [27]. The majority of the side-effects are caused by the complex pharmacokinetic profile of this compound, which results in bio-accumulation in small number of patients and sub-sequent toxicicity [26, 27]. This toxicity can be minimized by sustained monitoring of perhexiline plasma levels and adjusting the drug-dosing to result in blood levels below 600 ng/ml [44–46]. It is noteworthy that the anti-tumor activity of perhexiline has been demonstrated in mouse models of several cancers, including breast cancer [34], neuroblastoma [40] and chronic lymphocytic leukemia [36] with no obvious toxicity.
Normal cells are able to compensate for decreased lipid degradation through increased glucose consumption, and therefore inhibition of lipid utilization is not toxic [47]. In fact, certain tissues essentially require glucose as an energy source, and in certain instances inhibition of lipid oxidation is beneficial. This is the case in chronic ischemic cardiomyopathy, a condition with limited oxygen supply to heart tissue [25].
In summary, we propose that prostate cancer cells essentially require lipid degradation for energy production and to sustain proliferation. Failure to adapt to inhibition of lipid degradation leads to profound changes in total metabolome and eventually activation of the cell death response in prostate cancer cells.
Targeting cancer-cell specific metabolic vulnerabilities with approved compounds is an exciting approach, and should speed the development of new combinatorial therapies. Clinical trials using metabolic inhibitors in combination with established cancer-specific drugs are on-going, including compounds such as metformin (for details see: https://clinicaltrials.gov/), and scientific community will likely see more of those in the future. Our data provide evidence to combine anti-androgens with compounds targeting lipid metabolism.
MATERIALS AND METHODS
Evaluation of ECI2 expression in patient samples
RNA extracted from surgical specimens was obtained from Roswell Park Cancer Institute. These samples are covered by an approval (BDR 034313) from the Office of Research Subject Protection. The Salford prostate TMA consisting of diagnostic needle core biopsies from 144 patients attending the Urology clinic of Salford Royal NHS Foundation Trust was covered by MCRC Biobank Ethics 10_NOCL_02, Manchester, UK. Detailed protocols are provided in Supplementary Materials.
Cell lines, maintenance and treatments
Cells were obtained from ATCC and maintained according to ATCC guidelines. To simulate androgen deprivation, cells were kept in phenol-red free media supplemented with charcoal-stripped serum. Knockdown of ECI2 was performed with RNAiMAX reagent (Thermo Fisher Scientific) according to manufacturer’s instructions. ECI2 targeting siRNAs were obtained from Qiagen (SI04202282 and SI04201848). Perhexiline was purchased from Sigma (catalogue number: SML0120-10MG). More detailed protocols for cell lysate preparation, Oil Red O staining (obtained from Sigma, catalogue number: O1391-250ML) and Lipid Tox staining (obtained from Life Technologies, catalogue number: H34475) are available in Supplementary Materials.
Analysis of viability, growth rate and caspase activity
Cells were plated on 384-well plates one day before treatment. Caspase 3/7 activation was detected using IncuCyte® Caspase-3/7 Apoptosis Assay Reagent (EssenBiosciences, catalogue number 4440) and using the IncuCyte instrument (EssenBiosciences) according to manufacturer’s instructions. Growth rates were determined using the IncuCyte instrument according to manufacturer’s instructions. In brief: in order to determine growth rate of cells, cells were reverse-transfected into 384-well plate, and the plate was placed into IncuCyte instrument. Using this instrument, cells were imaged as indicated in the figures and confluency was determined using the built-in software that comes along with the instrument. In order to determine growth rate of cells after compound treatment, cells were plated one day before the treatments, and on the next day, cells were treated as indicated in the figure and growth rate was followed by life-cell imaging.
Metabolomics, RNA-seq and mayo cohort analysis
Detailed protocols and analysis of Gas-chromatography mass spectrometry, 1H NMR spectroscopy of cell culture media, lipidomics and RNA-seq, ChIP-seq and analysis of Mayo cohort samples for met- and prostate cancer specific mortality-free survival are provided in Supplementary Materials. RNA-seq data has been made available: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=wxydygwubzcxxax&acc=GSE75035 for editor/reviewers, and will be made publicly available once the manuscript has been accepted. The processed files are compressed folders containing multiple output files from CuffDiff runs estimating differentially expressed transcripts between the indicated ECI2 siRNA treated cells versus cells treated with Scrambled siRNAs (see Trapnell et al., 2012 for more info [48]).
Abbreviations
AR – Androgen receptor; AURKB – Aurora kinase B; CCND3 – Cyclin D3; CDC6 – Cell division cycle 6; CDC25A – Cell division cycle 25A; CDC25B – Cell division cycle 25B; CDK1 – Cyclin dependent kinase 1; CDKN1A – Cyclin dependent kinase inhibitor 1A; CDKN2C – Cyclin-dependent kinase inhibitor 2C; ChIP – Chromatin immunoprecipitation; ChIP-seq – Chromatin immunoprecipitation coupled to sequencing; Cl-PARP – Cleaved poly(ADP-ribose) polymerase; CYP2D6 – Cytochrome P450 family 2 subfamily D member 6; ECI2 – Enoyl-CoA delta isomerase 2; GAPDH – Glyceraldehyde-3-phosphate dehydrogenase; LC3 – Microtubule associated protein 1 light chain 3 alpha; MAD2L1 – MAD2 mitotic arrest deficient-like 1; MELK – Maternal Embryonic Leucine Zipper Kinase; p62 – Sequestosome 1; TBP – TATA-box binding protein; WASF3 – WAS Protein Family Member 3; WEE1 – WEE1 G2 checkpoint kinase.
Authors’ contributions
HMI designed studies, performed cell line experiments, prepared cell line samples for mRNA/metabolomics profiling and wrote the manuscript. MB and CH performed the immunohistochemistry studies. AU analyzed RNA-seq and ChIP-seq data. GDT performed mass spectrometry. CHL analyzed NMR data. SB showed androgen-dependent regulation. IJG performed patient mRNA profiling. KB, RD, EW and JWS performed lipidomic profiling. MP, NE and ED analyzed CDC6 expression. NC, HH and JLM provided access to clinical samples. All authors reviewed the manuscript prior to submission and provided comments. IGM supervised the study.
CONFLICTS OF INTEREST
None.
FUNDING
This work was supported by the Norwegian Cancer Society (IGM; HMI), the Norwegian Research Council, Helse Sor-Ost and the University of Oslo through the Centre for Molecular Medicine (Norway), which is a part of the Nordic EMBL (European Molecular Biology Laboratory) partnership (IGM), the National Cancer Institute Cancer Center Support Grant CA77739 from the National Cancer Institute Cancer Center Support Grant, for the Pathology Resource Network at Roswell Park Cancer Center CA016056 (JLM) and NIH-NCI grant CA166440 (HVH). In addition IGM, MB, CH and NC are supported by CE013_2-004: FASTMAN Centre, Movember Prostate Cancer Centre of Excellence. Funding sources had no involvement (beyond funding) in the work described.
REFERENCES
1. Mills IG. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nature reviews Cancer. 2014; 14:187–198.
2. Rathkopf D, Scher HI. Androgen receptor antagonists in castration-resistant prostate cancer. Cancer journal. 2013; 19:43–49.
3. Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, MacArthur S, Stark R, Warren AY, Mills IG, Neal DE. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer cell. 2013; 23:35–47.
4. Geisbrecht BV, Zhang D, Schulz H, Gould SJ. Characterization of PECI, a novel monofunctional Delta(3), Delta(2)-enoyl-CoA isomerase of mammalian peroxisomes. The Journal of biological chemistry. 1999; 274: 21797–21803.
5. Chang KH, Ercole CE, Sharifi N. Androgen metabolism in prostate cancer: from molecular mechanisms to clinical consequences. British journal of cancer. 2014; 111:1249–1254.
6. Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng S, Carles J, Mulders PF, Basch E, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. The New England journal of medicine. 2013; 368:138–148.
7. Barfeld SJ, Itkonen HM, Urbanucci A, Mills IG. Androgen-regulated metabolism and biosynthesis in prostate cancer. Endocrine-related cancer. 2014; 21:T57–66.
8. Dimitrakopoulou-Strauss A, Strauss LG. PET imaging of prostate cancer with 11C-acetate. Journal of nuclear medicine. 2003; 44:556–558.
9. Zadra G, Photopoulos C, Loda M. The fat side of prostate cancer. Biochimica et biophysica acta. 2013; 1831:1518–1532.
10. Moon H, Hill MM, Roberts MJ, Gardiner RA, Brown AJ. Statins: protectors or pretenders in prostate cancer? Trends in endocrinology and metabolism. 2014; 25:188–196.
11. Baron A, Migita T, Tang D, Loda M. Fatty acid synthase: a metabolic oncogene in prostate cancer? Journal of cellular biochemistry. 2004; 91:47–53.
12. Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer research. 2004; 64:2070–2075.
13. Mizushima N. Autophagy: process and function. Genes & development. 2007; 21:2861–2873.
14. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011; 7:279–296.
15. Huang L, Min JN, Masters S, Mivechi NF, Moskophidis D. Insights into function and regulation of small heat shock protein 25 (HSPB1) in a mouse model with targeted gene disruption. Genesis. 2007; 45:487–501.
16. Kuner R, Falth M, Pressinotti NC, Brase JC, Puig SB, Metzger J, Gade S, Schafer G, Bartsch G, Steiner E, Klocker H, Sultmann H. The maternal embryonic leucine zipper kinase (MELK) is upregulated in high-grade prostate cancer. Journal of molecular medicine. 2013; 91:237–248.
17. Fernando HS, Sanders AJ, Kynaston HG, Jiang WG. WAVE3 is associated with invasiveness in prostate cancer cells. Urologic oncology. 2010; 28:320–327.
18. Teng Y, Ren MQ, Cheney R, Sharma S, Cowell JK. Inactivation of the WASF3 gene in prostate cancer cells leads to suppression of tumorigenicity and metastases. British journal of cancer. 2010; 103:1066–1075.
19. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009; 37:1–13.
20. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009; 4:44–57.
21. Jung YS, Qian Y, Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cellular signalling. 2010; 22:1003–1012.
22. Ayad NG. CDKs give Cdc6 a license to drive into S phase. Cell. 2005; 122:825–827.
23. Erho N, Crisan A, Vergara IA, Mitra AP, Ghadessi M, Buerki C, Bergstralh EJ, Kollmeyer T, Fink S, Haddad Z, Zimmermann B, Sierocinski T, Ballman KV, et al. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PloS one. 2013; 8:e66855.
24. Lionetti V, Stanley WC, Recchia FA. Modulating fatty acid oxidation in heart failure. Cardiovascular research. 2011; 90:202–209.
25. Kolwicz SC Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circulation research. 2013; 113:603–616.
26. Sorensen LB, Sorensen RN, Miners JO, Somogyi AA, Grgurinovich N, Birkett DJ. Polymorphic hydroxylation of perhexiline in vitro. British journal of clinical pharmacology. 2003; 55:635–638.
27. Ashrafian H, Horowitz JD, Frenneaux MP. Perhexiline. Cardiovascular drug reviews. 2007; 25:76–97.
28. Itkonen HM, Engedal N, Babaie E, Luhr M, Guldvik IJ, Minner S, Hohloch J, Tsourlakis MC, Schlomm T, Mills IG. UAP1 is overexpressed in prostate cancer and is protective against inhibitors of N-linked glycosylation. Oncogene. 2015; 34:3744–3750.
29. Itkonen HM, Minner S, Guldvik IJ, Sandmann MJ, Tsourlakis MC, Berge V, Svindland A, Schlomm T, Mills IG. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer research. 2013; 73:5277–5287.
30. Mallik I, Davila M, Tapia T, Schanen B, Chakrabarti R. Androgen regulates Cdc6 transcription through interactions between androgen receptor and E2F transcription factor in prostate cancer cells. Biochimica et biophysica acta. 2008; 1783:1737–1744.
31. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis. 2016; 5:e189.
32. Itkonen HM, Gorad SS, Duveau DY, Martin SE, Barkovskaya A, Bathen TF, Moestue SA, Mills IG. Inhibition of O-GlcNAc transferase activity reprograms prostate cancer cell metabolism. Oncotarget. 2016; 7:12464–12476. doi: 10.18632/oncotarget.7039.
33. Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, Eyob H, Kajimura S, Tward A, Krings G, Nomura DK, Goga A. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nature medicine. 2016; 22:427–432.
34. Ren XR, Wang J, Osada T, Mook RA Jr, Morse MA, Barak LS, Lyerly HK, Chen W. Perhexiline promotes HER3 ablation through receptor internalization and inhibits tumor growth. Breast cancer research. 2015; 17:20.
35. Balgi AD, Fonseca BD, Donohue E, Tsang TC, Lajoie P, Proud CG, Nabi IR, Roberge M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PloS one. 2009; 4:e7124.
36. Liu PP, Liu J, Jiang WQ, Carew JS, Ogasawara MA, Pelicano H, Croce CM, Estrov Z, Xu RH, Keating MJ, Huang P. Elimination of chronic lymphocytic leukemia cells in stromal microenvironment by targeting CPT with an antiangina drug perhexiline. Oncogene. 2016; 35:5663–5673.
37. Rodriguez-Enriquez S, Hernandez-Esquivel L, Marin-Hernandez A, El Hafidi M, Gallardo-Perez JC, Hernandez-Resendiz I, Rodriguez-Zavala JS, Pacheco-Velazquez SC, Moreno-Sanchez R. Mitochondrial free fatty acid beta-oxidation supports oxidative phosphorylation and proliferation in cancer cells. The international journal of biochemistry & cell biology. 2015; 65:209–221.
38. Foster BJ, Grotzinger KR, McKoy WM, Rubinstein LV, Hamilton TC. Modulation of induced resistance to adriamycin in two human breast cancer cell lines with tamoxifen or perhexiline maleate. Cancer chemotherapy and pharmacology. 1988; 22:147–152.
39. Ramu A, Fuks Z, Gatt S, Glaubiger D. Reversal of acquired resistance to doxorubicin in P388 murine leukemia cells by perhexiline maleate. Cancer research. 1984; 44:144–148.
40. Vella S, Penna I, Longo L, Pioggia G, Garbati P, Florio T, Rossi F, Pagano A. Perhexiline maleate enhances antitumor efficacy of cisplatin in neuroblastoma by inducing over-expression of NDM29 ncRNA. Scientific reports. 2015; 5:18144.
41. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, Yamada SD, Peter ME, Gwin K, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nature medicine. 2011; 17:1498–1503.
42. Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Lisanti MP, Sotgia F. Ketone bodies and two-compartment tumor metabolism: stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell cycle. 2012; 11:3956–3963.
43. Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell metabolism. 2012; 15:4–5.
44. Horowitz JD, Sia ST, Macdonald PS, Goble AJ, Louis WJ. Perhexiline maleate treatment for severe angina pectoris—correlations with pharmacokinetics. International journal of cardiology. 1986; 13:219–229.
45. Cole PL, Beamer AD, McGowan N, Cantillon CO, Benfell K, Kelly RA, Hartley LH, Smith TW, Antman EM. Efficacy and safety of perhexiline maleate in refractory angina. A double-blind placebo-controlled clinical trial of a novel antianginal agent. Circulation. 1990; 81:1260–1270.
46. Horowitz JD, Chirkov YY. Perhexiline and hypertrophic cardiomyopathy: a new horizon for metabolic modulation. Circulation. 2010; 122:1547–1549.
47. Fang YH, Piao L, Hong Z, Toth PT, Marsboom G, Bache-Wiig P, Rehman J, Archer SL. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle’s cycle. Journal of molecular medicine. 2012; 90:31–43.
48. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012; 7:562–578.