
INTRODUCTION
Type 2 diabetes mellitus (T2DM) accounts for ~ 90% of diabetes and constitutes a great challenge for healthcare systems. T2DM is a principle contributor to heart disease, neuropathy, eye failure, and nephropathy [1]. Skin infections and other diabetes-associated skin disorders are among the most common complications, affecting 30 to 70% of people with diabetes [2]. Certain skin disorders are clearly more frequent in diabetic patients, but little is known about the molecular mechanisms underlying this association [3]. Due to the impaired wound healing in diabetic skin, chronic ulcers are one of the most severe cutaneous lesions faced by people with T2DM [4]. Hence, a better understanding of the mechanisms underlying the greater susceptibility of T2DM patients to developing skin disorders should lead to a better management or prevention of such complications.
There is a consensus that the mechanical competence of the dermis is reduced in diabetes [3]. A study led by Bermudez et al. found that db/db diabetic mice skin is biomechanically inferior to nondiabetic skin [5]. It has been suggested that altered collagen expression may contribute to this reduced mechanical competence; however, results of the expression of dermal collagen are not consistent [5, 6]. Furthermore, a recent study showed an increased number of skin inflammatory cells in patients with type 1 or type 2 diabetes, indicating a more permissive barrier in DM [7].
In this study, we hypothesized that T2DM is associated with dysregulated gene networks, which underlie the higher risk of developing cutaneous diseases and skin infections. With the rapid development of high-throughput sequencing technologies and precision medicine, scientists are now increasingly able to understand genetic and environmental factors that may contribute to complex diseases. We performed differential expression and pathway enrichment analyses on 74 T2DM cases and 148 non-DM controls from the Genotype-Tissue Expression (GTEx) database [8]. The GTEx project provides a wealth of gene expression and associated extensive clinical data [9]. We have previously performed extensive bioinformatics analyses using the GTEx database and successfully demonstrated that type 2 diabetic skeletal muscle is characterized by insulin resistance [10], which is considered to be the primary defect involved in T2DM [11]. To the best of our knowledge, our analysis is the first, and currently the largest, RNA-Seq-based transcriptome study in the skin of T2DM patients. RNA-Seq results provided 182 significant differentially expressed (DE) genes in the skin of T2DM. These dysregulated gene expression profiles could help in determining molecular targets for the prevention and the development of potential therapeutic avenues of T2DM-associated skin disorders.
RESULTS
Differential expression analysis identified 182 significant DE genes in the human skin of T2DM
We examined expression changes of 56,318 transcribed genes (on the basis of Gencode V19 annotation, TrueSeq V1) in skin samples (lower leg, N=357) from the GTEx RNA-Seq database. To reduce the bias due to covariates in the generalized linear regression model, we used an optimal matching algorithm to balance gender, race, and age between T2DM and control groups (Supplementary Table 1), which minimized the average of distances among matched units [12]. The jitter plot in Figure 1A shows a similar overall distribution of propensity scores in matched control (N=148) and T2DM (N=74). After removing unmatched controls (N=106), we performed generalized linear regression of normalized read counts for each expressed gene against disease status, adjusting for known and hidden surrogate variables [13]. The volcano plot in Figure 1B shows significant T2DM-related gene expression changes in the skin. At a stringent FDR level of 0.2, we identified 182 significant DE genes in the type 2 diabetic skin (Supplementary Table 2).
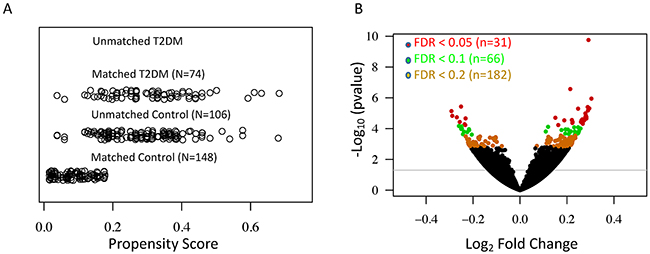
Figure 1: Differential expression analyses reveal a large number of significant T2DM-associated genes in the human skin. (A) Jitter plot shows the distribution of propensity scores in the T2DM and control groups. (B) Volcano plot shows the –log10 (P-value) and log2 fold change for all expressed genes. Differentially expressed genes in the T2DM at the FDR level of 0.05, 0.1, and 0.2 are indicated by red, green and yellow respectively.
Validation by microarray dataset
To validate our results of significant T2DM-related gene expression changes, we compared our findings with the microarray-based gene expression GTEx dataset. The GTEx study has demonstrated a strong correlation (Pearson’s r = 0.83) between RNA-Seq and Microarray platforms [8]. We pulled out 58 skin samples (lower leg) with microarray-based transcriptome profiles, then applied the same exclusion criteria and preprocesses, resulting in 12 T2DM cases and 24 Non-DM controls. We performed a similar linear regression-based approach to model gene expression values against T2DM status. As shown in Figure 2, Log2 Fold Changes (LFCs) of our DE genes were significantly correlated with LFCs of common genes (n=135) in the microarray dataset (Pearson’s Correlation = 0.53). These results suggest a consistency of T2DM-related gene expression changes between these two platforms.
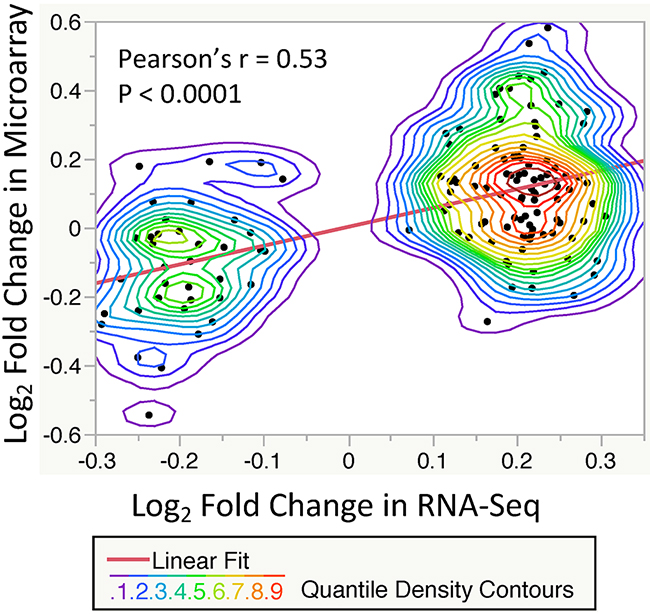
Figure 2: Correlation of significant T2DM-related gene expression changes (FDR < 0.2, n = 135) between the RNA-Seq and Microarray datasets. Each point showing the log2 fold change between T2DM and control subjects. A significant correlation is observed with P-value < 0.0001 and Pearson’s r = 0.53 (Linear fit line slope = 0.55). The nonparametric density contour lines are quantile contours in 5% intervals.
Pathway enrichment analysis revealed altered immune function and pathways related to infectious diseases in T2DM
To obtain a functional overview of these significant T2DM-associated DE genes (FDR<0.2), we conducted overrepresentation tests on up- and down-regulated genes separately. As shown in Table 1, up-regulated genes were significantly overrepresented in two categories: the immune response and infectious disease pathways (q-value < 0.05). Genes in two significant T2DM-associated KEGG pathways, Toll-like receptor signaling pathway (hsa04620, Figure 3A) and Measles pathway (hsa05162, Figure 3B), were visualized through “pathview” R package [14]. To determine whether the FDR cutoff affects the pathway enrichment analysis, we performed overrepresentation test on 66 DE genes with FDR < 0.1. As shown in Supplementary Table 3, up-regulated DE genes were significantly enriched in the similar immune response and infectious diseases pathways as observed with FDR < 0.2. These results indicate an activation of infectious disease-related immune functions in type 2 diabetic skin. Moreover, gene set enrichment analysis, an approach which determines whether a group of genes has significant concordant differences, showed coordinated up-regulation of genes in all of these immune response and infectious disease pathways (Supplementary Table 4).
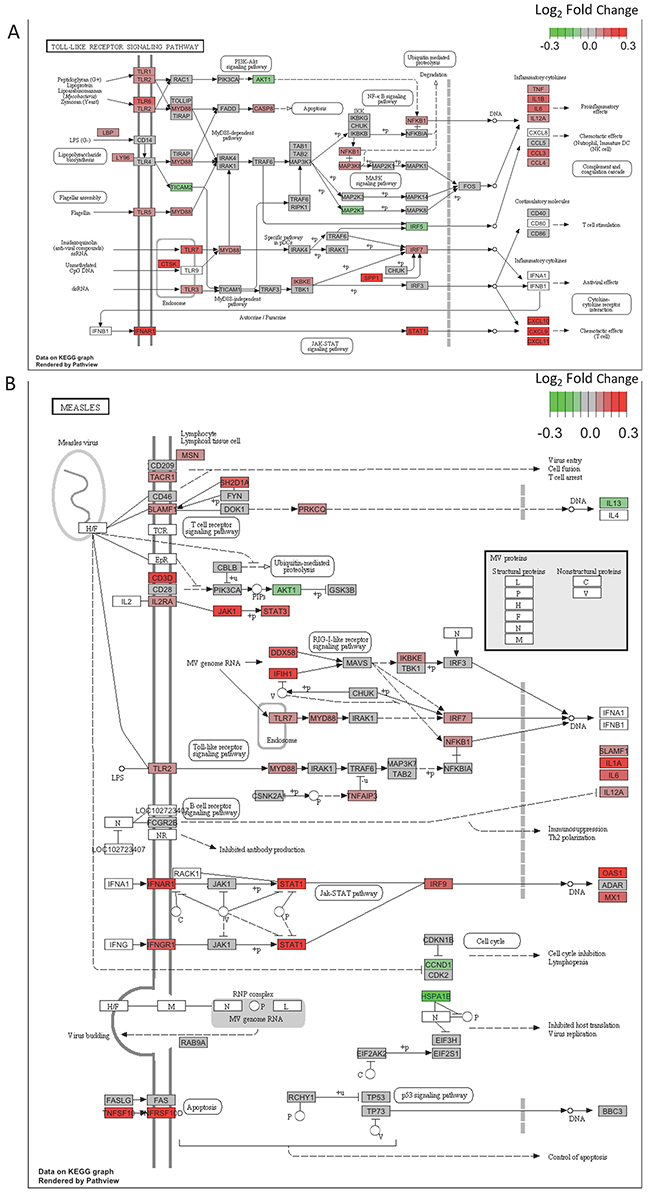
Figure 3. (A) DE genes are significantly overrepresented in the Toll-like receptor signaling pathway. Pathview is used to visualize gene expression changes. Color key indicates log2 fold change. Up-regulated genes appear in red, and down-regulated genes appear in green. (B) Genes in the Measles pathway show a concordant up-regulation in T2DM. Pathview is used to visualize gene expression changes. Color key indicates log2 fold change. Up-regulated genes appear in red, and down-regulated genes appear in green.
Table 1. Significantly enriched pathways on upregulated DE genes (FDR < 0.2) in T2DM
ID |
Description |
P-value |
q-value |
Gene ID |
Count |
|---|---|---|---|---|---|
Immune System |
|||||
hsa04062 |
Chemokine signaling pathway |
1.15E-07 |
1.40E-05 |
STAT1/CCL20/CXCL9/CXCL10/CXCL11/CXCL13/ADCY1/LYN/STAT2/CCL18/JAK3 |
11 |
hsa04620 |
Toll-like receptor signaling pathway |
1.41E-04 |
4.26E-03 |
CTSK/STAT1/CXCL9/CXCL10/CXCL11/SPP1 |
6 |
hsa04060 |
Cytokine-cytokine receptor interaction |
8.06E-04 |
1.63E-02 |
CCL20/TNFSF10/CXCL9/CXCL10/CXCL11/CXCL13/TNFSF15/CCL18 |
8 |
Infection |
|||||
hsa05164 |
Influenza A |
5.98E-06 |
3.00E-04 |
RSAD2/IFIH1/STAT1/TNFSF10/CXCL10/STAT2/OAS1/OAS3/OAS2 |
9 |
hsa05162 |
Measles |
7.44E-06 |
3.00E-04 |
IFIH1/STAT1/TNFSF10/STAT2/OAS1/OAS3/OAS2/JAK3 |
8 |
hsa05168 |
Herpes simplex infection |
4.80E-04 |
1.16E-02 |
IFIH1/STAT1/SP100/STAT2/OAS1/OAS3/OAS2 |
7 |
Although there were no enriched pathways among down-regulated genes, cell adhesion molecules (CAMs) pathway was the most significant pathway with P-value = 0.069 (q-value = 0.417). The expression of cell adhesion molecules, NCAM1 (neural cell adhesion molecule 1) and L1CAM (L1 cell adhesion molecule), were significantly decreased in T2DM (Supplementary Table 2). Moreover, the expression of the collagen-related genes including PCOLCE2 (procollagen C-endopeptidase enhancer 2), COL9A3 (collagen type IX alpha 3), and COL22A1 (collagen type XXII alpha 1) were significantly changed in T2DM (Supplementary Table 2). These results indicate altered lymphocyte trafficking and structural changes in dermal collagen in the skin of T2DM.
DISCUSSION
Our study shows for the first time T2DM-related whole-genome gene expression changes in the human skin. Although it is well known that patients with T2DM are at particular risk of developing skin infections and disorders, the molecular mechanisms leading to this complication are still poorly understood. The clearest trend in our RNA-Seq analysis is the significant enrichment of up-regulated genes in the immune response and infectious disease pathways.
T2DM is associated with increased systemic inflammation, which has been suggested to induce insulin resistance [15], the key feature of T2DM. Although a recent study indicates that increased inflammation was found in the skin of patients with T2DM [7], the molecular insights into the inflamed skin of T2DM remain unclear. Here we showed that three KEGG immune-related pathways including chemokine signaling pathway (hsa04062), toll-like receptor signaling pathway (hsa04620), and cytokine-cytokine receptor interaction (hsa04060) were significantly up-regulated in the skin of T2DM patients (Table 1). Notably, among significantly up-regulated DE genes in immune response pathways, chemokines including CCL20, CXCL9, CXCL10, CXCL11, CXCL13, and CCL18 (Table 1 and Supplementary Table 2) have been suggested to induce leukocyte chemotaxis and activation [16]. Specifically, chemokines CXCL9, CXCL10, and CXCL11 were involved in the T cell recruitment and infiltration in inflammatory skin diseases [17]. Our expression profiles indicated that JAK/STAT molecules including JAK3, STAT1, and STAT2 were up-regulated in the skin of T2DM (Table 1 and Supplementary Table 2). The JAK/STAT signaling pathway is associated with cellular functions such as proliferation, differentiation, and migration [18]. Importantly, the JAK-STAT pathway has been shown to dysregulate the immune response in the chronic inflammatory skin disorders [19]. Moreover, tumor necrosis factor superfamily members including TNFSF10 and TNFSF15, which play essential roles in stimulating T and B lymphocytes [20], were up-regulated in T2DM (Table 1 and Supplementary Table 2). Increased expression of chemokines, JAK/STAT molecules, and TNFSF genes indicates inflammatory dysregulation of leukocytes in the skin of T2DM. Altered expression of immune-related genes may underlie the greater susceptibility of T2DM for developing infectious diseases.
A number of studies have shown that patients with T2DM are associated with a higher risk of infectious diseases [21–24]. About fifty percent of the diabetic patients involved in Shah’s study had at least one hospitalization or physician claim for infection in each cohort year [23]. Another prospective cohort study in Australia showed that the risk of hospitalization for an infectious disease increased more than two-fold in diabetic patients [22]. Furthermore, infection-related mortality has been shown to be higher in patients with diabetes [24]. Although the hyperglycemic environment is suggested to cause immune dysfunction that increases the risk for diabetic patients acquiring infectious diseases, the molecular mechanisms underlying the relationship between glycaemia and infections are still poorly understood [25]. We identified three infectious disease pathways that were significantly enriched among up-regulated DE genes (Table 1). Notably, three genes, OAS1, OAS2, and OAS3 of the 2'-5'-oligoadenylate synthetase family, were significantly up-regulated in T2DM and presented in all of these infectious disease pathways (Table 1 and Supplementary Table 2). The 2'-5'-oligoadenylate synthetase and its downstream effector enzyme RNase L are known to be involved in the host defense mechanisms against viral infection [26]. The activity of 2'-5'-oligoadenylate synthetase was found to be persistently activated in type 1 diabetes [27]. Moreover, another antiviral gene, IFIH1 (interferon induced with helicase C domain 1), was significantly up-regulated in patients with T2DM and involved in all infectious disease pathways (Table 1 and Supplementary Table 2). IFIH1 is a member of the RIG-I-like receptor (RLR) family involved in the recognition of viral RNA and mediates the virus-induced innate immune response [28]. It has been shown that rare variant or reduced expression of IFIH1 protects against type 1 diabetes [29, 30]. However, the influences of these antiviral genes in T2DM are not clear. Excessive expression of OAS family genes and IFIH1 may limit the magnitude of the response to viruses in the skin of T2DM, leading to a higher risk of viral infection.
Importantly, we observed the similar immune activation in response to infections in the human skeletal muscle of T2DM [10]. Three infectious disease pathways, Influenza A (hsa05164), Measles (hsa05162) and Herpes simplex infection (hsa05168), were significantly up-regulated in both muscle and skin of type 2 diabetic patients. In addition, 152 DE genes in the skin (FDR < 0.2), which are also expressed in the muscle, showed a consistent T2DM-associated expression changes between the muscle and skin (Supplementary Figure 1). These results indicated the burden of infection in both human skin and muscle of T2DM, and are consistent with high rates of skin and soft-tissue infections observed in patients with type 2 diabetes [21].
Although no pathways were significantly enriched among down-regulated genes, Cell Adhesion Molecules (CAMs) pathway was placed at the top of all down-regulated pathways from overrepresentation tests (q-value = 0.417). CAMs are cell surface proteins involved in mediating leukocyte migration in the immune system [31]. Notably, the expression of NCAM1 was significantly down-regulated in the skin of T2DM (Supplementary Table 2). NCAM1 is expressed in NK cells and serves as a host protective component in the local innate immune response against viral infections [32]. Moreover, the expression of another cell adhesion molecule L1CAM was significantly down-regulated in T2DM (Supplementary Table 2). L1CAM is expressed on the surface of leukocytes and regulates the adhesion of leukocytes through L1 and L1/NCAM homophilic binding [33]. Decreased expression of cell adhesion molecules in T2DM may underlie a greater susceptibility to the development of infectious diseases. In addition, two collagen-related genes, PCOLCE2 and COL9A3 were significantly down-regulated in T2DM, while COL22A1 was significantly up-regulated (Supplementary Table 2). Altered expression of collagen-related genes may affect the physical integrity of the epidermis, leading to greater susceptibility of T2DM patients to infectious diseases.
In conclusion, our RNA-Seq analyses of the human skin from 74 T2DM cases and 148 matched controls revealed comprehensive molecular and network defects associated with T2DM. We acknowledge that gene expression studies in the human postmortem tissues alone do not allow establishing the causal relationship with the type 2 diabetes [33]. Nerveless, skin and soft tissue infections are prominent in T2DM patients with chronic hyperglycemia, and hyperglycemia has been shown to affect immune responses and increase susceptibility to infections in patients with T2DM [34]. Therefore, it is reasonable for us to hypothesize that the up-regulation of genes related to immune response and infectious disease pathways partly reflect the adaptive changes caused by the type 2 diabetic states such as hyperglycemia. Skin, as the most visible and largest organ of the integumentary system, can be considered as the first warning signal for T2DM and a good predictor marker for evaluating the therapy efficiency in patients with T2DM. This analytical study provides novel molecular targets for developing therapeutics in the prevention and management of T2DM-associated skin disorders. In addition, given the easy accessibility of skin, genes whose expression levels significantly changed in T2DM can be employed as potential diagnostic biomarkers for T2DM.
MATERIALS AND METHODS
GTEx database
The GTEx database (v6, October 2015 release) contains 357 skin samples from lower leg with RNA-Seq transcriptome profiles. Detailed information on sample collection, RNA sequencing, and the data processing pipeline can be found in the GTEx Consortium paper [8]. We excluded cases with type 1 diabetes, unknown T2DM status, and races other than black or white, leaving 254 non-diabetic samples and 74 T2DM cases. To reduce effects of cofounders in our statistical model, MatchIt (v2.4) in R was used to balance three covariates (age, gender, and race) between Non-DM controls and T2DM cases with “optimal” matching and 2:1 optimal ratio (Supplementary Table 1).
Identification of significantly differentially expressed genes in T2DM
Differentially expressed genes were identified as described previously [10]. Briefly, we used the “svaseq” function from the sva R package to identify hidden cofounding factors [13]. In addition to gender, 5 surrogate variables were added to the formula in DESeq2 [35]. The residuals for normalized read counts, after gender and surrogate variables correction, were tested against the “T2DM” status using the following negative binomial (NB) generalized linear regression model (GLM):
Kij ~ NB (μij, αj) (1)
μij = si qij (2)
Where Kij is the read count for gene j in sample i, fitted with a negative binomial distribution. αj is a gene-specific dispersion parameter. μij represents fitted mean, containing a sample-specific size factor si and a covariate-dependent part qij [35]. In Equation 3, β0 is the regression intercept for gene j, εij is the error term. β1j, β2j, and γkj (k=1, …, 5) denote the regression coefficients of T2DM, gender, and kth surrogate variables for gene j respectively. The Wald test P-values were adjusted for multiple testing using the Benjamini-Hochberg (BH) algorithm. We defined significant DE genes at the level of FDR < 0.2.
To validate our results, we extracted the microarray-based GTEx dataset from the skin of lower leg, which contains 12 subjects with T2DM and 46 Non-DM controls [36]. MatchIt (v2.4) in R was used to balance three covariates (age, gender, and race) with “optimal” matching and 2:1 optimal ratio, resulting 12 T2DM and 24 Non-DM controls. For each gene signature, we performed differential expression analysis by using the limma R package [37]. Multiple testing P-values were adjusted using the BH method.
Detecting significantly enriched KEGG pathways in T2DM
We performed overrepresentation tests on significant DE genes (FDR < 0.2) in T2DM by using clusterProfiler (v3.0.2) in R [38]. The function “enrichKEGG” was used to test whether up- (n=122) or down-regulated (n=60) DE genes are significantly overrepresented in given pathways from Kyoto Encyclopedia of Genes and Genomes (KEGG PATHWAY database). q-values were reported for FDR control. For pathway-based analyses, we defined significant T2DM-associated biological functions at the level of q-value less than 0.05. Selected significant T2DM-associated pathways were visualized through the “pathview” R package [14].
Gene set enrichment analysis (GSEA) was performed by using clusterProfiler (v3.0.2) in R [38], which implements the algorithm developed by the Broad Institute [39]. Specifically, we constructed a pre-ranked gene list of all expressed genes ordered by log2 fold change from DESeq2 package. Enrichment score (ES) and significance level of ES (nominal P value) were calculated by 1000 phenotype-based permutation test. Pre-defined pathways from KEGG PATHWAY database with minimal gene size of 10 and maximal of 500 was used in GSEA. q-values were calculated for FDR control. Significant pathways with q-value less than 0.05 were reported (Supplementary Table 3).
Statistical analysis and dataset access
All statistical computing was performed in the R (v3.3, https://www.r-project.org/) and JMP (v12, SAS Institute). The GTEx dataset can be downloaded from dbGaP, study accession no. phs000424.v6.p1.
Author contributions
C.W. conducted data analysis, C.W., C-T.L., X.C. and J.S. wrote the manuscript and contributed to the interpretation of data. C.W. and C-T.L. are the guarantors of this work and take responsibility for the accuracy of the data analysis.
ACKNOWLEDGMENTS
The authors thank to the GTEx program and the GTEx Consortium. This study would not be possible without the wealth of this enormous database and resources. We thank Gang Xu in the department of biostatistics, University of Miami for assistance with statistical analysis. The authors thank Shang-Yi A. Tsai, Ph.D. at the National Institutes of Health for the interpretation of data and revision. We thank Valerie Gramling, Ph.D. and April Mann, M.A. of the Writing Center at the University of Miami for writing assistance, language editing, and proofreading.
CONFLICTS OF INTEREST
No potential conflicts of interest relevant to this article were reported.
FUNDING
This work was supported in part by grants from National Natural Science Foundation of China 81200251 (J. S.), Key Platform Plan for Medicine and Health of Zhejiang Province 2015ZDA003 (J.S.), and Excellent Young Scientist Foundation of Zhejiang Provincial People’s Hospital ZRY2016A002 (X.C.).
REFERENCES
1. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005; 54:1615-1625.
2. Makrantonaki E, Jiang D, Hossini AM, Nikolakis G, Wlaschek M, Scharffetter-Kochanek K, Zouboulis CC. Diabetes mellitus and the skin. Reviews in endocrine & metabolic disorders. 2016.
3. Quondamatteo F. Skin and diabetes mellitus: what do we know? Cell and tissue research. 2014; 355:1-21.
4. Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005; 366:1736-1743.
5. Bermudez DM, Herdrich BJ, Xu J, Lind R, Beason DP, Mitchell ME, Soslowsky LJ, Liechty KW. Impaired biomechanical properties of diabetic skin implications in pathogenesis of diabetic wound complications. The American journal of pathology. 2011; 178:2215-2223.
6. Rodgers KE, Ellefson DD, Espinoza T, Hsu YH, diZerega GS, Mehrian-Shai R. Expression of intracellular filament, collagen, and collagenase genes in diabetic and normal skin after injury. Wound Repair Regen. 2006; 14:298-305.
7. Tellechea A, Kafanas A, Leal EC, Tecilazich F, Kuchibhotla S, Auster ME, Kontoes I, Paolino J, Carvalho E, Nabzdyk LP, Veves A. Increased skin inflammation and blood vessel density in human and experimental diabetes. Int J Low Extrem Wounds. 2013; 12:4-11.
8. Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, Johnson R, Segre AV, Djebali S, et al. Human genomics. The human transcriptome across tissues and individuals. Science. 2015; 348:660-665.
9. Keen JC, Moore HM. The Genotype-Tissue Expression (GTEx) Project: Linking Clinical Data with Molecular Analysis to Advance Personalized Medicine. J Pers Med. 2015; 5:22-29.
10. Wu C, Xu G, Tsai SA, Freed WJ, Lee CT. Transcriptional profiles of type 2 diabetes in human skeletal muscle reveal insulin resistance, metabolic defects, apoptosis, and molecular signatures of immune activation in response to infections. Biochemical and biophysical research communications. 2017; 482:282-288.
11. DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes care. 2009; 32:S157-163.
12. Rosenbaum PR. Optimal Matching for Observational Studies. J Am Stat Assoc. 1989; 84:1024-1032.
13. Leek JT. svaseq: removing batch effects and other unwanted noise from sequencing data. Nucleic acids research. 2014; 42.
14. Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013; 29:1830-1831.
15. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. The Journal of clinical investigation. 2006; 116:1793-1801.
16. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014; 32:659-702.
17. Flier J, Boorsma DM, van Beek PJ, Nieboer C, Stoof TJ, Willemze R, Tensen CP. Differential expression of CXCR3 targeting chemokines CXCL10, CXCL9, and CXCL11 in different types of skin inflammation. The Journal of pathology. 2001; 194:398-405.
18. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. Journal of cell science. 2004; 117:1281-1283.
19. Bao L, Zhang H, Chan LS. The involvement of the JAK-STAT signaling pathway in chronic inflammatory skin disease atopic dermatitis. JAKSTAT. 2013; 2:e24137.
20. Croft M, Duan W, Choi H, Eun SY, Madireddi S, Mehta A. TNF superfamily in inflammatory disease: translating basic insights. Trends Immunol. 2012; 33:144-152.
21. Dryden M, Baguneid M, Eckmann C, Corman S, Stephens J, Solem C, Li J, Charbonneau C, Baillon-Plot N, Haider S. Pathophysiology and burden of infection in patients with diabetes mellitus and peripheral vascular disease: focus on skin and soft-tissue infections. Clin Microbiol Infect. 2015; 21:S27-32.
22. Hamilton EJ, Martin N, Makepeace A, Sillars BA, Davis WA, Davis TM. Incidence and predictors of hospitalization for bacterial infection in community-based patients with type 2 diabetes: the fremantle diabetes study. PloS one. 2013; 8:e60502.
23. Shah BR, Hux JE. Quantifying the risk of infectious diseases for people with diabetes. Diabetes care. 2003; 26:510-513.
24. Bertoni AG, Saydah S, Brancati FL. Diabetes and the risk of infection-related mortality in the U.S. Diabetes care. 2001; 24:1044-1049.
25. Peleg AY, Weerarathna T, McCarthy JS, Davis TM. Common infections in diabetes: pathogenesis, management and relationship to glycaemic control. Diabetes/metabolism research and reviews. 2007; 23:3-13.
26. Rebouillat D, Hovanessian AG. The human 2',5'-oligoadenylate synthetase family: interferon-induced proteins with unique enzymatic properties. J Interferon Cytokine Res. 1999; 19:295-308.
27. Bonnevie-Nielsen V, Martensen PM, Justesen J, Kyvik KO, Kristensen B, Levin K, Beck-Nielsen H, Worsaa A, Dyrberg T. The antiviral 2',5'-oligoadenylate synthetase is persistently activated in type 1 diabetes. Clin Immunol. 2000; 96:11-18.
28. Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M Jr, Akira S, Yonehara S, Kato A, Fujita T. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005; 175:2851-2858.
29. Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009; 324:387-389.
30. Downes K, Pekalski M, Angus KL, Hardy M, Nutland S, Smyth DJ, Walker NM, Wallace C, Todd JA. Reduced expression of IFIH1 is protective for type 1 diabetes. PloS one. 2010; 5.
31. Muller WA. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol. 2011; 6:323-344.
32. Ulm C, Saffarzadeh M, Mahavadi P, Muller S, Prem G, Saboor F, Simon P, Middendorff R, Geyer H, Henneke I, Bayer N, Rinne S, Lutteke T, et al. Soluble polysialylated NCAM: a novel player of the innate immune system in the lung. Cellular and molecular life sciences. 2013; 70:3695-3708.
33. Hubbe M, Kowitz A, Schirrmacher V, Schachner M, Altevogt P. L1 adhesion molecule on mouse leukocytes: regulation and involvement in endothelial cell binding. Eur J Immunol. 1993; 23:2927-2931.
34. Casqueiro J, Casqueiro J, Alves C. Infections in patients with diabetes mellitus: A review of pathogenesis. Indian journal of endocrinology and metabolism. 2012; 16:S27-36.
35. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550.
36. Jin W, Goldfine AB, Boes T, Henry RR, Ciaraldi TP, Kim EY, Emecan M, Fitzpatrick C, Sen A, Shah A, Mun E, Vokes V, Schroeder J, et al. Increased SRF transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. The Journal of clinical investigation. 2011; 121:918-929.
37. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research. 2015; 43:e47.
38. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012; 16:284-287.
39. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:15545-15550.