
INTRODUCTION
Aberrant growth signals in malignant tumors, including non small cell lung cancer (NSCLC) are frequently due to the deregulation of signaling cascades of growth factors and their receptors, such as epidermal growth factor receptor (EGFR) and its ligands [1]. The tumor microenvironment actively contributes to these events by providing cellular and molecular effectors which enhance the dysregulation of cancer cell signaling [2].
Prostaglandin E2 (PGE2), an inflammatory mediator, initiates multiple cellular responses, including tumor cell growth and progression. Increased PGE2 synthesis was observed in different malignancies such as colon, breast, lung, head and neck, prostate and bladder cancer [3, 4]. Notably, cycloxigenase-2 (COX-2) and microsomal prostaglandin E synthase-1 (mPGES-1), the two inducible key enzymes in PGE2 biosynthesis, were found overexpressed in NSCLC, correlating with the reduced survival in patients with stage I disease [5–7]. In addition to the large bulk of literature on prostanoids and colon cancer, several studies have shown that NSAID and aspirin reduced the risk to develop lung cancer [8, 9].
We and others have previously reported the importance of PGE2 in several processes of tumor cell adaptation to the microenvironment, such as cell survival, growth, migration, invasion, and angiogenesis [10–15]. In addition, PGE2 can transactivate EGFR-mediated signaling networks that confer an aggressive phenotype to tumor cells [16–18]. More recently, PGE2 has also been identified as a tumor-induced immunosuppressive factor, able to mediate the reprogramming of the tumor microenvironment [19], or as a direct modulator of macrophage activity by transactivation of CSF-1R [20]. All together, these data highlight the complex effects exerted by PGE2 on stromal/immune and cancer cells in creating a pro-tumorigenic microenvironment and in supporting tumor progression.
Using different experimental models, several reports have demonstrated EGFR activation by PGE2 and its receptors (EP receptors) coupled to different downstream effectors, including PKA, PKC, SRC and PI3K [17, 18, 21, 22]. The best-characterized mechanisms by which PGE2/EP signaling transactivates EGFR involve the autocrine and/or paracrine release of soluble EGF-like ligands [23]. Ligand shedding-independent transactivation of EGFR by direct intracellular phosphorylation has also been proposed [17, 18, 24, 25]. In this context, EGFR-supported transactivation is strongly dependent on intracellular signaling pathways, such as Ca2+, PKC and the non-receptor tyrosine kinase c-SRC [26].
Ligand binding to EGFR induces a variety of signaling cascades from the plasma membrane to different subcellular compartments [27]. Notably, ligand-activated EGFR can be targeted to the nucleus, where it acts as transcription factor and chromatin regulator and affects gene expression, DNA replication, and DNA damage repair promoting tumor progression, aggressiveness and resistance to therapies [28, 29]. In lung adenocarcinoma, nuclear EGFR expression has been associated with poor clinical outcome and chemo-resistance [30].
Despite the experimental evidence on the functional interaction between PGE2 and EGFR, the role of PGE2 in EGFR nuclear translocation is not known. Since we have previously demonstrated that PGE2 induces angiogenesis by promoting fibroblast growth factor receptor-1 (FGFR1) nuclear translocation [31] and that PGE2 transactivates EGFR leading to tumor progression [15, 17], we have tested whether PGE2 coupling with EP receptors induces EGFR nuclear shuttling in NSCLC cells. Here we report the molecular mechanisms by which PGE2 regulates EGFR nuclear translocation and the contribution of this signaling cascade to sustain tumor growth.
RESULTS
PGE2 promotes EGFR nuclear translocation and cell growth in human NSCLC cells
Using A549 and GLC82 NSCLC cells, we investigated whether PGE2 promoted EGFR nuclear internalization. EGF was used as a positive control. Cells were treated with EGF 25 ng/ml (10–120 min), and EGFR nuclear translocation was determined by cell fractionation and immunoblotting. Upon EGF treatment, EGFR translocated to the nucleus with a peak at 10 min and declined to baseline at 60 min (Figure 1A for A549 cells and 1D for GLC82 cells). To assess whether PGE2 was able to induce EGFR nuclear translocation, we treated tumor cells for the same time points with PGE2 1 μM. PGE2 induced EGFR nuclear accumulation, which was detectable starting at 30 min, reaching a plateau at 60 min and declining toward the baseline at 120 min after treatment (Figure 1B and 1E). Immunofluorescence staining followed by confocal microscopy analysis showed that in control conditions, EGFR was confined to the cell membrane (Figure 1C and 1F and upper panels). After 10 min of EGF 25 ng/ml treatment, EGFR was mobilized from the cell membrane and localized within the nucleus, an event reproduced by 60 min exposure to PGE2 1 μM (Figure 1C and 1F central and bottom panels, respectively). 3D reconstruction of confocal laser scanning microscopy stacks confirmed the nuclear translocation of EGFR upon EGF or PGE2 treatment (Supplementary Figure 1A and 1B).
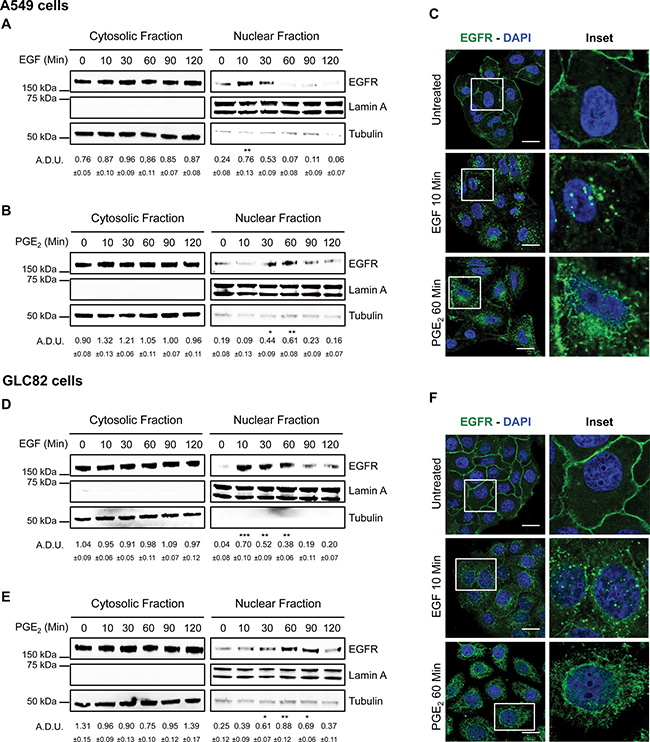
Figure 1: PGE2 induces EGFR nuclear translocation. Immunoblotting analysis of EGFR expression in cytosolic and nuclear fraction in overnight starved A549 (A, B, C) and GLC82 (D, E, F). Cells were exposed for 10–120 min to 25 ng/ml EGF (A, D) or 1 μM PGE2 (B, E). Tubulin and Lamin A were used as loading control for cytosolic and nuclear fraction respectively. Immunoblotting quantification was expressed in A.D.U. (arbitrary density unit) and as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs Ctrl. EGFR in the cytoplasmic and nuclear fractions was normalized to Tubulin or Lamin A respectively. Confocal analysis of EGFR localization in A549 (C) and GLC82 (F) exposed to 25 ng/ml EGF (10 min, middle panel) or 1 μM PGE2 (60 min, bottom panel). EGFR was stained in green and DAPI (blue) was used to counterstain the nuclei. Confocal images were captured in the middle section of the nuclei using 63× objective. Scale bars, 20 μm. Boxed areas are shown in detail in the inset.
Next, we investigated whether the PGE2-mediated EGFR nuclear internalization was associated with increased cell growth. In A549 cells exposed for a time frame of 2–24 h to the treatments, EGF promoted the expression of a panel of well-known nuclear EGFR-target genes involved in cell proliferation, cell cycle progression and inflammation, such as cyclin D1 (CCND1), c-Myc (MYC) cyclooxygenase-2 (PTGS2), and inducible nitric oxide synthase (NOS2) (Supplementary Figure 2A), maximal activation occurred at 2 h. PGE2 mimicked EGF activity on nuclear EGFR-target gene expression with a maximal effect at 4 h in both A549 and GLC82 cells (Figure 2A and 2B). Other nuclear EGFR-target genes, such as Aurora A (AURKA), Breast cancer resistant protein (BCRP), B-Myb (MYBL2) and Thymidylate synthase (TYMS), were not regulated by EGF or PGE2 (Supplementary Figure 2B, 2C and 2D).
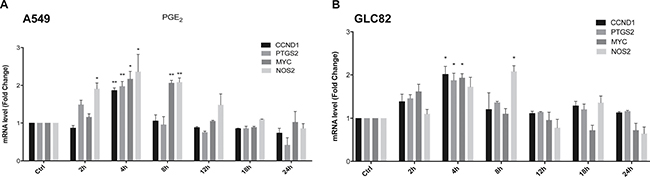
Figure 2: PGE2 regulates nuclear EGFR target genes. A549 (A) and GLC82 (B) cells were starved overnight and then treated with 1 μM PGE2 for 2, 4, 8, 12, 18, 24 h. RNA was isolated and analyzed by qRT-PCR for a panel of nuclear EGFR target genes. The data are presented as mean of fold change ± SEM of three independent experiments, relative to non-treated cells (Control), which were assigned to 1. *p < 0.05, **p < 0.01 vs Ctrl.
To demonstrate that the tumor gene reprogramming promoted by PGE2 was mediated by nuclear EGFR, the expression of EGFR was genetically ablated by CRISPR/Cas9 in A549 (Figure 3A) and GLC82 cells (Supplementary Figure 3A), and then two clones, knockout for EGFR (EGFR −/− #1, #2), were transfected with EGFR plasmids bearing a wild type (WT) or a mutated nuclear localization sequence, NLSm12 and dNLS, respectively [32]. In NLSm12 and dNLS cells, EGFR nuclear translocation by either EGF or PGE2 was significantly reduced compared to cells expressing WT EGFR or to parental cells (Figure 3B and 3C). EGFR-NLS clones maintained the EGF-induced EGFR canonical signaling, such as receptor phosphorylation on Tyr 1068 and AKT activation, as did the EGFR WT clones (Figure 3D and 3E). Further, A549 and GLC82 cells transfected with constructs encoding for WT and mutant EGFR exhibited a comparable level of EGFR expression (Figure 3F and Supplementary Figure 3B), yet only cells expressing WT EGFR showed significant cell proliferation when exposed to EGF or PGE2, while cells expressing EGFR-NLS mutants did not proliferate in response to EGF or PGE2 (Figure 4A left and right and Supplementary Figure 4A left and right). Additionally, a clonogenic in vitro assay showed that PGE2 and EGF increased the number of clones in parental and EGFR WT A549 and GLC82 cells by approximately 50%, whereas in EGFR-NLS mutants cells PGE2 or EGF did not promote clonal outgrowth (Figure 4B left and right and Supplementary Figure 4B left and right). Furthermore, qRT-PCR analysis of nuclear EGFR-target genes indicated that PGE2 promoted the expression of CCND1, MYC, PTGS2 and NOS2 only in A549 and GLC82 cells bearing EGFR WT, while on the contrary, in EGFR-NLS mutant cells, PGE2 did not induce gene expression (Figure 4C and Supplementary Figure 4C).
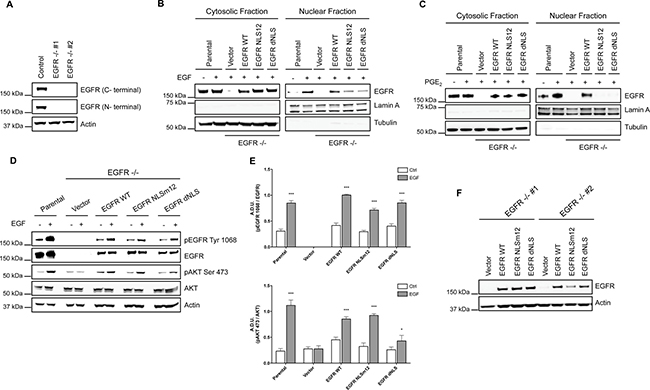
Figure 3: NSCLC cell models to study PGE2-induced EGFR nuclear translocation. (A) Immunoblotting analysis of EGFR expression in A549 wild type cells and two clones knockout for EGFR, generated by CRISPR/Cas9 (EGFR −/− #1, #2). Actin was used as loading control. (B, C) EGFR knockout cells were transiently transfected with Vector or EGFR-WT or EGFR mutated in NLS (NLSm12 or dNLS) plasmids for 48 h. Then EGFR nuclear translocation in response to 25 ng/ml EGF for 10 min (B) or 1 μM PGE2 for 60 min (C) was analyzed by immunoblotting upon cell fractionation. Parental cells were included as a control. Tubulin and Lamin A were used as loading control for cytosolic and nuclear fraction respectively. (D) Immunoblotting analysis of EGFR phosphorylation on tyrosine 1068 and AKT on serine 473, upon EGF treatment in parental and EGFR knockout cells expressing Vector, EGFR-WT and NLS mutant plasmids. (E) Immunoblotting quantification of pEGFR Tyr 1068, normalized to EGFR and pAKT Ser 473, normalized to AKT, were expressed in A.D.U. (arbitrary density unit) and as mean ± SEM. *p < 0.05, ***p < 0.001 vs Ctrl. (F). Expression of EGFR in EGFR −/− #1, #2 transfected with Vector, EGFR-WT and NLS mutant plasmids for 72 h.
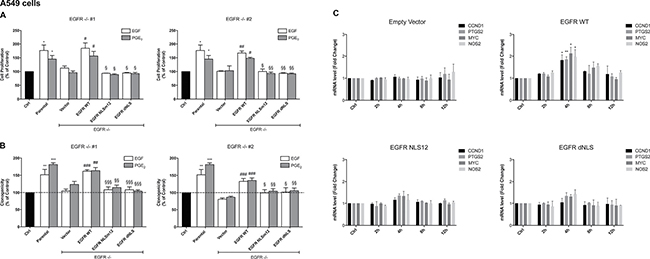
Figure 4: PGE2 promotes cell proliferation, clonogenicity and gene regulation via nuclear EGFR. (A, B, C) Parental A549 cells or EGFR −/− #1, #2 cells transfected with Vector or EGFR-WT or NLS mutant plasmids, were seeded and incubated for 24 h. Next, cells were harvested and seeded for MTT, clonogenic assay and RNA isolation. (A) Cell growth was assessed by MTT assay after 48 h treatment with 25 ng/ml EGF or 1 μM PGE2. Data are presented as mean ± SEM of triplicate cultures, expressed as % of control. *p < 0.05 vs Ctrl; #p < 0.05, ##p < 0.01 vs Vector; §p < 0.05, §§p < 0.01 vs EGFR WT. (B) Clonal outgrowth was assessed by counting number of clones (>50 cells) 12 days after treatment with 25ng/ml EGF or 1μM PGE2. Data are presented as mean ± SEM of triplicates, expressed as % of control. **p < 0.01, ***p < 0.001 vs Ctrl; ##p < 0.01, ###p < 0.001 vs Vector; §p < 0.05, §§p < 0.01, §§§p < 0.001 vs EGFR WT. (C) RNA was isolated after 2, 4, 8, 12 h treatment with 1μM PGE2 and analyzed by qRT-PCR for regulated nuclear EGFR target genes. The data are presented as fold change ± SEM of three independent experiments, relative to non-treated cells (Control), which were assigned to 1. *p < 0.05, **p < 0.01 vs Ctrl.
These results document that PGE2 acts as a potent promoter of NSCLC growth and progression by inducing EGFR nuclear translocation and by increasing the expression of nuclear EGFR target genes involved in cell proliferation, cell cycle progression and inflammation.
PGE2 requires EP3 receptor to induce EGFR nuclear translocation
To characterize the EP receptor subtype involved in EGFR nuclear translocation, we used specific EP receptor agonists at 1 μM for 60 min: Butaprost as EP2 agonist, Sulprostone as EP3 agonist, and L-902,688 as EP4 agonist. In A549 cells, only the EP3 agonist promoted EGFR internalization indicating its relevance for PGE2-mediated EGFR nuclear translocation (Figure 5A). Confocal imaging analysis and 3D reconstruction demonstrated EGFR trafficking and nuclear localization upon EP3 agonist treatment recapitulating PGE2 effect (Figure 5B and Supplementary Figure 5). Similar results were obtained in GLC82 cells (Supplementary Figure 6A and 6B). Consistently, the selective antagonist of EP3, L798-106 (10 μM) or siRNA-mediated EP3 silencing (si-EP3) abolished PGE2-induced EGFR nuclear translocation, as corroborated by confocal analysis (Figure 5C, 5D, 5E). In si-EP3 cells, EGFR nuclear translocation did not occur upon PGE2 treatment and EGFR was confined at the cell membrane as in untreated cells (Figure 5E and Supplementary Figure 7). As a control, EGF-induced EGFR nuclear translocation was not modified in cells with siRNA-ablated EP3 receptor expression (Supplementary Figure 8). These results demonstrate that PGE2-mediated EGFR nuclear translocation requires the EP3 receptor.
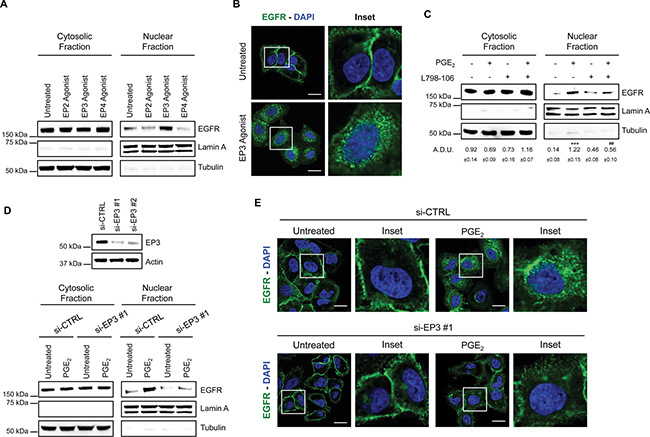
Figure 5: PGE2 promotes EGFR nuclear translocation via EP3 receptor. (A) Immunoblotting analysis of EGFR expression in cytosolic and nuclear fraction in A549 exposed for 60 min to 1 μM EP2, EP3 and EP4 agonists. Tubulin and Lamin A were used as loading control for cytosolic and nuclear fraction, respectively. (B) Confocal analysis of EGFR localization in A549 exposed to EP3 agonist for 60 min. EGFR was stained in green, DAPI (blue) was used to counterstain the nuclei. Confocal images were captured in the middle section of the nuclei using 63x objective. Scale bars, 20 μm. Boxed areas are shown in detail in the inset. (C) Immunoblotting analysis of EGFR expression in cytosolic and nuclear fraction in A549 cells pretreated with or without EP3 antagonist (L798-106; 1 μM) for 30 min before challenging with 1μM PGE2 for 60 min. Immunoblotting quantification was expressed in A.D.U. (arbitrary density unit) and as mean ± SEM. ***p < 0.001 vs Ctrl; ###p < 0.001 vs PGE2. (D) A549 cells were transfected with siRNA control or siRNAs against EP3 receptor for 24 h. After that, cells were serum starved overnight and treated with 1 μM PGE2 for 60 min. EGFR level in cytoplasmic and nuclear fraction was assessed using western blot with indicated antibodies. Knockdown efficiency was verified by immunoblotting with EP3 antibody, actin was used as loading control. Data are shown only for si-EP3#1, similar data were obtained with si-EP3#2. (E) 48 h post transfection, cells were treated with PGE2 as indicated in the panels, fixed and stained for EGFR (green) and DAPI (blue). Pictures were acquired in the middle section of nuclei at 63× magnification. Scale bars, 20 μm. Boxed areas are shown in detail in the inset.
EGFR kinase activity is essential for its nuclear translocation
To explore whether EGFR nuclear translocation was functionally dependent on its phosphorylation, A549 cells were incubated with PGE2 at increasing time points (5–60 min) and EGFR, ERK1/2 and AKT phosphorylation were determined by immunoblotting. EGFR phosphorylation and the downstream signaling pathways were activated in a time-dependent manner with a maximum between 5 and 15 min of PGE2 treatment (Figure 6A). We next assessed the requirement of EGFR tyrosine kinase activity for its internalization by incubating NSCLC cells with the EGFR selective tyrosine kinase inhibitor (TKI) AG1478 at 10 μM before exposure to EGF or PGE2. AG1478 treatment substantially reduced EGFR nuclear translocation in response to either EGF or PGE2 (Figure 6B and 6C), indicating that the tyrosine kinase domain of EGFR is required for nuclear translocation. These results were confirmed in GLC82 cells (Figure 6D).
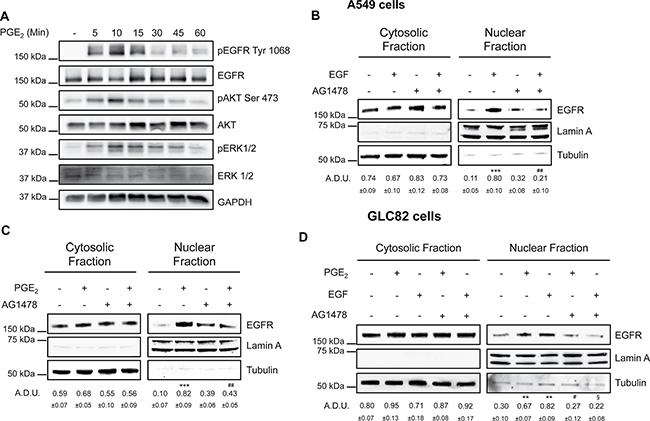
Figure 6: EGFR kinase domain is necessary for its nuclear translocation. (A) Immunoblotting analysis of EGFR phosphorylation on Tyr1068, and AKT phosphorylation on Ser473 in A549 exposed to 1 μM PGE2 for 5–60 min. GAPDH was used as loading control (B, C) Immunoblotting analysis of EGFR expression in cytosolic and nuclear fraction in A549 exposed for 10 min to 25 ng/ml EGF (B), or 60 min to 1 μM PGE2 (C), with or without pre-incubation with AG1478 (10 μM) for 30 min. (D) Immunoblotting analysis of EGFR expression in cytosolic and nuclear fraction in GLC82 exposed for 10 min to 25 ng/ml EGF, or 60 min to 1μM PGE2, with or without pre-incubation with 10 μM AG1478 for 30 min. Tubulin and Lamin A were used as loading control for cytosolic and nuclear fraction respectively. Immunoblotting quantification was expressed in A.D.U. (arbitrary density unit) and as mean ± SEM. **p < 0.01, ***p < 0.001 vs Ctrl; #p < 0.05, ##p < 0.01vs PGE2; §p < 0.05 vs EGF.
PKA, AKT and PKC are not required for PGE2-induced nuclear translocation of EGFR
To further explore the molecular mechanism, we investigated the functional contribution of potential PGE2-EP3 downstream signaling pathways. EP3 receptor consists of multiple isoforms generated by alternative splicing, which upon binding of PGE2 trigger different downstream effectors, including protein kinase A (PKA), protein kinase C (PKC), SRC and phosphoinositide 3 kinase (PI3K) known to mediate EGFR activation [33]. To examine which of the protein kinases downstream of EP3 might be critical for EGFR nuclear localization, we assessed the effect of PGE2 treatment on EGFR nuclear translocation in the presence of selective inhibitors targeting PKA (H89), PI3K/AKT (LY294002) and PKC (Go6983). None of the selective protein kinases inhibitors affected PGE2/EP3-induced EGFR nuclear translocation in A549 (Figure 7A and 7B) and GLC82 cells ( C) excluding functional contribution of PKA, PI3K/AKT and PKC to PGE2-mediated EGFR nuclear translocation.
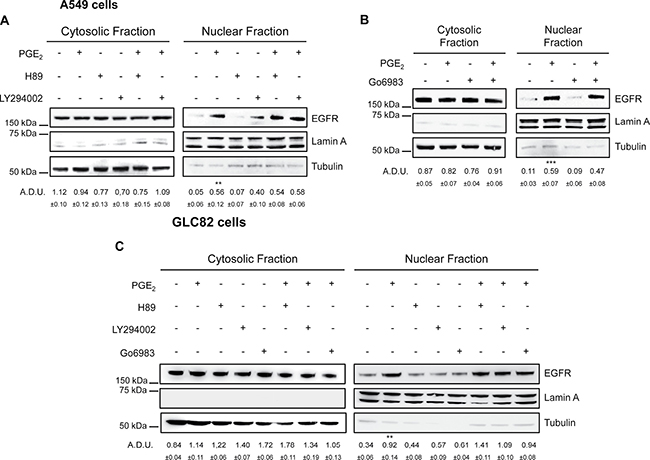
Figure 7: EGFR nuclear translocation in response to PGE2 does not involve PKA, AKT and PKC. (A, B, C) A549 and GLC82 cells were starved overnight and then treated with 1 μM H89 or 10 μM LY294002 or 10 μM Go6983 for 30 min before challenge with 1 μM PGE2 for 60 min. Immunoblotting analysis of EGFR expression on cytoplasmic and nuclear fractions was then performed. Tubulin and Lamin A were used as loading control for cytosolic and nuclear fraction respectively. Immunoblotting quantification was expressed in A.D.U. (arbitrary density unit) and as mean ± SEM. **p < 0.01, ***p < 0.001 vs Ctrl.
PGE2/EP3 induces EGFR nuclear translocation via SRC family kinases
In addition to the kinases mentioned above, PGE2 can also activate SRC Family Kinases (SFK) via EP3 receptor [34, 35]. Notably, this pathway has been shown to serve as a signaling mediator between G protein coupled receptors (GPCRs) and EGFR, as well as downstream effectors of the EGFR [17, 18, 36, 37]. To assess the role of SRC in PGE2 induced EGFR nuclear translocation and its relation with EGFR activation, NSCLC cells were treated with PGE2 in the presence of pharmacological inhibitors of SRC and EGFR. The SFK inhibitors PP1 or SU6656 abolished PGE2-induced EGFR nuclear translocation (Figure 8A, 8B, 8C) whereas did not influence EGF activity (Supplementary Figure 9A and 9B). Inhibition of EGFR activity by AG1478, did not affect PGE2-mediated SRC phosphorylation (Figure 8D left and right). To confirm the central role of SFK in EGFR translocation, a constitutively active SRC (pcSRC-Y527F) was overexpressed in A549 cells. The forced activation of c-SRC, documented by enhanced pSRC phosphorylation, led to an increase in nuclear EGFR localization (Supplementary Figure 9C). Thus, PGE2/EP3 signaling acts via SRC to promote EGFR nuclear translocation. However, SRC activation by PGE2 does not involve the tyrosine kinase activity of EGFR suggesting that SRC is activated by PGE2 upstream of EGFR, subsequently leading to EGFR activation and nuclear localization.
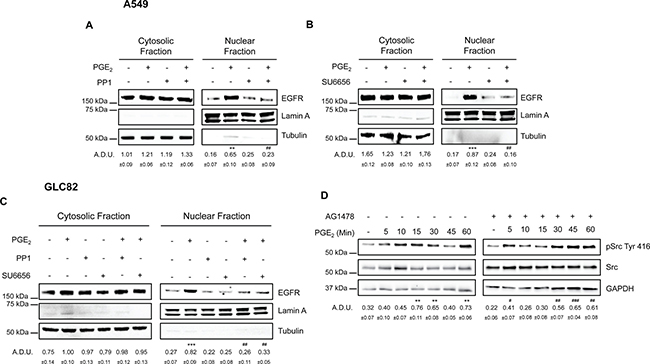
Figure 8: SRC family kinases play a pivotal role in PGE2 induced EGFR nuclear translocation. Immunoblotting analysis of EGFR expression in cytosolic and nuclear fraction in A549 (A, B) and GLC82 (C) exposed for 60 min to 1 μM PGE2 with or without 10 μM PP1 or SU6656. Tubulin and Lamin A were used as loading control for cytosolic and nuclear fraction respectively. Immunoblotting quantification was expressed in A.D.U. (arbitrary density unit) and as mean ± SEM. **p < 0.01, ***p < 0.001 vs Ctrl; ##p < 0.01 vs PGE2. (D) Immunoblotting analysis of SRC phosphorylation on Tyr 416 in A549 exposed for 0–60 min to 1 μM PGE2 with or without 10 μM AG1478. GAPDH was used as loading control. Immunoblotting quantification of pSRC Tyr 416, normalized to SRC, was expressed in A.D.U. and as mean ± SEM. **p < 0.01 vs Ctrl; #p < 0.05, ##p < 0.01, ###p < 0.001 vs Ctrl exposed to AG1478.
SRC/ADAMs signaling mediates PGE2-induced release of EGFR ligands
Since EP-SRC signaling has been reported to activate EGFR by inducing the release of EGFR ligands from the cell membranes [16, 21, 38], we investigated whether PGE2 promoted the shedding of EGF-like ligands in NSCLC cells. Cleavage of EGFR ligands is mediated mainly by A disintegrin and metalloproteinases, ADAMs, in particular ADAM10 and ADAM17 represented the major sheddases in mammals [39]. Notably, matrix metalloproteinases (MMPs), such as MMP-2 and MMP-9 are reported to be important regulators in GPCR-induced EGFR ligands shedding [26, 40]. Treatment of A549 or GLC82 cells with the broad-spectrum metalloproteinase inhibitor GM6001 (10 μM or 25 μM) before adding PGE2 blocked EGFR nuclear accumulation, indicating that ADAMs-MMPs activation was required for PGE2-induced EGFR nuclear translocation (Figure 9A and Supplementary Figure 10A). To explore the contribution of MMP-2, MMP-9, ADAM17 and ADAM10 in mediating the putative PGE2-induced EGFR ligands release in NSCLC cells, we assessed their basal expression using qRT-PCR. A549 and GLC82 cells expressed low levels of MMP-2 and MMP-9, whereas ADAM17 and ADAM10 were highly expressed (Supplementary Figure 10B). Additionally, to examine whether PGE2 might induces MMPs activation, we performed a gelatin zymography. No lytic activity was observed in both A549 and GLC82 cells exposed to PGE2 treatment for 30 min (Supplementary Figure 10C) indicating that ADAMs mediate PGE2-induced EGFR ligands cleavage.
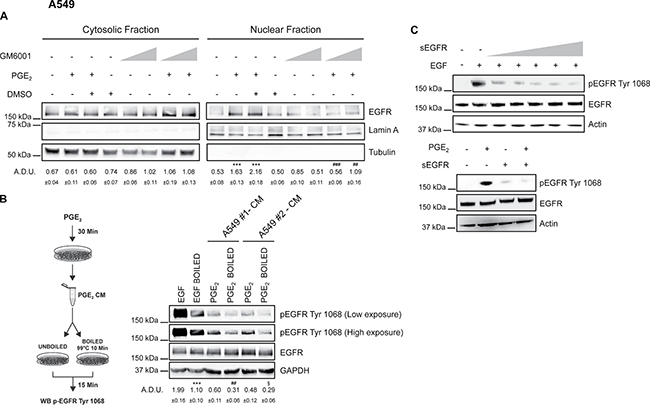
Figure 9: PGE2 acts via shedding of EGF-like ligands to promote EGFR nuclear translocation. A549 were starved overnight and then pre-treated with 10 μM or 25 μM GM6001 before challenge with 1 μM PGE2 for 60 min. DMSO, matching the solvent concentration of 25 μM GM6001, was used as a control. (A) Immunoblotting analysis of EGFR expression in cytosolic and nuclear fraction in A549 treated as described above. Tubulin and Lamin A were used as loading control for cytosolic and nuclear fraction respectively. Immunoblotting quantification was expressed in A.D.U. (arbitrary density unit) and as mean ± SEM. ***p < 0.001 vs Ctrl; ##p < 0.01, ###p < 0.001 vs PGE2+DMSO. (B) Overnight starved A549 cells were treated or not with 1 μM PGE2 for 30 min. Conditioned medium (CM) was collected from each group and subjected or not to heat inactivation at 99°C for 10 min. Serum-starved A549 cells were stimulated with boiled or unboiled CM for 15 min and then analyzed by immunoblotting of whole cell lysate. Boiled or unboiled medium supplemented with 25ng/ml EGF of 1μM PGE2 was used as technical control (data not shown). Immunoblotting analysis of EGFR phosphorylation on Tyr1068 was then performed. A549#1 and A549#2 represent two biological replicates. Low and high exposure were acquired to show protein modulation. GAPDH was used as loading control. Immunoblotting quantification of pEGFR Tyr 1068 (High exposure), normalized to EGFR, was expressed in A.D.U. and as mean ± SEM. ***p < 0.001 vs EGF; ##p < 0.01 vs PGE2 A549#1; §p < 0.05 vs .PGE2 A549#2. (C) A549 cells (left panel) were incubated for 10 min with 5 ng/ml EGF and increasing concentrations of soluble EGFR (sEGFR) (0, 1, 5, 10, 25, 50 μg/ml). Immunoblotting analysis of EGFR phosphorylation on Tyr1068 and total EGFR expression in A549 exposed to 1 μM PGE2 and 50μg/ml sEGFR was performed (right panel). Actin was used as loading control.
Consistent with the hypothesis that EGFR ligands might be released following PGE2 treatment by SRC and subsequent ADAMs activation, conditioned medium (CM) of A549 cells treated with PGE2 for 30 min was collected and added to untreated A549 to assess EGFR phosphorylation. Putative EGF-like ligands were inactivated in the CM either by heat inactivation and/or by soluble EGFR (sEGFR) as a decoy receptor [19, 41]. CM from two different cell clones, A549#1 and A549#2, was collected and processed for heat inactivation. EGFR phosphorylation was dramatically reduced by heat inactivation of CM suggesting that PGE2 activated EGFR by the release of EGF-like ligands in the medium (Figure 9B). Consistently, when sEGFR was added at the maximally effective concentration of 50 μg/ml to A549 and GLC82 exposed to PGE2, EGFR phosphorylation was abolished (Figure 9C left and right and Supplementary Figure 10D). Taken together, these experiments indicate that PGE2 activates EGFR by the release of EGF-like ligands in NSCLC cells.
PGE2 requires EGFR ligands to promote EGFR nuclear internalization
EGFR is activated by seven ligands, including EGF, HB-EGF, TGFα, AREG, EREG, EPGN and BTC which are all produced as membrane-bound precursor proteins and released by different proteases such as ADAMs [39]. To identify the EGFR ligand(s) involved in PGE2-mediated EGFR nuclear translocation, we assessed their basal expression in NSCLC cells using quantitative RT-PCR (qRT-PCR). Both A549 and GLC82 cells expressed variable levels of EGFR ligands (Figure 10A and Supplementary Figure 11A). To assess whether PGE2 induced the expression of EGFR ligands, A549 and GLC82 were exposed to the prostanoid for a time frame of 2–24 h, and mRNA levels were determined by qRT-PCR. Upon PGE2 treatment, EGFR ligands were not regulated in both NSCLC cells (Figure 10B and Supplementary Figure 11B). The expression of the most expressed ligands for each cell line was individually ablated by siRNA-mediated knockdown, and PGE2-dependent EGFR nuclear translocation was assessed. In A549 cells, PGE2-induced EGFR nuclear translocation was significantly inhibited by AREG depletion. A reduction of EGFR nuclear accumulation was also observed in EREG silenced cells, whereas in TGFα and HB-EGF knockdown cells, EGFR internalization was marginally affected (Figure 10C), indicating that AREG and EREG were the main ligands mediating PGE2-induced EGFR nuclear translocation in A549. Similarly, in GLC82 cells, AREG knockdown significantly decreased PGE2-induced EGFR nuclear translocation, although significant reduction of EGFR internalization was also observed in EREG and EGF depleted cells (Supplementary Figure 11C). Taken together these data indicate that several EGFR ligands mediate PGE2 activity depending on the cell type, with AREG and EREG being the main involved.
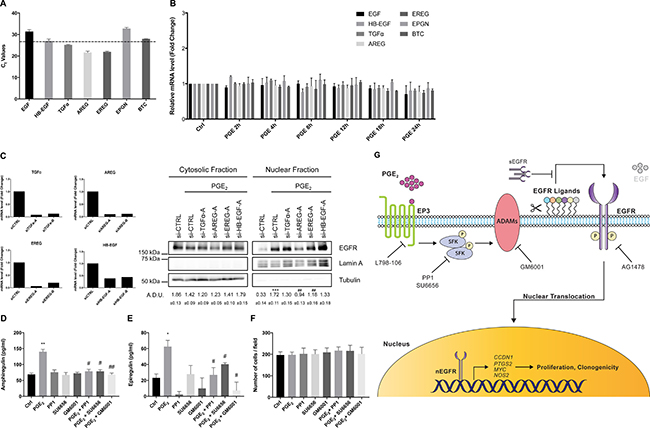
Figure 10: EGFR ligands mediate PGE2-dependent EGFR nuclear translocation. (A) qRT-PCR analysis of basal mRNA expression for EGFR ligands in A549. Results are presented as mean of Ct values ± SEM of two independent experiments. (B) A549 cells were starved overnight and then treated with 1 μM PGE2 for 2, 4, 8, 12, 18 and 24 h. RNA was isolated and analyzed by qRT-PCR for EGFR ligands. The data are presented as mean of fold change ± SEM of three independent experiments, relative to non-treated cells (Control), which were assigned to 1. (C) mRNA expression analysis of EGFR ligands by qRT-PCR in A549 silenced for 48 h for AREG, EREG, TGF-alpha and HB-EGF with two different oligos (siRNA A and B) (left panel). EGFR expression analysis by immunoblotting of cytosolic and nuclear fraction in A549 silenced for EGFR ligands (right panel). Tubulin and Lamin A were used as loading control for cytosolic and nuclear fraction respectively. Immunoblotting quantification was expressed in A.D.U. (arbitrary density unit) and as mean ± SEM. ***p < 0.001 vs siCTRL; ##p < 0.01 vs siCTRL+PGE2. Similar data were obtained with siRNA-B (Data not shown). (D, E) ELISA for AREG (D) and EREG (E) in conditioned media from A549 exposed for 60 min to PGE2 (1 μM), PP1 or SU6656 (10 μM) or GM6001 (10 μM), or PGE2 + PP1, PGE2 + SU6656 and PGE2 + GM6001. Data are reported as pg/ml. *p < 0.05, **p < 0.01 vs Ctrl (control condition); #p < 0.05, ##p < 0.01 vs PGE2. (F) Cell number in wells exposed to conditions described above for 60 min to obtain the conditioned media. (G) Schematic representation of a working model for PGE2-induced EGFR nuclear translocation. L798-106, EP3 receptor inhibitor; SFK, SRC family kinases; PP1 and SU6656, SRC family kinases inhibitors; GM6001, broad spectrum ADAM and MMP inhibitor; sEGFR, soluble EGFR; AG1478, EGFR Tyrosine kinase inhibitor; nEGFR, nuclear EGFR.
An ELISA assay for AREG and EREG corroborated the contribution of these EGFR ligands to PGE2 activity (Figure 10D and 10E, Supplementary Figure 11D and S11E). In tumor cells treated with PGE2 for 60 min the levels of AREG and EREG increased, whereas in the presence of c-SRC inhibitors (PP1 or SU6656) or ADAM-MMPs inhibitor (GM6001), AREG and EREG levels declined towards the baseline (Figure 10D and 10E, Supplementary Figure 11D and 11E). This result also supports the notion that c-SRC acts downstream of PGE2 to mediate ADAMs activation and EGFR ligands release. Cell numbers were not affected by these treatments (Figure 10F and Supplementary Figure 11F).
In summary, we have identified PGE2 as an inducer of EGFR nuclear translocation in human NSCLC cells. PGE2 coupling with EP3 receptor orchestrates a complex mechanism involving the activation of SFK and of ADAMs to release EGFR ligands, in particular AREG and EREG. Once activated by its ligands, EGFR translocates to the nucleus where it promotes transcription of genes implicated in cell cycle progression and inflammation leading to increased cell proliferation and clonogenicity (Figure 10G).
DISCUSSION
The findings presented in this study support a new model for the function of PGE2 in tumor growth control and adaptation to the microenvironment, in which the prostanoid regulates EGFR activity by inducing its nuclear internalization. Collectively, our data uncover a key mechanism by which tumor cells attain central hallmarks of cancer by PGE2-mediated EGFR nuclear localization.
EGFR is a tyrosine kinase receptor located at the cell surface. In addition to the classical signaling, the full-length EGFR can be shuttled from the plasma membrane to the nucleus in which it serves as co-transcriptional factor and tyrosine kinase [42, 43]. Nuclear EGFR contributes to promote an aggressive phenotype of cancer cells and correlates with poor prognosis and chemo-resistance in different cancer types, including NSCLC [44].
In this report, we identify PGE2 as a novel regulator of EGFR nuclear translocation that induces EGFR-mediated tumor cell progression. We delineate the molecular mechanisms and signaling pathways by which PGE2 induces EGFR nuclear import and promotes nuclear EGFR-mediated gene transcription in lung adenocarcinoma cells, demonstrating a role for the prostanoid as a critical mediator of EGFR oncogenicity (Figure 10G).
Transcriptional regulation of genes involved in cell proliferation, tumor progression, inflammation and chemo-resistance are among the main functions of nuclear EGFR [43]. We have analyzed a panel of nuclear EGFR target and we find that cyclin D1, COX-2, iNOS and c-Myc mRNA levels are upregulated by PGE2 as well as by EGF. The kinetic by which the prostanoid promotes EGFR nuclear translocation and gene transcription appears delayed compared to EGF, suggesting that additional effectors are involved. Among the genes upregulated by EGFR internalization, increased expression of COX-2, the key enzyme in PGE2 biosynthesis, indicates a positive feedback loop between PGE2/EGFR and COX-2, an instrumental regulatory circuit for the amplification of malignant tumor progression. EGFR nuclear translocation positively correlates with features of tumor aggressiveness: A549 and GLC82 cells expressing wild type EGFR increase their clonogenicity and proliferation in response to PGE2 or EGF in contrast to cells expressing a mutant EGFR lacking its nuclear localization sequence.
PGE2 exerts its pleiotropic effects by binding to four GPCR (EP1-4) [45]. Using selective EP agonists and antagonists and RNA interference experiments we demonstrate that EP3 is required for PGE2-mediated EGFR nuclear translocation in NSCLC cells. This interplay between EP3 and EGFR has been reported previously [46, 47]. In airway epithelial cancer cells, EP3 receptor promotes EGFR-mediated IL-8 production and tumor progression via EGFR ligand shedding [46]. Here, we document that in NSCLC cells PGE2-activated EP3 promotes nuclear EGFR translocation by activating SRC family kinases (SFK), which in turn activate ADAMs to cleave and shed EGFR ligands (Figure 10G).
EP3 receptor has multiple isoforms, and its activation can be coupled to adenylyl cyclase [48] and to Ca2+-mobilization/PKC activation [49, 50]. EP3 receptor can also stimulate cAMP production leading to PKA activation [51, 52]. In addition PKC [53] and PI3K/AKT [50, 54] are known to act as downstream effectors of EP3 and their contribution to EGFR phosphorylation and nuclear translocation has also been reported [55, 56]. In our setting, PGE2/EP3-mediated EGFR trafficking into the nucleus requires EGFR’s kinase activity, in contrast to conflicting reports on the importance of an active kinase domain in EGFR nuclear translocation [57–61]. Our experiments reveal that neither PKA, AKT nor PKC are involved in PGE2-mediated EGFR nuclear translocation. Conversely, SFK inhibitors markedly impair PGE2-mediated EGFR translocation into the nucleus. Thus, we demonstrate that PGE2/EP3 acts through SFK to induce EGFR activation and nuclear translocation, a finding consistent with the observation that in NSCLC cells EP3 is functionally connected to SFK [35]. SRC can acts as an upstream or downstream modulator of receptor tyrosine kinases [62]. In our setting, PGE2-induced c-SRC phosphorylation appears to be independent of EGFR activation indicating a direct link between SRC and PGE2 in promoting EGFR nuclear translocation. The kinetic of cSRC phosphorylation by PGE2 was biphasic, as we observed an early peak at 10–15 min and a second delayed peak at 60 min, suggesting that PGE2 functions as the initial trigger for a sustained amplification of malignant tumor progression.
PGE2 transactivates EGFR by inducing ADAMs-mediated proteolytic release of membrane-bound EGF-like ligands [16, 21, 38], and SFK members play a central role in the release of the ligands [63, 64]. SFK inhibitors block PGE2-induced EGFR nuclear translocation, as do ADAM and MMP inhibitors, suggesting that EGF-like ligands are shedded in NSCLC cells treated with PGE2. A dramatic reduction in the extent of EGFR phosphorylation occurs in cells, when conditioned medium from NSCLC cells is denatured or depleted of EGFR ligands with a soluble EGFR trap, demonstrating that PGE2 promotes EGFR activation and internalization through cleavage of membrane-bound EGFR ligands. Among the various EGFR ligands, we demonstrate that the shedding of Amphiregulin (AREG) and Epiregulin (EREG) play a central role in EGFR trafficking. Both ligands are known oncogenic factor [65–68]. In advanced NSCLC patients, increased AREG expression correlates with a poor response to therapy, and several studies have identified AREG as a biomarker for an efficient response to EGFR-targeted therapies [69–71]. Further, PGE2 has been reported to promote AREG induction and to stimulate growth of colon cancer cells [72]. Similarly, elevated EREG expression in NSCLC is associated with aggressive tumor phenotypes and unfavourable prognosis [73–75]. Additionally, in several tumor cell lines, COX-2 and EREG have been identified as metastasis associated genes [76, 77].
In summary, we have identified PGE2 as an inducer of EGFR nuclear translocation in human NSCLC cells. We propose the following mechanistic sequence of events: tumor stroma and/or tumor cells release PGE2, which couples with EP3 receptor and orchestrates a complex mechanism culminating in EGFR activation and translocation into the nucleus. We show that upon PGE2/EP3 interaction, SFKs activate ADAMs proteases, which in turn mediate the shedding of EGFR ligands, such as the oncogenic AREG and EREG, and then EGFR activation and nuclear internalization. Within the nucleus, EGFR induces the expression of iNOS, COX-2, c-Myc and cyclin D1, thus reprogramming important tumor growth parameters, including tumor cell proliferation and malignant progression (Figure 10G). The delayed gene transcription, observed in NSCLC cells exposed to PGE2, represents a clear functional evidence of the involvement of the release of EGFR ligands in this mechanism. Thus, it appears that, through EGFR nuclear translocation, PGE2 was able to amplify a robust oncogenic response, sustained by a wide variety of inflammatory and pro-proliferative genes.
The presence of receptor tyrosine kinase into the nucleus opens a new field of research. Here we demonstrate the significance of the nuclear translocation induced by PGE2-mediated GPCR signaling and its biological functions. These mechanisms regulating tumor growth and malignant progression may offer attractive opportunities for the design and development of innovative cancer therapies.
MATERIALS AND METHODS
Cell culture and cultured conditions
The human NSCLC cancer cell line A549 (CCL-185), was obtained from American Type Culture Collection and the GLC82 NSCLC cell line was kindly provided by Dr. Mario Chiariello (Istituto Toscano Tumori, Siena, Italy). Cells were certified by STRA, (LGC Standards S.r.l., Sesto San Giovanni, Milan, Italy) and were maintained in DMEM for A549 and in RPMI-1640 (Euroclone, Milan, Italy) for GLC82 supplemented with 10% FBS and 2 mM Glutamine, 100 Units Penicillin and 0.1 mg/l Streptomycin (Sigma Aldrich, St. Louis, MO, USA) in a humidified incubator with 5% CO2 at 37°C. A549 and GLC82 were immediately expanded after delivery (up to 6 × 107 cells) and frozen down (1 × 106 per vial) such that both cell lines could be restarted after a maximum of 10 passages every 3 months from a frozen vial of the same batch of cells. Control of mycoplasma was done from a frozen vial.
Chemical and reagents
Recombinant human EGF and soluble EGFR were purchased from PeproTech (Rocky Hill, NJ, USA). PGE2, L-798106, PP1 and SU6656 were purchased from Sigma Aldrich. Butaprost (EP2 agonist), Sulprostone (EP3 agonist), L-902,688 (EP4 agonist), Tyrphostin AG-1478 and GM6001 were obtained from Cayman Chemicals (Ann Arbor, MI, USA). H89 and LY294002 were purchased from Calbiochem (Darmstadt, Germany). Go6983 was obtained from Tocris (Bristol, United Kingdom).
Antibodies
Anti-EGFR, anti-pEGFR Tyr 1068, anti-AKT, anti-pAKT Ser 473, anti-ERK1/2, anti-pERK1/2, anti-SRC, anti-pSRC Tyr 416 antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-Tubulin and anti-EP3 receptor antibodies were purchased from Santa Cruz (Heidelberg, Germany). Anti-Lamin A, anti-Actin and anti-GAPDH antibodies were obtained from Sigma Aldrich. Anti-EGFR (N-terminal) was purchased from Abcam (Cambridge, United Kingdom).
Whole cell extracts
Cells were washed 2× with cold Dulbecco’s Phosphate Buffered Saline (Sigma-Aldrich) and lysed as described previously [78].
Cell fractionation
Nuclear and cytoplasmic extracts were prepared with NE-PER™ nuclear and cytoplasmic extraction reagents (ThermoFisher Scientific, Waltham, MA, USA) following the manufacturer’s instructions.
Immunoblotting analysis
4 × 105 cells were plated in 60 mm dishes, serum deprived (0.1%. fetal calf serum, overnight), then treated as described in the text. Immunoblot analysis was performed as described previously [17]. Signals were detected by SuperSignal WestPico Chemiluminescent Substrate (ThermoFisher Scientific) using ChemiDoc system and Quantity one software (Bio-Rad, Hercules, CA, USA). All experiments were performed at least three times. For all experiments using whole cell lysate, GAPDH or Actin were used as loading control. Lamin-A and Tubulin were used as loading and purity controls for the nuclear and cytosolic fractions, respectively. Immunoblots were analyzed by densitometry using NIH Image J 1.48v software, and the results, expressed as arbitrary density units (A.D.U.), were normalized to GAPDH, Actin, Lamin-A or Tubulin.
Immunofluorescence microscopy analysis
Cells were plated on 12 mm ø glass coverslips, starved overnight and treated according to the experimental design. Cells were fixed and incubated with anti-EGFR antibody followed by AlexaFluor® 488-labeled secondary antibody (Invitrogen, Carlsbad, CA, USA) as previously described [78]. 6-diamidino-2- phenylindole (DAPI) 1 μg/ml (Sigma-Aldrich) was used to stain the nuclei. Cells were imaged with a confocal laser scanning microscope Leica SP5. Images for documenting EGFR nuclear translocation were acquired in the middle section of the nuclei with 63× magnification. Confocal stacks were 3D-reconstructed with Imaris Software (Bitplane, Zurich, Switzerland).
Transfection of siRNAs and plasmids
siRNAs used for transient knock-down experiments were purchased from Qiagen (Hilden, Germany) and Ambion (Carlsbad, CA, USA). Cells were transfected with 20 nM targeting siRNA or scrambled control siRNA using Lipofectamine® RNAiMAX (Invitrogen) according to manufacturer’s instructions. Cells were assayed 48–72 h after transfection. Knockdown efficiency was assessed by immunoblotting or quantitative RT-PCR analysis. Target sequences are listed in Supplementary Table 1.
For DNA transfection, cells were transfected with 1–10 μg plasmid using Lipofectamine® 2000 (Invitrogen) according to manufacturer’s instructions. EGFR WT and NLS mutant plasmids (NLSm12 and dNLS) were kindly provided by Prof. Mien-Chie Hung (University of Texas MD Anderson Cancer Center, Houston, TX, USA) [32]. pSpCas9(BB)-2A-GFP (PX458) (#48138) pEVX (#17675), and pSRCY527F (#17675) were from Addgene. Cells were analyzed 24–72 h post-transfection.
Knockout of EGFR by CRISPR/Cas9-mediated genome editing
A549 EGFR knockout cells were generated by a CRISPR/Cas9 approach as described [79]. The sgRNA with the sequence TCGTTCGGAAGCGCACGCTGCGG within the EGFR gene was obtained using the CRISPR Design Tool (http://tools.genome-engineering.org). sgRNA targeting EGFR was cloned into BbsI (NEB, Ipswich, MA, USA) digested pSpCas9(BB)-2A-GFP (PX458) plasmid (Addgene #48138) using the oligos: Forward CACCGTCGTTCGGAAGCGCACGCTG and Reverse AAACCAGCGTGCGCTTCCGAACGAC. Cells were transiently transfected with 1 μg PX458 using an Amaxa Nucleofector machine (Lonza, Basel, Switzerland) according to manufacturer’s instructions. 48 h post transfection, GFP-expressing cells were FACS sorted and re-seeded at limiting dilution in 96-well plates in order to obtain individual clones. GLC82 EGFR knockout cells were generated using CRISPR/Cas9 human gene knockout kit (OriGene, Rockville, MD, USA) following manufacturer’s instructions. EGFR ablation was assessed by immunoblot with two different anti-EGFR antibodies targeting C- and N-terminal residues respectively.
RNA isolation and quantitative RT-PCR
Total RNA was prepared using Tri Reagent® (Sigma-Aldrich) following manufacturer’s instructions. 1 μg RNA was reverse transcribed using ImProm- II™ Reverse Transcriptase (Promega, Madison, WI, USA) and quantitative RT-PCR was performed using SYBR-green PCR MasterMix (Applied Biosystems, Waltham, MA, USA) in a StepOne Plus PCR machine (Applied Biosystems). Fold change expression was determined by the comparative Ct method (ΔΔCt) normalized to 60S Ribosomal protein L19 expression. qRT-PCR data are represented as fold increase relative to non-treated cells (Control), which were assigned to 1. Primers for quantitative RT-PCR are listed in Supplementary Table 2.
Conditioned medium and EGFR ligands denaturation
5 × 105 A549 cells were plated into 60 mm dishes, incubated for 24 h and then starved overnight with medium supplemented with 0.1% FBS. Then, cells were incubated with or without 1 μM PGE2 for 30 min in a total volume of 4 ml. Conditioned medium (CM) was collected and boiled or not at 99°C for 10 min. As control for EGFR ligands denaturation efficiency, boiled or unboiled medium supplemented with EGF 25 ng/ml was used. Next, media of serum-starved A549 cells were replaced with either 2 ml of boiled or unboiled CM or with 2 ml of boiled or unboiled medium derived from controls. Cells were treated for 15 min and subsequently analyzed by immunoblotting.
Gelatin zymography
5 × 103 A549 and GLC82 cells were plated into 96-well plates in medium supplemented with 10% FBS. After adhesion, cells were washed with PBS and starved overnight in serum-free medium. 1 μM PGE2 was added to 50 μL of fresh serum-free media for 30 min. Next, conditioned medium (CM) was collected and mixed with loading buffer. Zymography was carried out in SDS/8% PAGE containing 0.1% gelatin as described [31].
ELISA
1 × 105 A549 and GLC82 cells were plated into 12-well plates and incubated until 80–90% confluency. Then, cells were starved overnight and treated as described in the text. CM was collected and Amphiregulin and Epiregulin levels were measured using an ELISA kit R&D Systems, Minneapolis, MN, USA for Amphiregulin and an ELISA kit MyBioSource, San Diego, CA, USA for Epiregulin, following the manufacturer’s instructions.
MTT assay
Cell proliferation was quantified by the Vybrant MTT cell proliferation assay as previously described [17]. Briefly, A549 and GLC82 EGFR knockout cells were transfected with EGFR WT and NLS mutant plasmids for 24 h. Next, transfected cells were seeded (3 × 103) into 96-well plates, starved overnight and treated with either 25 ng/ml EGF or 1 μM PGE2. 48 h post treatment, cell were exposed to MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma-Aldrich) for 4 h in fresh medium without phenol red. Absorbance at 540 nm was measured with Infinite 200 Pro SpectraFluor microplate absorbance reader (Tecan, Mannedorf, Switzerland). Data of three independent experiments are presented as % relative to untreated cells (Control), which were assigned to 100%.
Clonogenic assay
A549 and GLC82 EGFR knockout cells were transfected with EGFR WT and NLS mutant plasmids for 24 h. Transfected cells were seeded (5 × 102) into 6-well plates and incubated in medium supplemented with 10% FBS for 12 h. Then, cells were treated in triplicates with 25 ng/ml EGF or 1 μM PGE2 in 1% FBS medium. 10 days after treatment, cells were stained with Panreac kit (Darmstadt, Germany), and colonies (> 50 cells) were counted; data are expressed as % relative to untreated cells (Control), which were assigned to 100%.
Statistical analysis
Statistical analysis and graphs were generated using the GraphPad Prism software (San Diego, CA, USA). All statistical analysis was done by unpaired/paired Student’s t-test, p-value < 0.05 was considered significant.
ACKNOWLEDGMENTS
We thank Pascal Lorentz (BioOptics Facility, Department of Biomedicine, University of Basel) for support with confocal microscopy as well as Fengyuan Tang (Department of Biomedicine, University of Basel) and Maurizio Abbate for scientific and technical help. We thank Antonio Giachetti for critical discussion and manuscript revision. We are grateful to Prof. Mien-Chie Hung (MD Anderson Cancer Center, University of Texas) for providing EGFR plasmids and to Dr. Mario Chiariello (Istituto Toscano Tumori) for GLC82 cells.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work was supported by Associazione Italiana della Ricerca sul Cancro (AIRC) IG10731, Istituto Toscano Tumori (ITT), MIUR-PRIN (2015Y3C5KP), the Swiss National Science Foundation (310030B-163471) and the Swiss Cancer League (KFS-3479-08-2014).
REFERENCES
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674.
2. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013; 19:1423–1437.
3. Donnini S, Finetti F, Terzuoli E, Bazzani L, Ziche M. Targeting PGE2 Signaling in Tumor Progression and Angiogenesis. Forum on Immunopathological Diseases and Therapeutics. 2014; 5:2014.
4. Murphey LJ, Williams MK, Sanchez SC, Byrne LM, Csiki I, Oates JA, Johnson DH, Morrow JD. Quantification of the major urinary metabolite of PGE2 by a liquid chromatographic/mass spectrometric assay: determination of cyclooxygenase-specific PGE2 synthesis in healthy humans and those with lung cancer. Anal Biochem. 2004; 334:266–275.
5. Achiwa H, Yatabe Y, Hida T, Kuroishi T, Kozaki K, Nakamura S, Ogawa M, Sugiura T, Mitsudomi T, Takahashi T. Prognostic significance of elevated cyclooxygenase 2 expression in primary, resected lung adenocarcinomas. Clin Cancer Res. 1999; 5:1001–1005.
6. Ermert L, Dierkes C, Ermert M. Immunohistochemical expression of cyclooxygenase isoenzymes and downstream enzymes in human lung tumors. Clin Cancer Res. 2003; 9:1604–1610.
7. Yoshimatsu K, Altorki NK, Golijanin D, Zhang F, Jakobsson PJ, Dannenberg AJ, Subbaramaiah K. Inducible prostaglandin E synthase is overexpressed in non-small cell lung cancer. Clin Cancer Res. 2001; 7:2669–2674.
8. McCormack VA, Hung RJ, Brenner DR, Bickeboller H, Rosenberger A, Muscat JE, Lazarus P, Tjonneland A, Friis S, Christiani DC, Chun EM, Le Marchand L, Rennert G, et al. Aspirin and NSAID use and lung cancer risk: a pooled analysis in the International Lung Cancer Consortium (ILCCO). Cancer Causes Control. 2011; 22:1709–1720.
9. Olsen JH, Friis S, Poulsen AH, Fryzek J, Harving H, Tjonneland A, Sorensen HT, Blot W. Use of NSAIDs, smoking and lung cancer risk. Br J Cancer. 2008; 98:232–237.
10. Ziche M, Jones J, Gullino PM. Role of prostPCaglandin E1 and copper in angiogenesis. J Natl Cancer Inst. 1982; 69:475–482.
11. Xia D, Wang D, Kim SH, Katoh H, DuBois RN. Prostaglandin E2 promotes intestinal tumor growth via DNA methylation. Nat Med. 2012; 18:224–226.
12. Li Z, Zhang Y, Kim WJ, Daaka Y. PGE2 promotes renal carcinoma cell invasion through activated RalA. Oncogene. 2013; 32:1408–1415.
13. Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009; 30:377–386.
14. Finetti F, Donnini S, Giachetti A, Morbidelli L, Ziche M. Prostaglandin E primes the angiogenic switch via a synergic interaction with the fibroblast growth factor-2 pathway. Circ Res. 2009; 105:657–666.
15. Donnini S, Finetti F, Terzuoli E, Giachetti A, Iniguez MA, Hanaka H, Fresno M, Radmark O, Ziche M. EGFR signaling upregulates expression of microsomal prostaglandin E synthase-1 in cancer cells leading to enhanced tumorigenicity. Oncogene. 2012; 31:3457–3466.
16. Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS. Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med. 2002; 8:289–293.
17. Donnini S, Finetti F, Solito R, Terzuoli E, Sacchetti A, Morbidelli L, Patrignani P, Ziche M. EP2 prostanoid receptor promotes squamous cell carcinoma growth through epidermal growth factor receptor transactivation and iNOS and ERK1/2 pathways. FASEB J. 2007; 21:2418–2430.
18. Buchanan FG, Wang D, Bargiacchi F, DuBois RN. Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J Biol Chem. 2003; 278:35451–35457.
19. Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, Sahai E, Reis e Sousa C. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell. 2015; 162:1257–1270.
20. Digiacomo G, Ziche M, Dello Sbarba P, Donnini S, Rovida E. Prostaglandin E2 transactivates the colony-stimulating factor-1 receptor and synergizes with colony-stimulating factor-1 in the induction of macrophage migration via the mitogen-activated protein kinase ERK1/2. FASEB J. 2015; 29:2545–2554.
21. Tveteraas IH, Muller KM, Aasrum M, Odegard J, Dajani O, Guren T, Sandnes D, Christoffersen T. Mechanisms involved in PGE2-induced transactivation of the epidermal growth factor receptor in MH1C1 hepatocarcinoma cells. J Exp Clin Cancer Res. 2012; 31:72.
22. Hsieh HL, Sun CC, Wang TS, Yang CM. PKC-delta/c-Src-mediated EGF receptor transactivation regulates thrombin-induced COX-2 expression and PGE production in rat vascular smooth muscle cells. Biochim Biophys Acta. 2008; 1783:1563–1575.
23. Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999; 402:884–888.
24. Joo CK, Kim HS, Park JY, Seomun Y, Son MJ, Kim JT. Ligand release-independent transactivation of epidermal growth factor receptor by transforming growth factor-beta involves multiple signaling pathways. Oncogene. 2008; 27:614–628.
25. Drube S, Stirnweiss J, Valkova C, Liebmann C. Ligand-independent and EGF receptor-supported transactivation: lessons from beta2-adrenergic receptor signalling. Cell Signal. 2006; 18:1633–1646.
26. Cattaneo F, Guerra G, Parisi M, De Marinis M, Tafuri D, Cinelli M, Ammendola R. Cell-surface receptors transactivation mediated by g protein-coupled receptors. Int J Mol Sci. 2014; 15:19700–19728.
27. Han W, Lo HW. Landscape of EGFR signaling network in human cancers: biology and therapeutic response in relation to receptor subcellular locations. Cancer Lett. 2012; 318:124–134.
28. Wang YN, Yamaguchi H, Hsu JM, Hung MC. Nuclear trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene. 2010; 29:3997–4006.
29. Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, Bourguignon L, Hung MC. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001; 3:802–808.
30. Wang YN, Hung MC. Nuclear functions and subcellular trafficking mechanisms of the epidermal growth factor receptor family. Cell Biosci. 2012; 2:13.
31. Finetti F, Solito R, Morbidelli L, Giachetti A, Ziche M, Donnini S. Prostaglandin E2 regulates angiogenesis via activation of fibroblast growth factor receptor-1. J Biol Chem. 2008; 283:2139–2146.
32. Hsu SC, Hung MC. Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J Biol Chem. 2007; 282:10432–10440.
33. Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov. 2011; 10:47–60.
34. Kim JI, Lakshmikanthan V, Frilot N, Daaka Y. Prostaglandin E2 promotes lung cancer cell migration via EP4-betaArrestin1-c-Src signalsome. Mol Cancer Res. 2010; 8:569–577.
35. Yamaki T, Endoh K, Miyahara M, Nagamine I, Thi Thu Huong N, Sakurai H, Pokorny J, Yano T. Prostaglandin E2 activates Src signaling in lung adenocarcinoma cell via EP3. Cancer Lett. 2004; 214:115–120.
36. Zhang J, Kalyankrishna S, Wislez M, Thilaganathan N, Saigal B, Wei W, Ma L, Wistuba, II, Johnson FM, Kurie JM. SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines. Am J Pathol. 2007; 170:366–376.
37. Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem. 1999; 274:8335–8343.
38. Thomas SM, Bhola NE, Zhang Q, Contrucci SC, Wentzel AL, Freilino ML, Gooding WE, Siegfried JM, Chan DC, Grandis JR. Cross-talk between G protein-coupled receptor and epidermal growth factor receptor signaling pathways contributes to growth and invasion of head and neck squamous cell carcinoma. Cancer Res. 2006; 66:11831–11839.
39. Adrain C, Freeman M. Regulation of receptor tyrosine kinase ligand processing. Cold Spring Harb Perspect Biol. 2014; 6.
40. Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011; 278:16–27.
41. Lindzen M, Carvalho S, Starr A, Ben-Chetrit N, Pradeep CR, Kostler WJ, Rabinkov A, Lavi S, Bacus SS, Yarden Y. A recombinant decoy comprising EGFR and ErbB-4 inhibits tumor growth and metastasis. Oncogene. 2012; 31:3505–3515.
42. Brand TM, Iida M, Li C, Wheeler DL. The nuclear epidermal growth factor receptor signaling network and its role in cancer. Discov Med. 2011; 12:419–432.
43. Lee HH, Wang YN, Hung MC. Non-canonical signaling mode of the epidermal growth factor receptor family. Am J Cancer Res. 2015; 5:2944–2958.
44. Traynor AM, Weigel TL, Oettel KR, Yang DT, Zhang C, Kim K, Salgia R, Iida M, Brand TM, Hoang T, Campbell TC, Hernan HR, Wheeler DL. Nuclear EGFR protein expression predicts poor survival in early stage non-small cell lung cancer. Lung Cancer. 2013; 81:138–141.
45. Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999; 79:1193–1226.
46. Kim S, Lewis C, Nadel JA. Epidermal growth factor receptor reactivation induced by E-prostanoid-3 receptor- and tumor necrosis factor-alpha-converting enzyme-dependent feedback exaggerates interleukin-8 production in airway cancer (NCI-H292) cells. Exp Cell Res. 2011; 317:2650–2660.
47. Israel DD, Regan JW. EP prostanoid receptor isoforms utilize distinct mechanisms to regulate ERK 1/2 activation. Biochim Biophys Acta. 2009; 1791:238–245.
48. Namba T, Sugimoto Y, Negishi M, Irie A, Ushikubi F, Kakizuka A, Ito S, Ichikawa A, Narumiya S. Alternative splicing of C-terminal tail of prostaglandin E receptor subtype EP3 determines G-protein specificity. Nature. 1993; 365:166–170.
49. Yamaoka K, Yano A, Kuroiwa K, Morimoto K, Inazumi T, Hatae N, Tabata H, Segi-Nishida E, Tanaka S, Ichikawa A, Sugimoto Y. Prostaglandin EP3 receptor superactivates adenylyl cyclase via the Gq/PLC/Ca2+ pathway in a lipid raft-dependent manner. Biochem Biophys Res Commun. 2009; 389:678–682.
50. Morimoto K, Shirata N, Taketomi Y, Tsuchiya S, Segi-Nishida E, Inazumi T, Kabashima K, Tanaka S, Murakami M, Narumiya S, Sugimoto Y. Prostaglandin E2-EP3 signaling induces inflammatory swelling by mast cell activation. J Immunol. 2014; 192:1130–1137.
51. Rundhaug JE, Fischer SM. Molecular mechanisms of mouse skin tumor promotion. Cancers (Basel). 2010; 2:436–482.
52. Hatae N, Sugimoto Y, Ichikawa A. Prostaglandin receptors: advances in the study of EP3 receptor signaling. J Biochem. 2002; 131:781–784.
53. Kobayashi K, Murata T, Hori M, Ozaki H. Prostaglandin E2-prostanoid EP3 signal induces vascular contraction via nPKC and ROCK activation in rat mesenteric artery. Eur J Pharmacol. 2011; 660:375–380.
54. Zhang J, Zou F, Tang J, Zhang Q, Gong Y, Wang Q, Shen Y, Xiong L, Breyer RM, Lazarus M, Funk CD, Yu Y. Cyclooxygenase-2-derived prostaglandin E promotes injury-induced vascular neointimal hyperplasia through the E-prostanoid 3 receptor. Circ Res. 2013; 113:104–114.
55. Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Kehlbach R, Rodemann HP. Nuclear EGFR shuttling induced by ionizing radiation is regulated by phosphorylation at residue Thr654. FEBS Lett. 2010; 584:3878–3884.
56. Huang WC, Chen YJ, Li LY, Wei YL, Hsu SC, Tsai SL, Chiu PC, Huang WP, Wang YN, Chen CH, Chang WC, Chang WC, Chen AJ, et al. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J Biol Chem. 2011; 286:20558–20568.
57. Brand TM, Iida M, Luthar N, Starr MM, Huppert EJ, Wheeler DL. Nuclear EGFR as a molecular target in cancer. Radiother Oncol. 2013; 108:370–377.
58. Kim HP, Yoon YK, Kim JW, Han SW, Hur HS, Park J, Lee JH, Oh DY, Im SA, Bang YJ, Kim TY. Lapatinib, a dual EGFR and HER2 tyrosine kinase inhibitor, downregulates thymidylate synthase by inhibiting the nuclear translocation of EGFR and HER2. PLoS One. 2009; 4:e5933.
59. Wang Q, Villeneuve G, Wang Z. Control of epidermal growth factor receptor endocytosis by receptor dimerization, rather than receptor kinase activation. EMBO Rep. 2005; 6:942–948.
60. Wang Y, Pennock S, Chen X, Wang Z. Endosomal signaling of epidermal growth factor receptor stimulates signal transduction pathways leading to cell survival. Mol Cell Biol. 2002; 22:7279–7290.
61. Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2009; 315:683–696.
62. Bromann PA, Korkaya H, Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004; 23:7957–7968.
63. Ptasznik A, Gewirtz AM. Crosstalk between G protein-coupled receptors and tyrosine kinase signaling: Src take centre stage. Arch Immunol Ther Exp (Warsz). 2000; 48:27–30.
64. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010; 10:181–193.
65. Busser B, Sancey L, Brambilla E, Coll JL, Hurbin A. The multiple roles of amphiregulin in human cancer. Biochim Biophys Acta. 2011; 1816:119–131.
66. Hurbin A, Dubrez L, Coll JL, Favrot MC. Inhibition of apoptosis by amphiregulin via an insulin-like growth factor-1 receptor-dependent pathway in non-small cell lung cancer cell lines. Ann N Y Acad Sci. 2003; 1010:354–357.
67. Stoll SW, Stuart PE, Swindell WR, Tsoi LC, Li B, Gandarillas A, Lambert S, Johnston A, Nair RP, Elder JT. The EGF receptor ligand amphiregulin controls cell division via FoxM1. Oncogene. 2016; 35:2075–2086.
68. Yuan CH, Sun XM, Zhu CL, Liu SP, Wu L, Chen H, Feng MH, Wu K, Wang FB. Amphiregulin activates regulatory T lymphocytes and suppresses CD8+ T cell-mediated anti-tumor response in hepatocellular carcinoma cells. Oncotarget. 2015; 6:32138–53. doi: 10.18632/oncotarget.5171.
69. Addison CL, Ding K, Zhao H, Le Maitre A, Goss GD, Seymour L, Tsao MS, Shepherd FA, Bradbury PA. Plasma transforming growth factor alpha and amphiregulin protein levels in NCIC Clinical Trials Group BR.21. J Clin Oncol. 2010; 28:5247–5256.
70. Carvalho S, Lindzen M, Lauriola M, Shirazi N, Sinha S, Abdul-Hai A, Levanon K, Korach J, Barshack I, Cohen Y, Onn A, Mills G, Yarden Y. An antibody to amphiregulin, an abundant growth factor in patients’ fluids, inhibits ovarian tumors. Oncogene. 2016; 35:438–447.
71. Ishikawa N, Daigo Y, Takano A, Taniwaki M, Kato T, Hayama S, Murakami H, Takeshima Y, Inai K, Nishimura H, Tsuchiya E, Kohno N, Nakamura Y. Increases of amphiregulin and transforming growth factor-alpha in serum as predictors of poor response to gefitinib among patients with advanced non-small cell lung cancers. Cancer Res. 2005; 65:9176–9184.
72. Shao J, Lee SB, Guo H, Evers BM, Sheng H. Prostaglandin E2 stimulates the growth of colon cancer cells via induction of amphiregulin. Cancer Res. 2003; 63:5218–5223.
73. Sunaga N, Kaira K. Epiregulin as a therapeutic target in non-small-cell lung cancer. Lung Cancer (Auckl). 2015; 6:91–98.
74. Zhang J, Iwanaga K, Choi KC, Wislez M, Raso MG, Wei W, Wistuba, II, Kurie JM. Intratumoral epiregulin is a marker of advanced disease in non-small cell lung cancer patients and confers invasive properties on EGFR-mutant cells. Cancer Prev Res (Phila). 2008; 1:201–207.
75. Sunaga N, Kaira K, Imai H, Shimizu K, Nakano T, Shames DS, Girard L, Soh J, Sato M, Iwasaki Y, Ishizuka T, Gazdar AF, Minna JD, et al. Oncogenic KRAS-induced epiregulin overexpression contributes to aggressive phenotype and is a promising therapeutic target in non-small-cell lung cancer. Oncogene. 2013; 32:4034–4042.
76. Wang X, Colby JK, Rengel RC, Fischer SM, Clinton SK, Klein RD. Overexpression of cyclooxygenase-2 (COX-2) in the mouse urinary bladder induces the expression of immune- and cell proliferation-related genes. Mol Carcinog. 2009; 48:1–13.
77. Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Genes that mediate breast cancer metastasis to lung. Nature. 2005; 436:518–524.
78. Waldmeier L, Meyer-Schaller N, Diepenbruck M, Christofori G. Py2T murine breast cancer cells, a versatile model of TGFbeta-induced EMT in vitro and in vivo. PLoS One. 2012; 7:e48651.
79. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013; 8:2281–2308.