
INTRODUCTION
Despite considerable advancements in diagnosis and treatment of solid tumors, distant metastases remain the main cause of cancer-related mortality. The development of metastasis is a multi-step process. The successful initiation of metastatic growth, termed “metastatic colonization”, is the final and inefficient step for many types of cancer [1, 2]. The critical determinants of whether the disseminated carcinoma cells (the tumor cells disseminated to a distant organ) could succeed in forming significant metastases include not only the intrinsic properties of cancer cells themselves but also local tissue microenvironment and systemic environment such as the cancer-related inflammation.
During the metastatic process, single or small clusters of tumor cells shed from the primary tumors and passage to distant organs. Tumor cells that spread in the peripheral blood are characterized as circulating tumor cells (CTCs) [3]. CTCs are commonly detectable in cancer patients at varying frequency. In some patients, CTCs could reach higher counts in blood (102/ml~103/ml), resulting in the poorer prognosis and shorter survival of the patients [4–6]. CTCs promote tumor progression via multiple pathways, including modulating tumor microenvironment [7–9]. Accordingly, CTCs might be not only the source of metastatic cells in target organs, but also an important factor for promoting tumor cell metastasis. However, little is known about whether CTCs could influence the metastatic colonization of disseminated carcinoma cells by orchestrating tumor associated-inflammation.
The tumor-promoting effects of inflammation are now widely recognized and better understood [10, 11]. Polymorphonuclear leukocytes (PMNs or neutrophils) are the major part of the infiltrated inflammatory cells found in a wide variety of human cancers and animal models [12]. Neutrophils have been found to promote multiple tumor processes, including tumorigenesis, tumor growth, and metastasis. The role of neutrophils in the formation of lung metastasis has been well demonstrated [13]. Neutrophils could release MMP-9 [14], Bv8 [15], Arginase-1 (Arg-1) [16], and inducible nitric oxide synthase (iNOS) [17], which not only promote tumor angiogenesis [14, 15, 17], but also suppress the proliferation and cytotoxicity of CD8+ T cells [16], thus suppressing the anti-tumor immune response and promoting tumor growth and metastasis. Neutrophils can alter their polarization state in the tumor-bearing host, switching from suppressing to promoting roles in tumor metastasis [18, 19]. However, little is known about whether CTCs could induce the conversion of neutrophil function.
In this study, we investigated how CTCs promote tumor metastasis, and found that CTCs could initiate pro-metastatic inflammatory responses, promote neutrophilic infiltration in lung, and induce the conversion of neutrophil function to pro-metastasis, consequently promoting the metastatic colonization of disseminated carcinoma cells.
RESULTS
CTCs promote the metastatic colonization of disseminated carcinoma cells
Recent studies have demonstrated that the vast majority of CTCs are unable to form a metastatic clone [1, 2, 20]. In order to explore whether CTCs could influence the metastatic colonization of the disseminated carcinoma cells, we used B16F0 cells, a non-metastatic melanoma cell line, which could not extravasate (Figure 1A) to form visible metastatic nodules in the lungs (Figure 1B) after intravenous (i.v.) injection. B16F1 cells, a metastatic melanoma cell line, were used to establish the metastasis model. To provide CTCs, B16F0 cells were intravenously injected to the mice inoculated with B16F1 cells. Intriguingly, the intravenously injected B16F0 cells (circulating B16F0 cells) could promote the pulmonary metastasis of intravenously inoculated B16F1 cells (Figure 1C and Supplementary Figure 1A). Furthermore, the intravenous injection of B16F0 cells could not influence the extravasation of B16F1 cells (Figure 1D and Supplementary Figure 1B), indicating that the circulating B16F0 cells may promote the metastatic colonization of the disseminated B16F1 cells and thus contribute to the metastasis. Consistently, the growth of B16F1 cells after intramuscular inoculation could also be promoted by circulating B16F0 cells (Supplementary Figure 1C). Taken together, these results indicated that, in addition to seeding metastases by themselves, CTCs could promote tumor metastasis by facilitating the metastatic colonization of disseminated carcinoma cells.
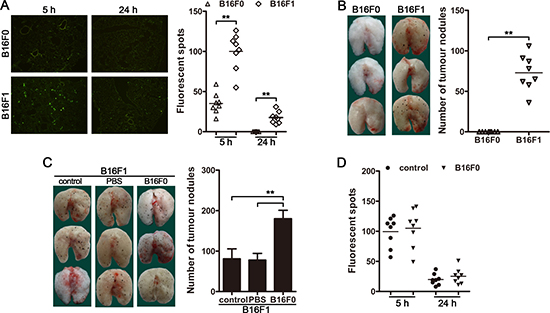
Figure 1: CTCs promote the metastatic colonization of disseminated carcinoma cells. (A) Mice were injected with CFSE-labeled B16F1 or B1F0 cells (5 × 105 cells/mice) via tail vein. The mice (n = 8 in each group) were sacrificed 5 h (for analysis of tumor cell arrest) or 24 h (for analysis of extravasation) after tumor cell injection. Tumor cells in frozen sections were visualized by fluorescence microscopy (left) and counted (right). (B and C) Mice (n = 8 in each group) were intravenously inoculated with B16F1 cells and/or B16F0 cells as described in Methods. Metastatic nodules on the surface of lung (left) were counted (right). PBS was used as control (C). (D) Mice were injected with CFSE-labeled B16F1 cells 12 h after non-labeled B16F0 cells injection via tail vein. The mice (n = 8 in each group) were sacrificed 5 h or 24 h after B16F1 cell injection. Tumor cells in frozen sections were counted. **p < 0.01.
Inflammation is involved in the promoting effect of CTCs on metastasis
Recent data have expanded the concept that inflammation, especially the chronic inflammation, is a critical component for promoting tumor growth and metastasis [10, 11, 21]. We next investigated whether inflammation might be involved in the promoting effect of CTCs on metastasis. For this purpose, we expressed IL-37, an anti-inflammatory cytokine [22] that suppresses the expression of multiple pro-inflammatory cytokines and may also has an anti-tumor effect [22–25], in B16F1-inoculated mice. The inoculation of B16F1 cells induced the inflammation in vivo, as evidenced by the increase of IL-6 and IL-1β in the serum of mice (Figure 2A). The in vivo expression of IL-37 resulted in a significant decrease in lung metastases in B16F1-bearing mice (Figure 2B), accompanied by the inhibition of B16F1 cell-induced inflammation (Figure 2A). To exclude the possibility that IL-37 may have a direct effect on tumor cells, we tested the effect of IL-37 on tumor cell proliferation and colonization. The proliferation in vitro and colony-formation in soft agar of B16F1 cells were not influenced by IL-37 (Figure 2C and 2D). Consistently, B16F1 cells did not express the gene of IL-37 receptor, Sigirr (Il1r8) and Il18r (Supplementary Figure 2A). Collectively, these results validated that inflammation is required for tumor metastasis, and that IL-37 could effectively inhibit tumor metastasis by suppressing the tumor-associated inflammatory response.
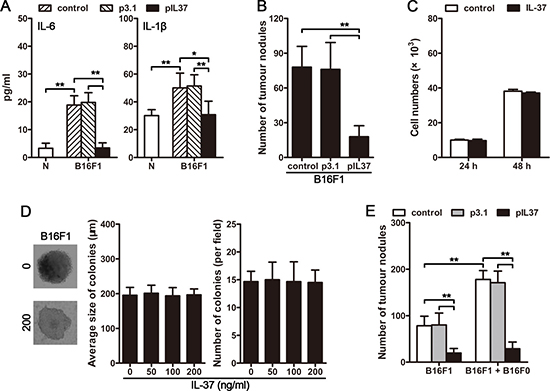
Figure 2: CTC-induced systemic inflammation is essential for the promoting effect of CTCs on tumor metastasis. (A) Mice were intravenously inoculated with B16F1 cells, and received pIL37 plasmid treatment. Serum levels of IL-6 and IL-1β were detected by ELISA on day 4 after primary inoculation. (B) Mice (n = 9 in each group) were intravenously inoculated with B16F1 cells, and received the treatment by i.v. injection of pIL37 plasmid. Metastatic nodules on the surface of lung were counted. (C) B16F1 cells were cultured in the absence or presence of IL-37 (200 ng/ml) for 24 h or 48 h. Then, CCK-8 cell proliferation assay was performed. (D) B16F1 cells were cultured in soft agar for 3 weeks in the absence or presence of IL-37 at the indicated concentration. The representative colonies in the absence (0) or presence of 200 ng/ml IL-37 (200) were photographed (left). The average size of colonies was calculated (middle), and the colonies were counted (right). (E) Mice received the i.v. injection of B16F1 cells with or without B16F0 cells. The mice (n = 9 in each group) were treated with i.v. injection of pIL37 plasmid. Metastatic nodules on the surface of lung were counted. Data are pooled from three independent experiments with a total of six samples in each group (B, C). *p < 0.05, **p < 0.01.
To further ascertain whether CTCs were involved in inducing systemic inflammation that promotes the metastatic colonization of the disseminated carcinoma cells, we next investigated whether circulating B16F0 cells could induce a systemic inflammatory response. The results showed that, after intravenous inoculation of B16F1 cells, the circulating B16F0 cells could enhance the inflammatory response in vivo, as evidenced by the elevated levels of IL-6 and IL-1β in the serum (Supplementary Figure 2B). Moreover, the B16F0-enhanced inflammation could be reversed by in vivo expression of IL-37 (Supplementary Figure 2B). Accordingly, the promoting effect of circulating B16F0 cells on the metastatic colonization of disseminated B16F1 cells was also suppressed by IL-37 (Figure 2E). Together, these observations suggested that CTCs could induce systemic inflammation and consequently promoting the metastatic colonization of disseminated carcinoma cells.
Neutrophils are required for CTCs to promote tumor metastasis
Neutrophils, as a key component in inflammatory response, play a crucial role in inflammation-driven tumorigenesis and tumor progression [26–31]. Intriguingly, the circulating B16F0 cells could promote neutrophil infiltration in lung, and this effect could also be partially impaired by IL-37 (Figure 3A). Moreover, after intravenous inoculation of B16F1 cells, the circulating B16F0 cells could further increase the infiltration of neutrophils in lung (Figure 3B). We then depleted neutrophils in vivo (Supplementary Figure 3) to decrease neutrophil infiltration in lung (Figure 3B). In this situation, CTCs were unable to promote tumor metastasis (Figure 3C), suggesting that CTCs could not promote tumor metastasis in the absence of neutrophils, even if CTCs could induce systemic inflammation.
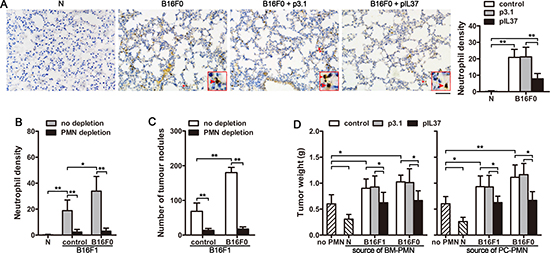
Figure 3: Neutrophils are involved in the pro-metastasis effect of CTCs. (A) Mice (n = 9 in each group) were inoculated with B16F0 cells intravenously, and received pIL37 plasmid treatment. Lung tissue sections were prepared and subjected to immunohistochemical staining to analyze the infiltration of neutrophils on d12 after primary inoculation (left, Bar, 50 μm). Neutrophil density (number of neutrophils per microscopic field) was determined (right). Naive mice (N) were used as control. (B and C) Mice (n = 9 in each group) received the i.v. injection of B16F1 cells with or without B16F0 cells. Neutrophils were depleted in vivo as indicated. (B) The infiltrated neutrophils in lung tissue were analyzed. The neutrophil density was determined after immunohistochemical staining. (C) The metastatic nodules on the surface of lung were counted. (D) Neutrophils were isolated from the bone marrow (BM) or peritoneal cavity (PC) of naive mice, B16F1-bearing mice, cir-B16F0-mice with or without treatment with i.v. injection of pIL37 plasmid. The neutrophils were used for co-inoculation with B16F1 cells to naive mice in the right hind thigh. Tumors (n = 9 in each group) were dissected and weighted on d12 after tumor cell inoculation. *p < 0.05, **p < 0.01.
We then further analyzed the effect of CTCs on neutrophil function. For this purpose, mice only received i.v. injection of B16F0 cells, thus preparing the mice with circulating tumor cells. The function of neutrophils could be altered in bone marrow, and the altered function could be maintained after the process of chemotaxis [32]. We therefore isolated neutrophils from the bone marrow and peritoneal cavity of naive mice, B16F1-bearing mice, and circulating B16F0-bearing mice (cir-B16F0-mice), respectively. In co-inoculation test, tumor growth was suppressed by the neutrophils from naive mice. However, the neutrophils from B16F1-bearing mice and cir-B16F0-mice could significantly promote the growth of tumor (Figure 3D). The conversion of neutrophil function in cir-B16F0-mice indicated that the function of neutrophils could be converted in the presence of circulating tumor cells. Moreover, CTCs failed to induce the tumor-promoting function of neutrophils in B16F1-bearing mice and cir-B16F0-mice, if IL-37 was expressed in vivo (Figure 3D). Taken together, the above results demonstrated that CTC-induced systemic inflammation could not only promote the infiltration of neutrophils in the target tissues, but also convert neutrophil function to promote tumor growth and metastasis.
CTCs promote the expression of tumor-promoting genes in neutrophils
In order to further explore the effect of CTCs on the protumor function of neutrophils, we analyzed the gene expression of neutrophils from cir-B16F0-mice with or without in vivo expression of IL-37. Compared with the neutrophils from naive mice, the neutrophils from cir-B16F0-mice expressed higher levels of genes that are related to tumor-promoting function of neutrophils, including Mmp9, Bv8, Arg1 and Nos2 (Figure 4A and Supplementary Figure 4), suggesting that CTCs could alter the expression of these genes to augment the tumor-promoting function of neutrophils.
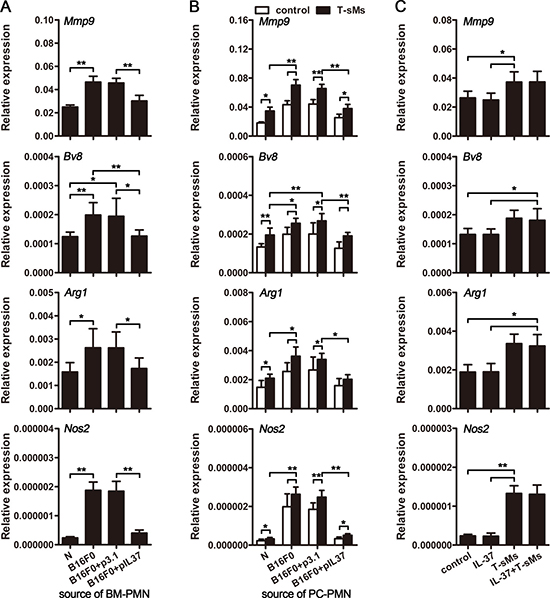
Figure 4: CTCs induce the pro-tumor function of neutrophils. (A and B) Mice (n = 9 in each group) were intravenously inoculated with B16F0 cells and treated by i.v. injection of pIL37 plasmid. Neutrophils were isolated from the bone marrow (BM) of mice without further stimulation (A), or isolated from the peritoneal cavity (PC) of mice and stimulated with T-sMs (0.5 mg/ml) for 12 h (B). Naive mice (N) were used as control. The expression of Mmp9, Bv8, Arg1 and Nos2 genes was detected by real-time RT-PCR. (C) Neutrophils were isolated from the BM of naive mice, and stimulated with IL-37 (200 ng/ml) and/or T-sMs (0.5 mg/ml) for 12 h. The expression of Mmp9, Bv8, Arg1 and Nos2 genes was detected by real-time RT-PCR. Data are pooled from three independent experiments with a total of six samples in each group (C). *p < 0.05, **p < 0.01.
We then investigated whether CTCs could alter the response of neutrophils to the stimuli in tumor microenvironment by stimulating neutrophils with soluble molecules from tumor (T-sMs), which might represent complex stimuli in tumor milieu [32]. In the presence of T-sMs, the expression of Mmp9, Bv8, Arg1 and Nos2 was further increased (Figure 4B). Compared to the neutrophils from naive mice, the neutrophils from cir-B16F0-mice showed much higher expression of Mmp9, Bv8, Arg1 and Nos2 (Figure 4B). However, the up-regulation of gene expression and the increase of neutrophil responsiveness to T-sMs were abrogated by in vivo expression of IL-37 in cir-B16F0-mice (Figure 4A and 4B). In addition, IL-37 could not directly affects the gene expression of neutrophils from naive mice, when they were stimulated with or without T-sMs (Figure 4C), suggesting that IL-37 could suppress CTC-induced inflammatory response, and the overall inhibition of inflammation affects the gene expression and responsiveness of neutrophils in our models.
CTCs suppress TRAIL expression and degranulation of neutrophils
We then investigated the effect of CTCs on the antitumor function of neutrophils. Neutrophils could suppress tumor growth by releasing TRAIL and myeloperoxidase (MPO) in response to suitable stimuli [32–34]. We therefore analyzed TRAIL expression and degranulation (releasing MPO) of neutrophils from cir-B16F0-mice. The results showed that circulating B16F0 cells could decrease the expression of TRAIL at both mRNA level and protein level (Figure 5A and Supplementary Figure 5A). Besides, the expression of Rab27a, which is required for efficient degranulation of neutrophils [35], was also decreased in cir-B16F0-mice (Figure 5A and Supplementary Figure 5A). Furthermore, the expression of Trail and Rab27a was further reduced in neutrophils, when they were stimulated by T-sMs (Figure 5B). Compared with neutrophils from naive mice, the neutrophils from cir-B16F0-mice showed much lower expression of Trail and Rab27a (Figure 5B). Consistent with the decreased of Rab27a, the spontaneous and T-sM-induced degranulation of neutrophils from cir-B16F0-mice were impaired, as evaluated by the release of MPO (Figure 5C) and the expression of CD63, a marker related to degranulation [36], on the surface of cells (Supplementary Figure 5B).
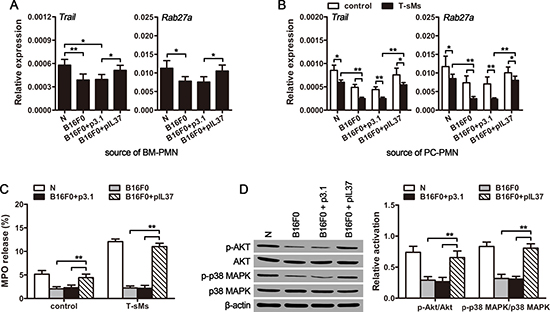
Figure 5: CTCs suppress the anti-tumor function of neutrophils. Neutrophils were isolated from the bone marrow (BM) (A) or peritoneal cavity (PC) (B–D) of cir-B16F0-mice (n = 8 in each group) with or without treatment with pIL37 plasmid, and stimulated with T-sMs (0.5 mg/ml) for 12 h (B) or 30 min (C and D). The mRNA level of Trail and Rab27a was detected by real-time RT-PCR (A and B). The release of MPO from neutrophils was detected after stimulation with T-sMs, as described in Methods (C). The phosphorylation of Akt and p38 MAPK was detected by Western blot after stimulation with T-sMs (D, left). The ratios of phospho-Akt to Akt (p-Akt/Akt) and phospho-p38 MAPK to p38 MAPK (p-p38 MAPK/p38 MAPK) were calculated after densitometric analysis of Western blots (D, right). Naive mice (N) were used as control. *p < 0.05, **p < 0.01.
To get further insights into the attenuation of neutrophil degranulation in cir-B16F0-mice, we next analyzed the activation of p38 MAPK and PI3K pathways that are required for the induced degranulation of neutrophils [37]. For this purpose, we stimulated neutrophils with T-sMs, which could activate p38 MAPK and PI3K pathways in neutrophils [32]. Intriguingly, the phosphorylation levels of p38 MAPK and Akt in neutrophils from cir-B16F0-mice were significantly lower than those in neutrophils from naive mice (Figure 5D). However, the effect of circulating B16F0 cells on TRAIL expression (Figure 5A and Supplementary Figure 5A) and degranulation (Figure 5C and Supplementary Figure 5B) of neutrophils was also inhibited by in vivo expression of IL-37, suggesting that CTC-induced inflammation contributed to the down-regulation of TRAIL expression and the attenuation of neutrophil degranulation.
CTCs induce inflammatory response by releasing TLR2/4 ligands
The function of neutrophils could be converted from tumor-suppressing to tumor promoting by G-CSF and IL-6 [32]. We then compared the expression of G-CSF and IL-6 in naive mice and cir-B16F0-mice. The mRNA levels of Csf3 (g-csf) and Il6 genes in lung (Figure 6A) and spleen (Supplementary Figure 6A) were much higher in cir-B16F0-mice. Consistently, compared to naive mice, the serum levels of G-CSF and IL-6 in cir-B16F0-mice were elevated (Figure 6B and Supplementary Figure 6B), which further demonstrated that the function of neutrophils could be converted to tumor-promoting by circulating tumor cells. The increased expression of Csf3 (g-csf) and Il6 in cir-B16F0-mice could be significantly suppressed by in vivo expression of IL-37 (Figure 6C and Supplementary Figure 6C) or blocking IL-1β in vivo (Supplementary Figure 6D and 6E). However, IL-37 could not directly influence the effect of G-CSF/IL-6 on neutrophils (Figure 6D). These results indicated that the increased production of proinflammatory cytokines in vivo was crucial for the effect of CTCs.
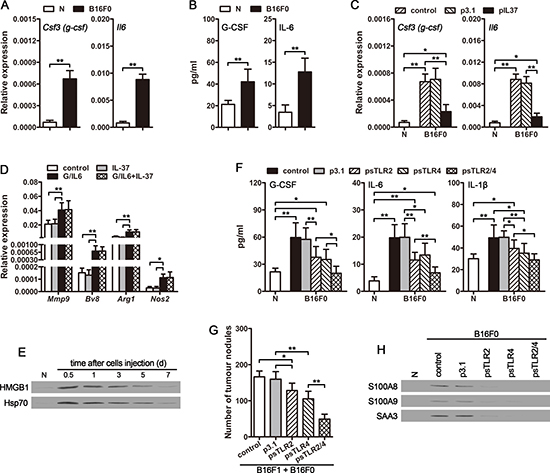
Figure 6: CTCs promote the expression of G-CSF and IL-6 in vivo. (A) The expression of Csf3 (g-csf) and Il6 genes in lung tissues of cir-B16F0-mice was detected by real-time RT-PCR on d4 after primary inoculation. (B) Serum levels of G-CSF and IL-6 in cir-B16F0-mice were detected by ELISA on d4 after primary inoculation. (C) Cir-B16F0-mice were untreated or treated by i.v. injection of pIL37 plasmid. The expression of Csf3 (g-csf) and Il6 genes in lung tissues was detected by real-time RT-PCR on d10 after primary inoculation. (D) Neutrophils were isolated from the bone marrow of naive mice and stimulated with G-CSF/IL-6 (50 ng/ml of each) and/or IL-37 (200 ng/ml) for 12 h. The expression of Mmp9, Bv8, Arg1 and Nos2 genes was detected by real-time RT-PCR. (E) Mice received i.v. injection of B16F0 cells (3 × 105). HMGB1 and HSP70 in the serum of mice were detected by Western blot at the indicated time points. (F) Cir-B16F0-mice were untreated or treated by i.v. injection of psTLR2 and/or psTLR4 plasmids. Serum levels of G-CSF, IL-6, and IL-1β were detected by ELISA on d10 after primary inoculation. (G) Mice received the i.v. injection of B16F1 cells with B16F0 cells. The mice were untreated or treated by i.v. injection of psTLR2 and/or psTLR4 plasmids. Metastatic nodules on the surface of lung were counted on d12 after B16F1 cell inoculation. (H) Cir-B16F0-mice were untreated or treated by i.v. injection of psTLR2 and/or psTLR4 plasmids. S100A8, S100A9 and SAA3 proteins in lung tissues were detected by Western blot on d10 after primary inoculation. Data are representative of three independent experiments (D, E and H). n = 9 in each group (A–C, F and G). Naive mice (N) were used as control (A–C, E, F and H). *p < 0.05, **p < 0.01.
To explore the inflammatory stimuli from CTCs that stimulate G-CSF and IL-6 expression, we focused on the tumor cell-derived TLR2 and TLR4 (TLR2/4) ligands, since IL-37 could suppress TLR4 ligand-induced inflammatory response [22, 38]. HMGB1 and HSP70, the representatives of TLR2/4 ligands released from tumor cells, have been implicated in promoting the production of proinflammatory cytokines [39, 40]. As shown in Figure 6E, the protein levels of HMGB1 and HSP70 were elevated in the serum of mice, and could be sustained for several days after intravenous injection of B16F0 cells. Intriguingly, the elevated serum levels of G-CSF, IL-6 and IL-1β in cir-B16F0-mice could be dramatically suppressed by the expression of soluble TLR2 (sTLR2) and soluble TLR4 (sTLR4) in vivo (Figure 6F). Consistently, the promotion effect of circulating B16F0 cells on the metastatic colonization of disseminated B16F1 cells was also suppressed by sTLR2 and sTLR4 (Figure 6G), further confirming that TLR2/4 ligands are the main inflammatory stimuli that are responsible for promoting tumor metastasis by CTCs.
Moreover, the expression of S100A8, S100A9 and SAA3, which are also the ligands for TLR2/4 and have been implicated in promoting G-CSF, IL-6 and IL-1β expression [41–4], in lung tissues were increased in cir-B16F0-mice (Figure 6H). The increased expression of these ligands was also suppressed by in vivo expression of sTLR2 and sTLR4 (Figure 6H), suggesting that CTC-derived TLR2/4 ligands could induce more endogenous TLR2/4 ligands expression in tissue, thus efficiently inducing systemic inflammation and promoting the metastatic colonization of disseminated carcinoma cells.
DISCUSSION
The metastatic colonization of disseminated carcinoma cells in distant organ is the most critical and rate-limiting step of metastasis, which is influenced by different factors. Inflammation has been recognized as a key player in cancer development, which can fuel both primary tumor growth and metastasis [10, 11, 21]. Our results confirmed that inflammation is indeed essential for the metastatic colonization. In our models, the inoculation with metastatic B16F1 cells could trigger an inflammatory response and induce the conversion of neutrophil function, which might be due to the existence of metastatic foci in the lung, or the B16F1 cells that did not extravasate, and retained as CTCs. By employing non-metastatic B16F0 cells, we confirmed that CTCs could trigger the systemic inflammation, which induced the conversion of neutrophil function, promoted neutrophil infiltration in the lung, and increased the expression of pro-metastatic proteins such as MMP-9, Bv8, S100A8, and S100A9 [13] in the lung. Moreover, the CTC-induced inflammation is the main mechanism underlying the promoting effect of CTCs on the metastatic colonization of disseminated carcinoma cells, since inhibiting the inflammatory response with IL-37 could efficiently suppress the effect of CTCs. Therefore, the anti-inflammatory therapy might be very important when the higher CTC count is detected.
Neutrophils are functionally plastic in the tumor microenvironment. They have been proposed to inhibit metastatic seeding in the lung by generating H2O2 [45]. In contrast, they also have been reported to be account for metastasis in the lung by supporting lung colonization of metastasis-initiating breast cancer cells [31]. Our data showed that neutrophils are required for CTCs to promote tumor metastasis. CTCs could suppress the anti-tumor function of neutrophils by down-regulating the expression of TRAIL and attenuating the degranulation of neutrophils. On the other hand, CTCs could up-regulate the expression of tumor-promoting genes, including Mmp9, Bv8, Arg1 and Nos2. Therefore, the functional phenotype of neutrophils could be converted by CTCs from tumor-suppressing to tumor-promoting. The tumor-promoting neutrophils are required for the initiation of angiogenesis in metastatic lesions [46], which is crucial for the formation of metastases. Thus, the conversion of neutrophil function was crucial for CTCs to promote tumor metastasis. Consistently, our data showed that depleting neutrophils in vivo could abrogate the promoting effect of CTCs on the metastatic colonization.
Neutrophils can be polarized into either an antitumoral (N1) or a protumoral (N2) phenotype, which depends on external cues in the environment [18, 32]. Our results showed that the conversion of neutrophil function was due to CTC-induced systemic inflammation, since inhibiting CTC-induced inflammatory response with IL-37 could suppress CTC-induced conversion of neutrophil function. More specifically, the CTC-induced production of G-CSF and IL-6 was accounted for CTC-induced neutrophil function alteration.
CTCs could be an important source of inflammatory stimuli, especially the ligands for TLR2 and TLR4 (TLR2/4) that have been known to induce the production of proinflammatory cytokines, such as G-CSF, IL-6, IL-1β, TNF-α [41, 44, 47]. After intravasation, most of tumor cells in circulation are damaged by the shear stress and mechanical stresses in blood, thus releasing intracellular molecules, many of which have been identified as the ligands for TLR2/4, including HMGB1, HSP60, HSP70, gp96, S100A8, S100A9, SAA3 and so on [48]. The half-life of the CTCs in blood of patients is only 1 to 2 hours [49]. The CTCs that are dying are replenished quickly, relying on the continuous entry of tumor cells into blood [49]. The continuous entry of tumor cells into blood and the rapid death of CTCs suggest the continuously release of TLR2/4 ligands into blood. CTC-released TLR2/4 ligands could induce the production of IL-1β, which has been implicated in promoting the expression of S100A8, S100A9 and SAA3 [49]. As shown in our data, CTC-released TLR2/4 ligands could further induce the production of S100A8, S100A9 and SAA3 that are also the ligands for TLR2/4 and have been reported to promote the production of G-CSF, IL-6 and IL-1β [41–44]. Thus, CTC-derived TLR2/4 signal could be amplified in vivo, and eventually efficient in inducing the expression of G-CSF and IL-6 in vivo.
In sum, we identify a role of CTCs in promoting tumor metastasis by inducing a systemic inflammatory response. The increased CTCs could promote neutrophil infiltration, induce the polarization of neutrophils to a pro-metastasis phenotype, and thus facilitate the metastatic colonization of disseminated carcinoma cells in distant organs. Therefore, the higher counts of CTCs could increase the risk of metastasis by inducing systemic inflammation, resulting in the poorer prognosis and shorter survival of the patients. On the other hand, our study also confirmed that the promoting effect of CTCs on tumor metastasis could be abrogated by either inhibiting the inflammatory response or blocking TLR2/4 signaling. Although the “N2” phenotype neutrophils are crucial for the promoting effect of CTCs on tumor metastasis, the protracted depletion of neutrophils is clinically untenable. Thus, inhibiting inflammatory response to reduce the production of cytokines such as G-CSF and IL-6, or blocking the ligands for TLR2/4, when the increased CTC counts is detected, might be very important for designing the therapeutic strategy to prevent tumor metastasis. More research on the metastatic axis of CTCs, tumor-associated systemic inflammation, neutrophil recruitment and function alteration may reveal effective targets to prevent and treat tumor metastasis.
MATERIALS AND METHODS
Animals and cell lines
BALB/c mice, 6-8 weeks old, were purchased from Center of Medical Experimental Animals of Hubei Province (Wuhan, China) for studies. All animal experiments were approved by the Animal Care and Use Committee of Tongji Medical College. C57BL/6 background B16F0 and B16F1 melanoma cells were purchased from China Center for Type Culture Collection (CCTCC, Wuhan, China) and cultured according to their guidelines.
Reagents and plasmids
Murine G-CSF and IL-6 were purchased from PeproTech (Rocky Hill, NJ). Recombinant human IL-37 was purchased from R&D Systems. Anti-Ly6G mAb was purchased from BioExpress (clone 1A8). Plasmids pIL37 and pCXCL1 are expression vectors carrying the cDNA encoding human IL-37 (IL-37b) and murine CXCL1, respectively. Plasmids psTLR2 and psTLR4 are expression vectors carrying the cDNA encoding the extracellular domain of murine Toll-like receptor 2 and 4, respectively. These plasmids were constructed by the insertion of cDNA into the mammalian expression vector pcDNA3.1 (Invitrogen, Carlsbad, CA) in our laboratory. All of the vectors were identified by in vivo expression (Supplementary Figure 7).
Assay of tumor cell arrest in lung and extravasation
Carboxyfluorescein succinimidyl ester (CFSE)-labeled B16F0 and B16F1 cells (5 × 105 cells/mice) were injected intravenously to mice. Lungs were harvested 5 and 24 h after tumor cell injection. Frozen sections were prepared and analyzed by fluorescence microscopy. Fluorescent spots were counted from randomly chosen fields in the sections of each mouse.
Animal experiments and treatment protocol
In tumor cell metastasis model, mice received i.v. injection of 3 × 105 B16F1 cells on d0. When indicated, B16F0 cells (3 × 105) were injected together with B16F1 cells, followed by another i.v. injection of B16F0 cells (3 × 105) on d6. The tumor nodules on the surface of lung were counted on d12 after inoculation of B16F1 cells.
To prepare the mice with circulating tumor cells, mice received i.v. injection of B16F0 cell (3 × 105) on d0 and d6 respectively. The mice were designated as cir-B16F0-mice, and used for other experiments on d12 after the first injection of B16F0 cells. When indicated, cir-B16F0-mice received i.v. injection of the indicated plasmid DNA (100 μg of each per injection), once every 2 days starting from d1 after primary inoculation. PBS and pcDNA3.1 (p3.1) were used as controls.
To analyze the effect of IL-37 on neutrophils in vivo, B16F1-bearing mice or cir-B16F0-mice received i.v. injection of pIL-37 plasmid (100 μg of each per injection) from d1 to d11, once every 2 days. Plasmid p3.1 was used as control. The mice were used for isolation of neutrophils on d12 after the first injection of tumor cells.
In co-inoculation test, mice received intramuscular injection of B16F1 cells (2 × 105) in the right hind thigh. When indicated, B16F1 cells were mixed with 1 × 106 neutrophils, which were isolated from naive mice (N) or B16F1-bearing mice or cir-B16F0-mice with or without the treatment by pIL-37 plasmid. Tumors were dissected and weighed on d12 after tumor cell inoculation.
In vivo gene transfection
Plasmids were prepared and analyzed as described previously [51]. Mice received the injection of plasmid DNA (100 μg) via the tail vein (i.v. injection) using the hydrodynamics-based gene delivery technique [51].
In vivo depletion of neutrophils
To deplete neutrophils, anti-Ly6G antibody (clone 1A8, BioExpress) was used [52]. Mice received i.p. injection of anti-Ly6G antibody at a dose of 300 μg in 500 μl PBS on -d1, d1, d4, d7, d10 of tumor inoculation [53]. The depletion of neutrophils was identified by flow cytometric analysis (Supplementary Figure 3).
Recruitment of neutrophils to peritoneal cavity
To recruit neutrophils to peritoneal cavity, CXCL1-expressing hepatocytes were injected to peritoneal cavity of mice as described previously [32]. 12 h later, the peritoneal cells were harvested for the isolation of neutrophils.
Isolation of neutrophils
Murine neutrophils were isolated from bone marrow cells, peripheral blood cells, or peritoneal cells as described previously [54]. Briefly, the cells were washed once in HBSS, layered over a Percoll gradient (54%/64%/72% for bone marrow cells, 54%/66%/78% for peripheral blood cells, and 54%/64%/80% for peritoneal cells), and centrifuged at 1060 × g for 30 min. The dense bands at 64%/72%, 66%/78%, or 64%/80% interface were collected as neutrophil fraction. The isolated cells were 90% neutrophils as assessed by flow cytometric analysis (Supplementary Figure 8).
Preparation of soluble molecules from tissues
Palpable tumors were dissected. The mixture of soluble molecules from tumor (T-sMs) was prepared by digesting the tissues with collagenase and removing debris by centrifugation. The concentration of T-sMs was defined by the concentration of protein, which was determined by using Coomassie Bradford reagent (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer’s instructions.
Other methods
Other methods were performed using standard protocols, including soft agar assay, cell proliferation assay, analysis of gene expression by conventional RT-PCR and real-time RT-PCR, Western blot assay, assay of degranulation, immunohistochemistry, cytokine neutralization, ELISA analysis, flow cytometric analysis. See Supplementary Methods for details.
Statistical analysis
Results were expressed as mean value ± SD and interpreted by one-way ANOVA. Differences were considered statistically significant when P <0.05.
ACKNOWLEDGMENTS
We thank Zhi-Hui Liang, Hui-Fen Zhu and Wen-Hong Lu for technical assistance.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest to disclose.
GRANT SUPPORT
This work was supported by National Natural Science Foundation of China (No. 81472704, 81272314, 30830095), and National Development Program (973) For Key Basic Research of China (No. 2009CB521806).
REFERENCES
1. Fidler IJ. Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125 I-5-iodo-2′-deoxyuridine. J Natl Cancer Inst. 1970; 45:773–782.
2. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002; 2:563–572.
3. Pantel K, Alix-Panabieres C, Riethdorf S. Cancer micrometastases. Nat Rev Clin Oncol. 2009; 6:339–351.
4. Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AG, Uhr JW, Terstappen LW. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res. 2004; 10:6897–904.
5. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, Hayes DF. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004; 351:781–791.
6. Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V, Klein C, Saini M, Bäuerle T, Wallwiener M, Holland-Letz T, Höfner T, Sprick M, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol. 2013; 31:539–544.
7. Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, Massague J. Tumor self-seeding by circulating cancer cells. Cell. 2009; 139:1315–1326.
8. Hamilton G, Rath B, Klameth L, Hochmair MJ. Small cell lung cancer: Recruitment of macrophages by circulating tumor cells. Oncoimmunology. 2016; 5:e1093277.
9. Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gerard A, Krummel MF. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature. 2016; 531:513–517.
10. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002; 420:860–867.
11. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008; 454:436–444.
12. Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer research. 2011; 71:2411–2416.
13. Wu CF, Andzinski L, Kasnitz N, Kröger A, Klawonn F, Lienenklaus S, Weiss S, Jablonska J. The lack of type I interferon induces neutrophil-mediated pre-metastatic niche formation in the mouse lung. Int J Cancer. 2015;137:837–847.
14. Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. P Natl Acad Sci USA. 2006; 103:12493–12498.
15. Shojaei F, Singh M, Thompson JD, Ferrara N. Role of Bv8 in neutrophil-dependent angiogenesis in a transgenic model of cancer progression. P Natl Acad Sci USA. 2008; 105:2640–2645.
16. Munder M, Schneider H, Luckner C, Giese T, Langhans CD, Fuentes JM, Kropf P, Mueller I, Kolb A, Modolell M, Ho AD. Suppression of T-cell functions by human granulocyte arginase. Blood. 2006; 108:1627–1634.
17. Sandhu JK, Privora HF, Wenckebach G, Birnboim HC. Neutrophils, nitric oxide synthase, and mutations in the mutatect murine tumor model. Am J Pathol. 2000; 156: 509–518.
18. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer cell. 2009; 16:183–194.
19. Liu Y, Cao X. Immunosuppressive cells in tumor immune escape and metastasis. J Mol Med (Berl). 2016; 94:509–522.
20. Brouwer A, De Laere B, Peeters D, Peeters M, Salgado R, Dirix L, Van Laere S. Evaluation and consequences of heterogeneity in the circulating tumor cell compartment. Oncotarget. 2016; 7:48625–48643. doi: 10.18632/oncotarget.8015.
21. Coffelt SB, de Visser KE. Systemic inflammation: Cancer’s long-distance reach to maximize metastasis. Oncoimmunology. 2016; 5:e1075694.
22. Nold MF, Nold-Petry CA, Zepp JA, Palmer BE, Bufler P, Dinarello CA. IL-37 is a fundamental inhibitor of innate immunity. Nat Immunol. 2010; 11:1014–1022.
23. Wang WQ, Zhao D, Zhou YS, Hu XY, Sun ZN, Yu G, Wu WT, Chen S, Kuang JL, Xu GG, Han ZC, Wang BM, Yang JX, et al. Transfer of the IL-37b gene elicits anti-tumor responses in mice bearing 4T1 breast cancer. Acta Pharmacol Sin. 2015; 36:528–534.
24. Zhao JJ, Pan QZ, Pan K, Weng DS, Wang QJ, Li JJ, Lv L, Wang DD, Zheng HX, Jiang SS, Zhang XF, Xia JC. Interleukin-37 mediates the antitumor activity in hepatocellular carcinoma: role for CD57+ NK cells. Sci Rep-Uk. 2014; 4:5177.
25. Gao W, Kumar S, Lotze MT, Hanning C, Robbins PD, Gambotto A. Innate immunity mediated by the cytokine IL-1 homologue 4 (IL-1H4/IL-1F7) induces IL-12-dependent adaptive and profound antitumor immunity. J Immunol. 2003; 170:107–113.
26. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJ, Ciampricotti M, Hawinkels LJ, Jonkers J, de Visser KE. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015; 522:345–348.
27. Huh SJ, Liang S, Sharma A, Dong C, Robertson GP. Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer research. 2010; 70:6071–6082.
28. Spicer JD, McDonald B, Cools-Lartigue JJ, Chow SC, Giannias B, Kubes P, Ferri LE. Neutrophils promote liver metastasis via Mac-1-mediated interactions with circulating tumor cells. Cancer research. 2012; 72:3919–3927.
29. Spiegel A, Brooks MW, Houshyar S, Reinhardt F, Ardolino M, Fessler E, Chen MB, Krall JA, DeCock J, Zervantonakis IK, Iannello A, Iwamoto Y, Cortez-Retamozo V, et al. Neutrophils suppress intraluminal NK-mediated tumor cell clearance and enhance extravasation of disseminated carcinoma cells. Cancer Discov. 2016.
30. Tazawa H, Okada F, Kobayashi T, Tada M, Mori Y, Une Y, Sendo F, Kobayashi M, Hosokawa M. Infiltration of neutrophils is required for acquisition of metastatic phenotype of benign murine fibrosarcoma cells: implication of inflammation-associated carcinogenesis and tumor progression. Am J Pathol. 2003; 163:2221–2232.
31. Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. 2015; 528:413–417.
32. Yan B, Wei JJ, Yuan Y, Sun R, Li D, Luo J, Liao SJ, Zhou YH, Shu Y, Wang Q, Zhang GM, Feng ZH. IL-6 cooperates with G-CSF to induce protumor function of neutrophils in bone marrow by enhancing STAT3 activation. J Immunol. 2013; 190:5882–5893.
33. Koga Y, Matsuzaki A, Suminoe A, Hattori H, Hara T. Neutrophil-derived TNF-related apoptosis-inducing ligand (TRAIL): a novel mechanism of antitumor effect by neutrophils. Cancer research. 2004; 64:1037–1043.
34. Cantarella G, Risuglia N, Dell’eva R, Lempereur L, Albini A, Pennisi G, Scoto GM, Noonan DN, Bernardini R. TRAIL inhibits angiogenesis stimulated by VEGF expression in human glioblastoma cells. British journal of cancer. 2006; 94:1428–1435.
35. Herrero-Turrion MJ, Calafat J, Janssen H, Fukuda M, Mollinedo F. Rab27a regulates exocytosis of tertiary and specific granules in human neutrophils. J Immunol. 2008; 181:3793–3803.
36. Abdel-Latif D, Steward M, Macdonald DL, Francis GA, Dinauer MC, Lacy P. Rac2 is critical for neutrophil primary granule exocytosis. Blood. 2004; 104:832–839.
37. Brzezinska AA, Johnson JL, Munafo DB, Ellis BA, Catz SD. Signalling mechanisms for Toll-like receptor-activated neutrophil exocytosis: key roles for interleukin-1-receptor-associated kinase-4 and phosphatidylinositol 3-kinase but not Toll/IL-1 receptor (TIR) domain-containing adaptor inducing IFN-beta (TRIF). Immunology. 2009; 127:386–397.
38. Bulau AM, Fink M, Maucksch C, Kappler R, Mayr D, Wagner K, Bufler P. In vivo expression of interleukin-37 reduces local and systemic inflammation in concanavalin A-induced hepatitis. TheScientificWorldJournal. 2011; 11:2480–2490.
39. Urbonaviciute V, Furnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, Bianchi ME, Kirschning C, Wagner H, Manfredi AA, Kalden JR, Schett G, Rovere-Querini P, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. The Journal of experimental medicine. 2008; 205:3007–3018.
40. Lee CH, Wu CL, Shiau AL. Toll-like receptor 4 signaling promotes tumor growth. J Immunother. 2010; 33:73–82.
41. De Filippo K, Neill DR, Mathies M, Bangert M, McNeill E, Kadioglu A, Hogg N. A new protective role for S100A9 in regulation of neutrophil recruitment during invasive pneumococcal pneumonia. Faseb J. 2014; 28:3600–3608.
42. Gao H, Zhang X, Zheng Y, Peng L, Hou J, Meng H. S100A9-induced release of interleukin (IL)-6 and IL-8 through toll-like receptor 4 (TLR4) in human periodontal ligament cells. Molecular immunology. 2015; 67:223–232.
43. Lee JM, Kim EK, Seo H, Jeon I, Chae MJ, Park YJ, Song B, Kim YS, Kim YJ, Ko HJ, Kang CY. Serum amyloid A3 exacerbates cancer by enhancing the suppressive capacity of myeloid-derived suppressor cells via TLR2-dependent STAT3 activation. Eur J Immunol. 2014; 44:1672–1684.
44. He RL, Zhou J, Hanson CZ, Chen J, Cheng N, Ye RD. Serum amyloid A induces G-CSF expression and neutrophilia via Toll-like receptor 2. Blood. 2009; 113:429–437.
45. Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer cell. 2011; 20:300–314.
46. Luo J, Feng XX, Luo C, Wang Y, Li D, Shu Y, Wang SS, Qin J, Li YC, Zou JM, Tian DA, Zhang GM, Feng ZH. 14,15-EET induces the infiltration and tumor-promoting function of neutrophils to trigger the growth of minimal dormant metastases. Oncotarget. 2016; 7:43324–43336. doi: 10.18632/oncotarget.9709.
47. Huang B, Zhao J, Unkeless JC, Feng ZH, Xiong H. TLR signaling by tumor and immune cells: a double-edged sword. Oncogene. 2008; 27:218–224.
48. Yu L, Wang L, Chen S. Exogenous or endogenous Toll-like receptor ligands: which is the MVP in tumorigenesis? Cell Mol Life Sci. 2012; 69:935–949.
49. Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, Beitsch PD, Leitch M, Hoover S, Euhus D, Haley B, Morrison L, Fleming TP, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10:8152–8162.
50. Hogmalm A, Bry M, Strandvik B, Bry K. IL-1beta expression in the distal lung epithelium disrupts lung morphogenesis and epithelial cell differentiation in fetal mice. Am J Physiol Lung Cell Mol Physiol. 2014; 306:L23–34.
51. Geng H, Zhang GM, Li D, Zhang H, Yuan Y, Zhu HG, Xiao H, Han LF, Feng ZH. Soluble form of T cell Ig mucin 3 is an inhibitory molecule in T cell-mediated immune response. J Immunol. 2006; 176:1411–1420.
52. Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. Journal of leukocyte biology. 2008; 83:64–70.
53. Mishalian I, Bayuh R, Levy L, Zolotarov L, Michaeli J, Fridlender ZG. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol Immunother. 2013; 62:1745–1756.
54. Nick JA, Young SK, Brown KK, Avdi NJ, Arndt PG, Suratt BT, Janes MS, Henson PM, Worthen GS. Role of p38 mitogen-activated protein kinase in a murine model of pulmonary inflammation. J Immunol. 2000; 164:2151–2159.
55. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009; 55:611–622.
56. Gong W, Zhang GM, Liu Y, Lei Z, Li D, Yuan Y, Huang B, Feng ZH. IFN-gamma withdrawal after immunotherapy potentiates B16 melanoma invasion and metastasis by intensifying tumor integrin alphavbeta3 signaling. Int J Cancer. 2008; 123:702–708.