
INTRODUCTION
Endometrial cancer (EC) is the fourth most common malignancy among women in the Western world [1, 2]. Surgery is the primary therapy for endometrial cancer followed by tailored adjuvant therapy [3]. Although recommendations for adjuvant treatment are currently based on clinical and histopathological prognostic factors, over- and undertreatment of EC patients remains. Therefore, recent studies focused on (epi) genetic characteristics to improve prediction of individual patients’ risk of recurrence.
The Cancer Genome Atlas (TCGA) identified, supported by other independent studies, promising prognostic molecular markers in EC: polymerase epsilon (POLE) exonuclease domain mutations, CTNNB1 mutations, mismatch repair (MMR) deficiency, and TP53 mutations [4–9]. Additionally, three large studies have demonstrated that L1 cell adhesion molecule (L1CAM; CD171) expression in more than 10% of tumour cells is a strong independent predictor for distant recurrences in EC [10–12]. As a result, based on an extensive integrated analysis of these molecular markers, two molecular integrated risk assignments, combining clinicopathological and molecular risk factors, have been proposed [6, 7]. In one of these assignments, similarly to the other, p53-mutant-like, L1CAM-positive, and substantial lymphovascular space invasion (LVSI) tumours were designated to have an unfavourable risk. MMR-deficient and CTNNB1-mutant were considered at intermediate risk, while POLE-mutant tumours and tumours with no specific molecular profile are designated favourable [5–7].
Molecular risk prediction in EC patients, once implemented, will be based on molecular analysis in a small portion of the tumour. In a cancer, however, multiple subclones can be present that differ in phenotype and/or genotype, resulting in a high degree of intratumour heterogeneity [13]. Such intratumour heterogeneity may interfere with correct risk prediction.
Studies on intratumour heterogeneity of the aforementioned molecular markers are sparse [13–22]. Some studies have reported low rates of subclonal p53 and MMR protein expression within one or multiple blocks of the same hysterectomy specimen [23–27]. Furthermore, in 92% of TP53-mutant cases, the mutation was present in the majority of tumour cells based on the cancer cell fraction [28]. In addition, previous studies have determined for a selected number of markers that molecular analysis on pre-operative endometrial cancer specimen are concordant with final hysterectomy specimen obtained at definitive surgical staging [10, 29–31]. In this study we aimed to investigate to which extent intratumour heterogeneity exists for the promising prognostic molecular markers and whether it affects the molecular risk assignment in EC.
RESULTS
Clinicopathological characteristics
In total, 49 ECs were included, of which the clinicopathological characteristics are shown in Table 1. The selected cases were representative of the endometrial cancer population with regard to age (median 65 years), FIGO stage (87.8% stage I+II) and tumour type (85.7% endometrioid), with a slight overrepresentation of grade 3 tumours (28.6%) [32]. The included ECs consisted of 10 POLE-mutant, 8 CTNNB1-mutant, 10 p53-mutant, 11 MMR deficient and 10 L1CAM-positive cases, and 9 cases without any of these markers. Nine ECs contained more than one molecular alteration.
Table 1: Clinicopathological patient characteristics (N = 49)
|
|
N |
% |
|---|---|---|---|
Age |
|
|
|
|
Median, range |
65 |
46-78 |
FIGO stage 2009 |
|
|
|
|
I, II |
43 |
87.8 |
|
III, IV |
6 |
12.2 |
Tumour type |
|
|
|
|
Endometrioid |
42 |
85.7 |
|
(Pseudoglandular) Serous |
7 |
14.3 |
Grade |
|
|
|
|
1, 2 |
35 |
71.4 |
|
3 |
14 |
28.6 |
Depth of myometrial invasion |
|
|
|
|
< 50% |
27 |
55.1 |
|
≥ 50% |
22 |
44.9 |
Lymphovascular space invasion |
|
|
|
|
Substantial |
7 |
14.3 |
|
Absent/mild |
42 |
85.7 |
Mutation status* |
|
|
|
|
POLE-mutant |
10 |
20.0 |
|
CTNNB1-mutant |
8 |
16.3 |
Altered protein expression* |
|
|
|
|
MMR deficiency |
11 |
22.4 |
|
>10% L1CAM |
10 |
20.0 |
|
Mutant-like-p53 |
10 |
20.0 |
Molecular risk assignment* |
|
|
|
|
favourable |
17 |
34.7 |
|
intermediate |
19 |
38.8 |
|
unfavourable |
13 |
26.5 |
* Based on tumour block used for selection.
Intratumour heterogeneity of the molecular markers
Concordance between the three tumour blocks for POLE and CTNNB1-mutational status, and p53, mismatch repair and L1CAM protein expression was found in 100% (48/48), 95.9% (47/49), 93.9% (46/49), 98.0% (48/49), and 91.8% (45/49) of tumours, respectively (Table 2). The ten discordances were identified in nine cases (9/49, 18.4%, Table 3). This intratumour heterogeneity was not reflected by differences in histological subtype, tumour grade or nuclear atypia within or between the tumour blocks.
Table 2: Intratumour concordance for the molecular markers
|
Subgroup |
|||||
|---|---|---|---|---|---|---|
POLE* |
CTNNB1 |
p53 |
MMR |
L1CAM |
Total |
|
Concordant |
48 (100.0) |
47 (95.9) |
46 (93.9) |
48 (98.0) |
45 (91.8) |
40 (81.6) |
Discordant |
0 (0.0) |
2 (4.1) |
3 (6.1) |
1 (2.0) |
4 (8.2) |
9 (18.4) |
* One case failed.
Table 3: Overview of the pathological characteristics and molecular markers in cases showing intratumour heterogeneity for at least one molecular marker
Case |
Tumour block |
FIGO stage |
Tumour type |
Grade |
POLE |
CTNNB1 |
MMR |
L1CAM |
p53 |
Sanger seq. TP53 |
Final assignment |
Difference in assignment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
1 |
a |
II |
EEC |
1 |
wt |
mut |
intact |
< 10% |
wt |
n.a. |
intermediate |
|
|
b |
|
EEC |
1 |
wt |
wt |
intact |
< 10% |
wt |
n.a. |
favourable |
yes |
|
c |
|
EEC |
1 |
wt |
wt |
intact |
< 10% |
wt |
n.a. |
favourable |
|
2 |
a |
IA |
EEC |
1 |
wt |
mut |
intact |
< 10% |
wt |
n.a. |
intermediate |
|
|
b |
|
EEC |
1 |
wt |
mut |
intact |
< 10% |
wt |
n.a. |
intermediate |
yes |
|
c |
|
EEC |
1 |
wt |
wt |
intact |
< 10% |
wt |
n.a. |
favourable |
|
3 |
a |
IB |
EEC |
2 |
wt |
wt |
MLH1/PMS2 loss |
< 10% |
wt |
n.a. |
intermediate |
|
|
b |
|
EEC |
2 |
wt |
wt |
MLH1/PMS2 loss* |
< 10% |
wt |
n.a. |
intermediate |
yes |
|
c |
|
EEC |
2 |
wt |
wt |
intact |
< 10% |
wt |
n.a. |
favourable |
|
4 |
a |
IIIA |
Serous |
3 |
wt |
wt |
intact |
> 10% |
mut |
n.a. |
unfavourable |
|
|
b |
|
Serous |
3 |
wt |
wt |
intact |
> 10% |
mut |
n.a. |
unfavourable |
no |
|
c |
|
Serous |
3 |
wt |
wt |
intact |
< 10% |
mut |
n.a. |
unfavourable |
|
5 |
a |
IA |
EEC |
1 |
wt |
wt |
intact |
> 10% |
mut |
n.a. |
unfavourable |
|
|
b |
|
EEC |
1 |
wt |
wt |
intact |
> 10% |
mut |
n.a. |
unfavourable |
no |
|
c |
|
EEC |
1 |
wt |
wt |
intact |
< 10% |
mut |
n.a. |
unfavourable |
|
6 |
a |
IB |
EEC |
1 |
mut |
wt |
intact |
< 10% |
wt |
n.a. |
favourable |
|
|
b |
|
EEC |
1 |
mut |
wt |
intact |
< 10% |
wt |
n.a. |
favourable |
no |
|
c |
|
EEC |
1 |
mut |
wt |
intact |
> 10% |
wt |
n.a. |
favourable |
|
7 |
a |
IA |
EEC |
3 |
mut |
wt |
intact |
< 10% |
wt |
mut |
favourable |
|
|
b |
|
EEC |
3 |
mut |
wt |
intact |
< 10% |
wt+ |
mut |
favourable |
uncertain |
|
c |
|
EEC |
3 |
n.a. |
wt |
intact |
> 10% |
mut |
mut |
uncertain |
|
8 |
a |
IB |
EEC |
1 |
wt |
mut |
intact |
< 10% |
wt |
mut |
intermediate |
|
|
b |
|
EEC |
1 |
wt |
mut |
intact |
< 10% |
wt+ |
mut |
unfavourable |
yes |
|
c |
|
EEC |
1 |
wt |
mut |
intact |
< 10% |
wt+ |
mut |
unfavourable |
|
9 |
a |
IB |
EEC |
3 |
wt |
wt |
MLH1/PMS2 loss |
< 10% |
wt |
wt |
intermediate |
|
|
b |
|
EEC |
3 |
wt |
wt |
MLH1/PMS2 loss |
< 10% |
wt+ |
wt |
intermediate |
yes |
|
c |
|
EEC |
3 |
wt |
wt |
MLH1/PMS2 loss |
< 10% |
wt+ |
mut |
unfavourable |
|
Abbreviations: seq, sequence; EEC, endometrioid; wt, wildtype; mut, mutation; n.a., not analysed,. *Subclonal loss.
wt+: wildtype with focal overexpression in <10% of the tumour.
Discordant CTNNB1 mutation status was observed in two cases (cases 1 and 2), discordant MLH1/PMS2 protein expression in one case (case 3), discordant L1CAM expression in four cases (cases 4-7), and three cases showed discordant p53-mutant-like expression (cases 7-9) (Table 2, Table 3). Competitive allele-specific PCR for POLE exonuclease domain mutations revealed six discordant cases. Reanalysis of these discordant cases using Sanger sequencing revealed concordant findings among the three tumour blocks for POLE mutation status. However, Sanger sequencing could not be performed for one tumour block due to low DNA yield (case 7, block c).
Sanger sequencing of CTNNB1 exon 3 revealed discordant results in cases 1 and 2. In case 1, tumour block 1a harboured a c.94G>GT mutation with a variant allele frequency (25%), whereas no CTNNB1 mutation was detected in block 1b and 1c. Case 1 was the only case from the initial group of nine cases without any of the markers that showed molecular alterations upon analysis of three tumour blocks. In case 2, a c.110C>CG mutation in CTNNB1 was detected at high allelic frequency (80-100%) in tumour block 2a and 2b, but not in block 2c (Figure 1). To exclude contamination or exchange of DNA, we confirmed that all DNA originated from the same patient.
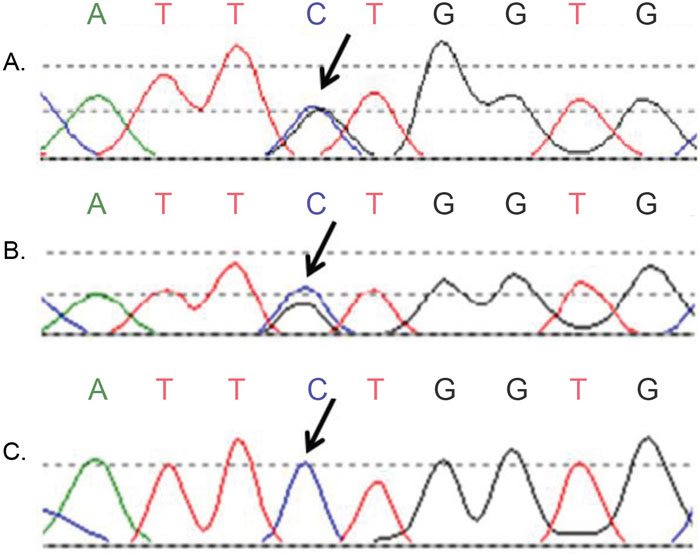
Figure 1: Discordant Sanger sequence results of CTNNB1 exon 3 (case 2). (A, B) Two tumour blocks showed a c.110C>CG mutation, indicated by the arrows; (C) The third tumour block of the same endometrial cancer patient was wildtype at the same position.
Case 3 demonstrated discordant MMR protein expression (Figure 2A-2C). Tumour block 3c showed retained MLH1 and PMS2 expression, whereas tumour block 3a and 3b showed subclonal (15%) and complete loss of MLH1 and PMS2 expression, respectively. Moreover, three cases with complete loss of MLH1 and PMS2 expression in all three tumour blocks also displayed subclonal loss of MSH6 expression (10%, 20% and 90% of the tumour respectively) in one tumour block.
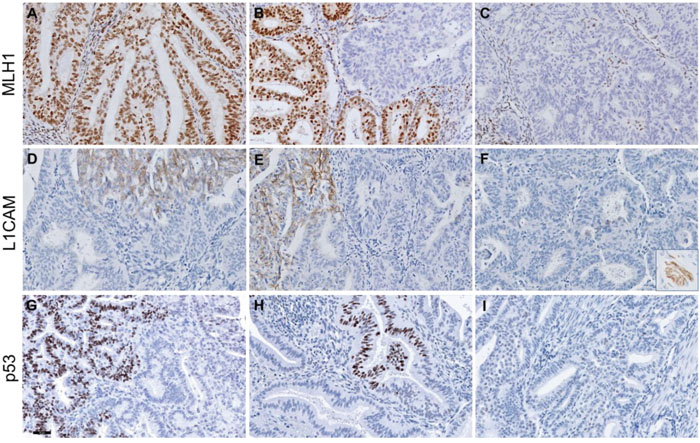
Figure 2: Representative figures of discordant MLH1, L1CAM and p53 protein expression in three endometrium carcinoma cases. Upper panels show discordant MLH1 expression: intact MLH1 staining (A), subclonal loss (B) and complete loss of MLH1 staining (C) in three blocks of case 3. Middle panels show discordant L1CAM staining in case 5: >10% L1CAM expression in two tumour blocks (D, F) and <10% in the third tumour block (E). The inset in (F) represents a L1CAM-positive nerve (positive control) within the same slide. Lower panels show wildtype+ p53 staining patterns with focal overexpression in <10% of the tumour in two tumour blocks (G, H) and wildtype p53 expression in the third block (I) in case 8. Scalebar represents 50 μm.
Cases 4-7 showed discordant L1CAM expression. In cases 4 and 5, more than 10% L1CAM expression was observed in two out of three tumour blocks (Figure 2D–2F). In cases 6 and 7, more than 10% L1CAM expression was observed in only one out of three tumour blocks.
Case 7 also showed discordant p53 expression: the first block demonstrated a p53-wildtype expression pattern, whereas the second block was scored ‘wildtype+’, because of focal p53 overexpression in a discrete geographical area of <10% of tumour cells. The third block showed a p53-mutant-like expression pattern in all tumour cells. Despite of the discordant p53 expression pattern found in the three blocks, the same pathogenic TP53 mutation (c. 637C>CT) was identified in all three tumour blocks. For case 8, a p53-wildtype+ expression pattern was found in tumour blocks 8b and 8c (Figure 2G, 2H), whereas a p53-wildtype expression pattern was seen in block 8a (Figure 2I). Case 9 also showed p53-wildtype expression in one tumour block (9a), while blocks 9b and 9c demonstrated p53-wildtype+ expression. Sanger sequencing revealed a TP53 pathogenic mutation (c.733G>GA) in all three blocks of case 8, while in case 9 only block 9c carried a pathogenic TP53 mutation (c.524G>GA) [33, 34]. Due to a limited surface area, both p53-mutant and -wildtype tissue may have been microdissected of block 9b, resulting in a TP53-wildtype status.
Molecular risk assignment
The intratumour heterogeneity found in the molecular markers affected the molecular risk assignment (Supplementary Figure 1) in five of 49 cases (10.2%, Table 3). Cases 1, 2 and 3 were assigned in the favourable or intermediate risk group depending on the tumour block used for molecular analysis. In these cases, risk assignment differed between the tumour blocks due to intratumour heterogeneity in CTNNB1 mutation status and MMR protein expression. The discordant L1CAM expression within cases 4 and 5 did not affect the risk assignment, because of the homogeneous p53-mutant-like expression pattern in the tumour. The presence of a mutation in the POLE exonuclease domain determined cases 6 and 7 to be at favourable risk. Therefore, the discordancy in L1CAM and p53 protein expression did not affect the risk assignment. However, in cases 8 and 9, the risk assignment was influenced by focal p53-mutant-like overexpression and the presence of a pathogenic TP53 mutation. These cases were assigned to the intermediate or unfavourable risk group depending on the tumour block used for molecular analysis.
DISCUSSION
This is the first study which comprehensively examined the prevalence of intratumour heterogeneity of well-established prognostic molecular markers and its impact on molecular risk assignment in EC. This series of 49 cases showed, depending on the marker analysed, no or low intratumour heterogeneity among three tumour blocks for POLE and CTNNB1 mutations, and p53, MMR and L1CAM expression (range of concordance rate: 92-100%). Similarly, previous studies showed that the molecular analysis on endometrial cancer preoperative specimens are concordant with final hysterectomy specimens obtained at definitive surgical staging [10, 29–31]. The intratumour heterogeneity in our study affected the integrated molecular risk assignment in only five cases. These findings suggest that testing one tumour tissue block from hysterectomy (or diagnostic) specimen is sufficient to provide a reliable molecular risk assessment in ECs.
Interestingly, we found discordant (subclonal) p53 mutant-like expression in three tumours. Although immunohistochemistry for p53 is routinely performed, only Feng et al. has described this p53 expression pattern [23]. As in the previous study, the majority of cases harboured a TP53 mutation, predicted to be pathogenic, in the p53-mutant tumour cells. The study of Feng et al. showed that subclonal p53-mutant-like expression may be related to differences in differentiation in half of the cases, suggesting that these TP53 mutations occurred at a later stage in tumour progression. No such histological differences were found in the present study within or between the tumour blocks of the same case showing subclonal p53-mutant-like expression. However, these three cases all showed an alteration in one of the other prognostic molecular markers, namely POLE mutation, CTNNB1 mutation or MMR deficiency. This supports the possibility of TP53 mutations as a late event, acting as a passenger alteration or aiding outgrowth of a more aggressive tumour. The concept is illustrated by case 7, a POLE-mutant tumour with subclonal p53-mutant-like expression due to a TP53 mutations at p.(R213*). As this particular substitution corresponds to the mutational signature associated with POLE mutations, it may be a secondary event [35]. The role of subclonal p53-mutant-like expression in endometrial carcinogenesis and the corresponding clinical outcome of these patients remains to be elucidated. Pathologists should be aware of this expression pattern and depending on the biological behaviour, subclonal p53 mutant-like expression should be implemented in the scoring system.
In comparison to the other molecular markers, intratumour heterogeneity was found most frequently for L1CAM expression (~8%). From a prognostic point of view, all but one of these cases with discordant L1CAM expression would have been classified as having an unfavourable risk regardless of L1CAM because of their p53-mutant-like expression or TP53 mutation. Furthermore, L1CAM expression may be a predictive marker: patients with high L1CAM expression may potentially benefit from L1CAM antibody-mediated therapy [36]. Whether the intratumour heterogeneity of L1CAM impacts the efficacy of such targeted therapy remains to be investigated.
The level of intratumour heterogeneity, found in the present study, may be an overestimation due to our selection of relatively large tumours with at least three tumour blocks. In practice, many endometrial cancers are small, resulting in only one or two tumour blocks. Therefore, limited intratumour heterogeneity can be anticipated in these cases. Contrastingly, additional sampling of larger tumours would result in higher intratumour heterogeneity. Moreover, this study contained only ECs without morphological heterogeneity. Intratumour heterogeneity may be larger when selecting for cases with mixed morphology. However, studies have shown that the different histotype components in mixed tumours are commonly clonally related, sharing the same molecular markers [37–39]. Regardless of whether the reported level of intratumour heterogeneity is an over- or underestimation, it will be comparable to the levels documented for other implemented molecular markers, such as HER2 in breast cancer (range 11-40%) [40–46].
In conclusion, intratumour heterogeneity of prognostic molecular markers in EC without morphologic heterogeneity is uncommon among three tumour fractions from hysterectomy specimen. The low levels of intratumour heterogeneity affected the molecular risk allocation in a limited number of cases. Therefore, prognostic molecular markers to guide adjuvant treatment decisions can be determined on a single representative sample of EC, facilitating their use in routine diagnostics.
MATERIALS AND METHODS
Case selection
ECs, previously molecularly profiled [7, 47], with a minimum of three available formalin-fixed paraffin-embedded (FFPE) tumour blocks were selected to give similar numbers of ±10 ECs with mutations in POLE and CTNNB1, alterations in MMR, p53 and L1CAM protein expression, and without any of these alterations. With this approach, our study set will be enriched for POLE-mutant ECs (20% of cases) in comparison to a random EC population (7-12% of cases) [48]. Only sporadic MMR-deficient tumours with proven MLH1 promotor hypermethylation were selected. Haematoxylin-eosin (H&E) stained slides of each selected case were reviewed to randomly select three tumour blocks containing the highest percentage of tumour tissue. As a result of this random selection, in 27 (55%) of cases the tumour block used for initial molecular profiling was also included in this study [7, 47]. H&E slides of the selected tumour blocks were evaluated for histological features (i.e. tumour grade, histotype and nuclear atypia) by a gynaecopathologist.
In total, 43 ECs were selected from the pathology archive (2001-2015) of the Leiden University Medical Centre, the Netherlands and 6 ECs from the PORTEC-1 and -2 studies [49, 50] All women underwent a hysterectomy either with or without bilateral salpingo-oophorectomy followed by tailored adjuvant therapy according to the national clinical guidelines [51]. The clinicopathological data were obtained from the pathology reports and trial databases [49, 50]. This study was approved by the institutional review board.
Immunohistochemical analysis
Immunohistochemistry on tissue slides (4 μM) was performed as previously described [10, 30, 52]. In brief, endogenous peroxidases were inactivated by 0.3% H2O2/methanol. Subsequently, antigen retrieval was achieved by microwave oven procedure in 10 mmol/l Tris-EDTA, pH 9.0. Sections were incubated overnight with primary monoclonal antibodies against L1CAM (clone 14.10, 1:500, Covance Inc.), p53 (clone DO-7; 1:1000; Neomarkers), MLH1 (clone ES05, 1:100; DAKO), MSH2 (clone FE11, 1:100, DAKO), MSH6 (clone EPR3945, 1:800, Genetex) or PMS2 (clone EP51, 1:25, DAKO). Sections stained for MLH1, MSH2 and PMS2 were incubated at room temperature with Envision FLEX+ Linker (DAKO) for 15 minutes. Thereafter, all sections were incubated and stained for 30 minutes using a secondary antibody (poly-HRP-GAM/R/R; DPV0110HRP; ImmunoLogic). Diamino-benzidine-tetrahydrochloride (DAKO) was used as a chromogen. Finally, the slides were counterstained with Mayer’s haematoxylin, dehydrated and mounted.
For immunohistochemical analysis, slides were randomly numbered to ensure the two independent observers were blinded for the paired three tumour slides, patient characteristics, and the initial assigned status of the p53, L1CAM and MMR protein expression. Discrepancies were resolved by consensus under a multihead microscope. p53 was scored mutant-like if the whole tumour or a distinct geographical area of >10% of the tumour showed strong positive nuclear staining in >50% of tumour cells (diffuse or subclonal mutant-like) [53]. Cases were scored wildtype with focal overexpression if a discrete geographical area of <10% of the tumour showed strong positive nuclear staining (wildtype+). Sanger sequencing for TP53 exon 5-8 mutations was performed as described previously for cases with ‘indefinite’ p53 expression (null staining), with subclonal mutant-like expression, with focal overexpression and ‘discordant’ cases in which the scores of the three tumour blocks differed [9]. The percentage of positive membranous L1CAM staining within the tumour was scored according to the scoring system for EC; 0%, <10%, 10-50%, or >50% [11]. Tumours were considered L1CAM positive if >10% L1CAM positivity was observed, based on prior studies [10, 11, 36]. Nerves from the deeper myometrium were used as internal positive controls and were identified in all cases. Tumours were considered MMR-deficient if tumour cells showed loss of nuclear staining of at least one of the MMR proteins either in the whole tumour or in a distinct geographical area (subclonal loss) with positive stromal cells, and MMR-proficient if tumour cells showed nuclear positivity for all mismatch repair proteins.
DNA isolation and mutation analysis
To obtain tumour DNA, five sections (10 μM) were used to microdissect fragments of tumour with the aim to reach tumour percentage of >70%. Briefly, the sections were deparaffinised in xylene, rehydrated through a graded ethanol series, and stained with haematoxylin. The area of tumour tissue, marked on the H&E stained slide by a gynaecopathologist, was manually microdissected. After overnight proteinase K digestion, DNA isolation was performed according to manufacturer’s instructions (NucleoSpin Tissue kit, Macherey-Nagel). Competitive allele-specific PCR (LGC Genomics) assays were used to screen for hotspot mutations in the POLE exonuclease domain [31]. Cases with discordant POLE mutation status among the three tumour blocks were reanalysed using Sanger sequencing to detect mutations in POLE exons 9 and 13 [31]. Sanger sequencing was also used to detect mutations in exon 3 of CTNNB1 [8]. For the discordant CTNNB1 cases, the Profiler Plus PCR Amplification kit (Applied Biosystems) was used to establish that the DNA isolated from three blocks have the same origin and to exclude contamination or exchange of DNA.
Molecular risk assignment
The three tumour blocks of each case were assigned to a favourable, intermediate or unfavourable risk group, based on molecular markers included in a simplified integrated risk assignment (Supplementary Figure 1) [5–7]. P53-mutant-like and L1CAM-positive tumours were assigned as having an unfavourable risk. CTNNB1-mutant and MMR-deficient tumours were assigned as having an intermediate risk. POLE-mutant tumours and tumours with no specific molecular profile were assigned as having a favourable risk.
Abbreviations
EC, endometrial cancer; FFPE, formalin-fixed paraffin-embedded; L1CAM, L1 cell adhesion molecule; MMR, mismatch repair; TCGA, The Cancer Genome Atlas.
FUNDING/ACKNOWLEDGMENTS
This study was supported by the Dutch Cancer Society (KWF-UL2012-5719).
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
Authors’ contributions
All authors participated to the critical revision of the manuscript for important intellectual content, manuscript writing, obtained funding, study supervision, acquisition of data, analysis and interpretation of data, data collection and analysis, drafting of the manuscript, oversight of all aspects of study, study concept and design, and technical support.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7-30.
2. Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D, Bray F. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. European journal of cancer. 2013; 49:1374-1403.
3. Colombo N, Preti E, Landoni F, Carinelli S, Colombo A, Marini C, Sessa C, Group EGW. Endometrial cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of oncology. 2013; 24 :vi33-38.
4. Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, Yau C, Laird PW, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013; 497:67-73.
5. Stelloo E, Bosse T, Nout RA, MacKay HJ, Church DN, Nijman HW, Leary A, Edmondson RJ, Powell ME, Crosbie EJ, Kitchener HC, Mileshkin L, Pollock PM, et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Modern pathology. 2015; 28:836-844.
6. Talhouk A, McConechy MK, Leung S, Li-Chang HH, Kwon JS, Melnyk N, Yang W, Senz J, Boyd N, Karnezis AN, Huntsman DG, Gilks CB, McAlpine JN. A clinically applicable molecular-based classification for endometrial cancers. Br J Cancer. 2015; 113:299-310.
7. Stelloo E, Nout RA, Osse EM, Jurgenliemk-Schulz IJ, Jobsen JJ, Lutgens LC, van der Steen-Banasik EM, Nijman HW, Putter H, Bosse T, Creutzberg CL, Smit VT. Improved Risk Assessment by Integrating Molecular and Clinicopathological Factors in Early-stage Endometrial Cancer-Combined Analysis of the PORTEC Cohorts. Clinical cancer research. 2016.
8. Liu Y, Patel L, Mills GB, Lu KH, Sood AK, Ding L, Kucherlapati R, Mardis ER, Levine DA, Shmulevich I, Broaddus RR, Zhang W. Clinical significance of CTNNB1 mutation and Wnt pathway activation in endometrioid endometrial carcinoma. Journal of the National Cancer Institute. 2014; 106.
9. Nout RA, Bosse T, Creutzberg CL, Jurgenliemk-Schulz IM, Jobsen JJ, Lutgens LC, van der Steen-Banasik EM, van Eijk R, Ter Haar NT, Smit VT. Improved risk assessment of endometrial cancer by combined analysis of MSI, PI3K-AKT, Wnt/beta-catenin and P53 pathway activation. Gynecologic oncology. 2012; 126:466-473.
10. Bosse T, Nout RA, Stelloo E, Dreef E, Nijman HW, Jurgenliemk-Schulz IM, Jobsen JJ, Creutzberg CL, Smit VT. L1 cell adhesion molecule is a strong predictor for distant recurrence and overall survival in early stage endometrial cancer: pooled PORTEC trial results. European journal of cancer. 2014; 50:2602-2610.
11. Zeimet AG, Reimer D, Huszar M, Winterhoff B, Puistola U, Azim SA, Muller-Holzner E, Ben-Arie A, van Kempen LC, Petru E, Jahn S, Geels YP, Massuger LF, et al. L1CAM in early-stage type I endometrial cancer: results of a large multicenter evaluation. Journal of the National Cancer Institute. 2013; 105:1142-1150.
12. van der Putten LJ, Visser NC, van de Vijver K, Santacana M, Bronsert P, Bulten J, Hirschfeld M, Colas E, Gil-Moreno A, Garcia A, Mancebo G, Alameda F, Trovik J, et al. L1CAM expression in endometrial carcinomas: an ENITEC collaboration study. Br J Cancer. 2016; 115:716-724.
13. Stanta G, Jahn SW, Bonin S, Hoefler G. Tumour heterogeneity: principles and practical consequences. Virchows Archiv. 2016; 469:371-384.
14. Campbell PJ, Pleasance ED, Stephens PJ, Dicks E, Rance R, Goodhead I, Follows GA, Green AR, Futreal PA, Stratton MR. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105:13081-13086.
15. Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal SA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010; 467:1109-1113.
16. Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, Downing JR. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008; 322:1377-1380.
17. Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, Lai JL, Philippe N, Facon T, Fenaux P, Preudhomme C. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002; 100:1014-1018.
18. Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002; 2:117-125.
19. Inukai M, Toyooka S, Ito S, Asano H, Ichihara S, Soh J, Suehisa H, Ouchida M, Aoe K, Aoe M, Kiura K, Shimizu N, Date H. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer research. 2006; 66:7854-7858.
20. Lee AJ, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA, Downward J, Szallasi Z, Tomlinson IP, Howell M, Kschischo M, Swanton C. Chromosomal instability confers intrinsic multidrug resistance. Cancer research. 2011; 71:1858-1870.
21. Gerlinger M, Swanton C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer. 2010; 103:1139-1143.
22. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011; 472:90-94.
23. Feng YZ, Shiozawa T, Horiuchi A, Shih HC, Miyamoto T, Kashima H, Suzuki A, Nikaido T, Konishi I. Intratumoral heterogeneous expression of p53 correlates with p53 mutation, Ki-67, and cyclin A expression in endometrioid-type endometrial adenocarcinomas. Virchows Archiv. 2005; 447:816-822.
24. Kato A, Sato N, Sugawara T, Takahashi K, Kito M, Makino K, Sato T, Shimizu D, Shirasawa H, Miura H, Sato W, Kumazawa Y, Sato A, et al. Isolated Loss of PMS2 Immunohistochemical Expression is Frequently Caused by Heterogenous MLH1 Promoter Hypermethylation in Lynch Syndrome Screening for Endometrial Cancer Patients. The American journal of surgical pathology. 2016; 40:770-776.
25. Pai RK, Plesec TP, Abdul-Karim FW, Yang B, Marquard J, Shadrach B, Roma AR. Abrupt loss of MLH1 and PMS2 expression in endometrial carcinoma: molecular and morphologic analysis of 6 cases. The American journal of surgical pathology. 2015; 39:993-999.
26. Graham RP, Kerr SE, Butz ML, Thibodeau SN, Halling KC, Smyrk TC, Dina MA, Waugh VM, Rumilla KM. Heterogenous MSH6 loss is a result of microsatellite instability within MSH6 and occurs in sporadic and hereditary colorectal and endometrial carcinomas. The American journal of surgical pathology. 2015; 39:1370-1376.
27. Watkins JC, Nucci MR, Ritterhouse LL, Howitt BE, Sholl LM. Unusual Mismatch Repair Immunohistochemical Patterns in Endometrial Carcinoma. The American journal of surgical pathology. 2016; 40:909-916.
28. Gibson WJ, Hoivik EA, Halle MK, Taylor-Weiner A, Cherniack AD, Berg A, Holst F, Zack TI, Werner HM, Staby KM, Rosenberg M, Stefansson IM, Kusonmano K, et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat Genet. 2016.
29. Talhouk A, Hoang LN, McConechy MK, Nakonechny Q, Leo J, Cheng A, Leung S, Yang W, Lum A, Kobel M, Lee CH, Soslow RA, Huntsman DG, et al. Molecular classification of endometrial carcinoma on diagnostic specimens is highly concordant with final hysterectomy: Earlier prognostic information to guide treatment. Gynecologic oncology. 2016.
30. Stelloo E, Nout RA, Naves LC, Ter Haar NT, Creutzberg CL, Smit VT, Bosse T. High concordance of molecular tumor alterations between pre-operative curettage and hysterectomy specimens in patients with endometrial carcinoma. Gynecologic oncology. 2014; 133:197-204.
31. Church DN, Stelloo E, Nout RA, Valtcheva N, Depreeuw J, ter Haar N, Noske A, Amant F, Tomlinson IP, Wild PJ, Lambrechts D, Jurgenliemk-Schulz IM, Jobsen JJ, et al. Prognostic significance of POLE proofreading mutations in endometrial cancer. Journal of the National Cancer Institute. 2015; 107:402.
32. Colombo N, Creutzberg C, Amant F, Bosse T, Gonzalez-Martin A, Ledermann J, Marth C, Nout R, Querleu D, Mirza MR, Sessa C, Group E-E-EECCW. ESMO-ESGO-ESTRO Consensus Conference on Endometrial Cancer: Diagnosis, Treatment and Follow-up. Int J Gynecol Cancer. 2016; 26:2-30.
33. Catalogue of somatic mutations in cancer. Mutation overview TP53 (c.733G>GA). http://cancer.sanger.ac.uk/cosmic/mutation/overview?id=6932, 2016.
34. Catalogue of somatic mutations in cancer. Mutation overview TP53 (c.524G>GA). http://cancer.sanger.ac.uk/cosmic/mutation/overview?id=10648, 2016.
35. Shinbrot E, Henninger EE, Weinhold N, Covington KR, Goksenin AY, Schultz N, Chao H, Doddapaneni H, Muzny DM, Gibbs RA, Sander C, Pursell ZF, Wheeler DA. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014; 24:1740-1750.
36. Van Gool IC, Stelloo E, Nout RA, Nijman HW, Edmondson RJ, Church DN, MacKay HJ, Leary A, Powell ME, Mileshkin L, Creutzberg CL, Smit VT, Bosse T. Prognostic significance of L1CAM expression and its association with mutant p53 expression in high-risk endometrial cancer. Modern pathology. 2016; 29:174-181.
37. Kobel M, Meng B, Hoang LN, Almadani N, Li X, Soslow RA, Gilks CB, Lee CH. Molecular Analysis of Mixed Endometrial Carcinomas Shows Clonality in Most Cases. The American journal of surgical pathology. 2016; 40:166-180.
38. Espinosa I, D'Angelo E, Palacios J, Prat J. Mixed, Ambiguous Endometrial Carcinomas: A Heterogenous Group of Tumors With Different Clinicopathologic and Molecular Genetic Features. The American journal of surgical pathology. 2016; 40:972-981.
39. Coenegrachts L, Garcia-Dios DA, Depreeuw J, Santacana M, Gatius S, Zikan M, Moerman P, Verbist L, Lambrechts D, Matias-Guiu X, Amant F. Mutation profile and clinical outcome of mixed endometrioid-serous endometrial carcinomas are different from that of pure endometrioid or serous carcinomas. Virchows Archiv. 2015; 466:415-422.
40. Bartlett AI, Starcyznski J, Robson T, Maclellan A, Campbell FM, van de Velde CJ, Hasenburg A, Markopoulos C, Seynaeve C, Rea D, Bartlett JM. Heterogeneous HER2 gene amplification: impact on patient outcome and a clinically relevant definition. Am J Clin Pathol. 2011; 136:266-274.
41. Ohlschlegel C, Zahel K, Kradolfer D, Hell M, Jochum W. HER2 genetic heterogeneity in breast carcinoma. J Clin Pathol. 2011; 64:1112-1116.
42. Allison KH, Dintzis SM, Schmidt RA. Frequency of HER2 heterogeneity by fluorescence
43. Chang MC, Malowany JI, Mazurkiewicz J, Wood M. 'Genetic heterogeneity' in HER2/neu testing by fluorescence
44. Seol H, Lee HJ, Choi Y, Lee HE, Kim YJ, Kim JH, Kang E, Kim SW, Park SY. Intratumoral heterogeneity of HER2 gene amplification in breast cancer: its clinicopathological significance. Modern pathology. 2012; 25:938-948.
45. Yang YL, Fan Y, Lang RG, Gu F, Ren MJ, Zhang XM, Yin D, Fu L. Genetic heterogeneity of HER2 in breast cancer: impact on HER2 testing and its clinicopathologic significance. Breast Cancer Res Treat. 2012; 134:1095-1102.
46. Murthy SS, Sandhya DG, Ahmed F, Goud KI, Dayal M, Suseela K, Rajappa SJ. Assessment of HER2/Neu status by fluorescence
47. van Gool IC, Bosse T, Church DN. POLE proofreading mutation, immune response and prognosis in endometrial cancer. Oncoimmunology. 2016; 5:e1072675.
48. Rayner E, van Gool IC, Palles C, Kearsey SE, Bosse T, Tomlinson I, Church DN. A panoply of errors: polymerase proofreading domain mutations in cancer. Nature reviews Cancer. 2016; 16:71-81.
49. Creutzberg CL, van Putten WL, Koper PC, Lybeert ML, Jobsen JJ, Warlam-Rodenhuis CC, De Winter KA, Lutgens LC, van den Bergh AC, van de Steen-Banasik E, Beerman H, van Lent M. Surgery and postoperative radiotherapy versus surgery alone for patients with stage-1 endometrial carcinoma: multicentre randomised trial. PORTEC Study Group. Post Operative Radiation Therapy in Endometrial Carcinoma. Lancet. 2000; 355:1404-1411.
50. Nout RA, Smit VT, Putter H, Jurgenliemk-Schulz IM, Jobsen JJ, Lutgens LC, van der Steen-Banasik EM, Mens JW, Slot A, Kroese MC, van Bunningen BN, Ansink AC, van Putten WL, et al. Vaginal brachytherapy versus pelvic external beam radiotherapy for patients with endometrial cancer of high-intermediate risk (PORTEC-2): an open-label, non-inferiority, randomised trial. Lancet. 2010; 375:816-823.
51. Comprehensive Cancer Centre the Netherlands. Guidelines for endometrial cancer. http://oncoline.nl/endometriumcarcinoom, 2017.
52. de Jong AE, van Puijenbroek M, Hendriks Y, Tops C, Wijnen J, Ausems MG, Meijers-Heijboer H, Wagner A, van Os TA, Brocker-Vriends AH, Vasen HF, Morreau H. Microsatellite instability, immunohistochemistry, and additional PMS2 staining in suspected hereditary nonpolyposis colorectal cancer. Clinical cancer research. 2004; 10:972-980.
53. McCluggage WG, Soslow RA, Gilks CB. Patterns of p53 immunoreactivity in endometrial carcinomas: 'all or nothing' staining is of importance. Histopathology. 2011; 59:786-788.