
INTRODUCTION
Ras proteins exist in two conformations: a GTP-bound active state and GDP-bound inactive state. The ratio of GTP and GDP bound to cellular Ras proteins is controlled by guanine nucleotide exchange proteins and GTPase-activating proteins, the enzymatic activity of which responds to extracellular stimuli (such as growth factors) [1–4]. Once activated, GTP-bound Ras interacts with target enzymes/effectors and triggers kinase cascades to regulate various cellular activities. Ras, through controlling the activity of these effectors, is able to influence cell behaviors. The best characterized effectors of Ras are various protein serine/threonine kinases, for example, Raf/MEK/MAPK, PI3K/Akt or Rho/Ral [2, 5]. Studies also demonstrated that Ras-governed pathways could induce apoptosis in several types of cancer cells [6–8]. In response to different apoptotic stimuli, Ras differentially used its downstream effector pathways and re-directed the cells to apoptosis. PI3K/Akt pathway was shown to be able to upregulate several ROS-related enzymes (such as NADPH oxidase), and play important roles in the regulation of endoplasmic reticulum (ER)-stress induced apoptosis [9, 10].
The uses of the high-throughput RNA-interference (siRNA) technique to selectively inhibit gene expressions, together with the knowledge of the full sequence of the genome, have made possible to identify intracellular targets of cancer cells, through large-scale functional genomic screens [11, 12]. Cancer cells often develop secondary dependencies on genes/factors for survival. Perturbation of these genes/factors results in oncogene-specific synthetic lethality. Such lethality often involves genes/factors within the same or parallel pathways that are required for an essentially vital function. Using siRNA screening, it has uncovered kinase-specific vulnerabilities in tumors harboring mutated ras [11, 12]. Furthermore, studies (including ours) demonstrated that loss of PKC (in particular the phorbol ester-dependent subgroup of PKC isoforms), together with Ras mutations, were synthetically lethal [13–17]. Since about 30–40% of human tumors contain an oncogenic ras and attempts to directly target mutated Ras proteins with small molecular weight inhibitors have proved to be unsuccessful [18], the research focus is now on identifying targeting pathways that function downstream of or parallel with oncogenic Ras. The inhibition of one of these pathways would specifically trigger apoptosis in the tumor cells harboring mutant ras, but be less or minimal toxic to surrounding normal tissues/cells.
Reactive oxygen species (ROS) are a diverse group of reactive, short-lived, oxygen containing species (such as superoxide radical, hydrogen peroxide and hydroxyl radical), and constantly generated in cells during normal aerobic metabolism [19–22]. Physiological levels of ROS are required for cell signaling cascades. The consequences of aberrant ROS increases in cells cause permanent structural changes in DNA (such as DNA strand breaks), initiation of lipid peroxidation, alterations of intracellular signaling pathways (such as ER stress-related proteins) or induction of apoptosis [23–25]. Cells possess an array of defense systems to suppress and scavenge excess ROS as well as repair related damages, in order to protect from being injured. When the defense systems cannot effectively suppress aberrant increases of ROS, a persistent oxidative or ER stress is triggered. Such stresses often cause the activation of the unfolded protein response (UPR) and in some cases, apoptosis is induced [26–28]. The proper balance of intracellular redox state is crucial for cell survival. Under normal growth conditions, a fraction of oxygen is converted to superoxide by NADH dehydrogenase and other enzymes of the mitochondrial respiratory chain [29, 30]. Superoxide is then converted to H2O2 by superoxide dismutase (SOD) [29, 30]. H2O2 at normal levels is able to diffuse through cell membranes and needed for regulating various cellular activities [31–33]. Persistently high amounts of ROS often cause damages in the mitochondrial transmembrane potential or activate UPR, which thereby could trigger cell death [26–28]. ROS-induced apoptosis was indicated to be via the activation of PI3K/Akt, p53 or other factors [15, 34, 35]. However, the mechanisms that control cell fate: cell survival versus death in response to oxidative or ER stress, have not been fully investigated.
The ER is a cytosolic compartment where proteins and lipids are synthesized. Chaperones resided in the ER facilitate proper protein folding, maintain proteins in a folded structure or prevent protein folding intermediates from aggregating [36–38]. BIP is one of the best characterized ER chaperone proteins and its synthesis can be stimulated by various stresses (including oxidative stress) that sometimes disrupt ER function and perturb homeostasis [39–41]. Studies suggested that increases in BIP expression are the indicator of ER stress. The ER compartment is very flexible and can accumulate partially folded proteins. Once accumulations of improper folded proteins exceed the limit of the ER, the UPR is activated, leading to diseases or apoptosis [42–45].
In the present study, using different ras loop mutant constructs, we intended to identify which Ras effector(s), together with suppression of PKC, was/were synthetically lethal in the cancer cells. The results suggested that Akt acted downstream of oncogenic Ras in the cancer cells and induced apoptosis after co-suppressing PKC α and β.
RESULTS
Pancreatic cancer cells expressing mu-K-ras underwent apoptosis after the co-suppression of PKC α and β
The inhibition of PKC triggered apoptosis in cultured cells expressing v-ras or cancer cells harboring mutated ras [13–17]. However, the underlying mechanisms by which oncogenic ras induces apoptosis remain unclear. For this reason, human pancreatic epithelial cells (HPNE) or its HPNE stably transfected with mu-K-ras as well as human pancreatic cell lines Panc-1 or MIA harboring mutated K-ras were used to test their sensitivities to PKC inhibition for the induction of apoptosis. Ras activation status in these cells was first analyzed using Active Ras Pull-Down and Detection kit (Figure 1A). The increased amount of the GTP-bound Ras was detected in all cells expressing aberrant K-ras, in comparison that only the baseline level of active Ras was revealed by the antibody in HPNE cells. Our previous reports demonstrated that murine fibroblasts ectopically expressing mutant ras were susceptible to apoptosis after the co-suppression of PKC α and β isoforms [13–17]. Therefore, the susceptibility of the pancreatic cancer cells to the co-inhibition of PKC α/β was studied. The effect of the knockdown of PKC α or β by the shRNAs was analyzed by immunoblotting. The shRNAs, but not the scRNAs, efficiently knocked down these two PKC isoforms (Figure 1B, right panels). The DNA fragmentation assay was then conducted (Figure 1B, left panel). Approximately 30% of HPNE/K-ras, Panc1 and MIA cells, but not HPNE cells, underwent apoptosis after the knockdown of PKC α/β.
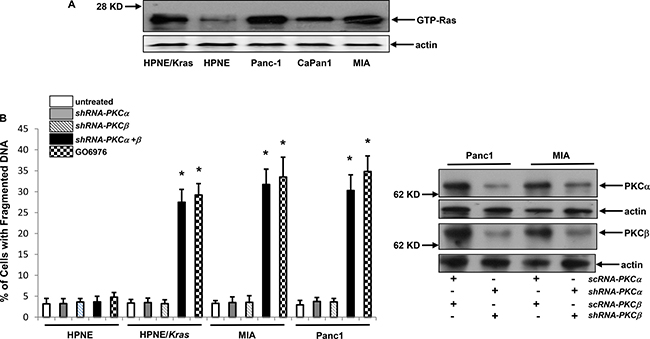
Figure 1: Mutant K-ras and loss of PKC α and β were synthetically lethal. (A) Ras GTP-binding activity was analyzed in the cells using active Ras pull-down and detection kit. Evenly loadings of the lysates were normalized by actin expression. (B) Cells were infected with sc or shRNA-PKC α or β for 48 h and the expressions of corresponding proteins were then analyzed by immunoblotting (right panels). After the infection of shRNA-PKC α, β or both as well as treatment of GO6976 (10 uM) for 48 h, DNA fragmentation assay was conducted to detect the induction of apoptosis in the cells (left panel). The error bars are SD from 5 independent experiments (n = 5, ρ < 0.05).
ROS is upregulated in response to the co-suppression of PKC α and β
ROS is required for various cellular activities. However, aberrant increases of ROS triggers oxidative stress, sometimes resulted in apoptosis [19–24]. To further test if the co-suppression of PKC α and β perturbed the redox state, ras effector loop mutant constructs were employed. V12S35 (the protein product of which is able to bind to and activate Raf), V12C40 (the protein product of which preferentially activates PI3K/Akt), or V12G37 (encoded protein interacts with RalGDS) was introduced into HPNE cells, respectively and the protein expressions of the Ras mutants were confirmed by immunoblotting (data not shown) [25]. The levels of ROS in response to co-knockdown of PKC α/β or treatment of GO6976 were measured in HPNE cells with or without overexpressing v-Kras loop mutant constructs (Figure 2A). The introduction of V12C40 mutant gene caused a slightly increased level of ROS in HPNE cells, which was significantly upregulated by the addition of GO6976 (Figure 2A, left panel) or co-knockdown of PKC α/β (Figure 2A, right panel). Such upregulation of ROS was suppressed by N-acetyl-L-cysteine (NAC, a ROS inhibitor) (data not shown). In comparison, other ras loop mutants did not cause the increases of ROS in the cells, even in the absence of PKC. To confirm the role of PI3K/Akt, human prostate epithelial PrEC and cancer DU145 cells that express active Akt were assayed for ROS accumulation in our experimental setting (Figure 2B). The amount of ROS in untreated DU145 cells was slight elevated, but significantly upregulated by GO6976 treatment, which was suppressed by NAC or KP-372-1 (an Akt inhibitor). GO6976 did not affect ROS level in PrEC cells. The data suggested a linear relationship between oncogenic Ras and PI3K/Akt pathway in the perturbation of the balance of the redox state in the cells after the co-inhibition of PKC α and β.
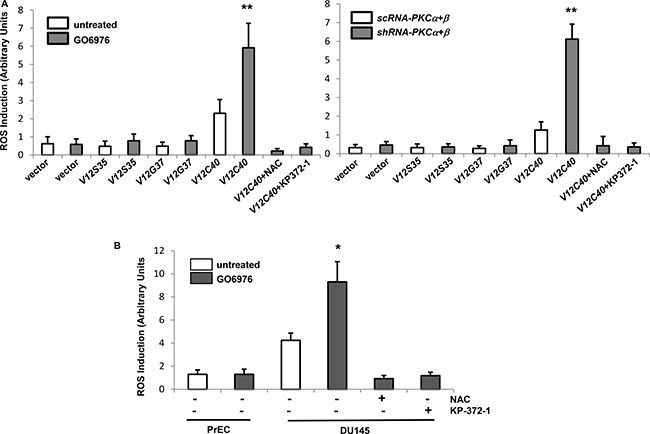
Figure 2: ROS was upregulated via PI3K/Akt pathway after the co-suppression of PKC α and β. (A) HPNE cells were stably transfected with the ras effector loop mutant constructs. After being treated with GO6976 or co-infected with sc or shRNA-PKC α and β, the amounts of ROS in the cells were measured. The error bars are SD from 5 independent experiments (n = 5, ρ < 0.005). (B) Prostate epithilieal PrEC and cancer DU145 cells were treated with NAC or KP-372-1 prior to GO6976 treatment, and the levels of ROS were measured. The error bars are SD from 5 independent experiments (n = 5, ρ < 0.05).
Oxidative regulators are activated in a PI3K/Akt-dependent fashion
NADPH oxidase is a membrane-bound enzymatic complex and one of the sources of intracellular superoxides [19–22]. Its cytosolic components (p67phox, p47phox and p40phox) translocated to the plasma membrane in response to oxidative stress [19–22]. Therefore, HPNE cells with or without transiently expressing V12C40 (Figure 3A, left panels) as well as pancreatic cancer MIA cells (Figure 3A, right panels) were treated with GO6976. The plasma membrane or cytosolic fractions were then isolated, and immunoblotted with the antibody for p67phox expression. p67phox was present in the cytosolic fraction of HPNE cells expressing vector alone regardless of GO6976 treatment. In comparison, a high amount of p67phox was present in the cytosolic fraction of untreated HPNE/V12C40 cells. After GO6976 treatment, a significant increased amount of p67phox appeared in the plasma membrane fraction of HPNE/V12C40 cells. The similar pattern of p67phox plasma membrane translocation was observed in GO6976-treated MIA cells. The expression of MnSOD (Mn2+ superoxide dismutase, one of ROS modulators) in HPNE, MIA and DU145 cells was then analyzed by immunoblotting (Figure 3B). A slight increase of MnSOD in untreated HPNE/V12C40 cells was detected. After GO6976 treatment, MnSOD expression was dramatically upregulated, which was blocked by the addition of NAC, indicating the role of ROS in the induction of MnSOD. The expression of another ROS regulator HO-1 was also examined under the same experimental conditions, and similar results were obtained (data not shown). Overall, the data further indicated that the co-suppression of PKC α/β perturbed the redox machinery.
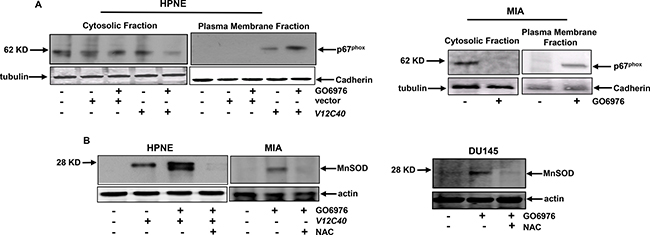
Figure 3: ROS regulators were activated after co-suppression of PKC α and β. (A) After the treatments, the cytosolic or plasma membrane fraction from the cells were isolated and immunoblotted for p67PHOX expression. The evenly loadings were normalized by tubulin or cadherin. (B) MnSOD expression in the cells after the treatments was analyzed by immunoblotting.
p73 was activated in an Akt-dependent fashion after co-suppressing PKC α and β
p73 is a member of p53 tumor suppressor family and involved in the regulation of apoptosis upon genotoxic stress or exposure to apoptotic stimuli [16]. It was reported that active p73 could re-locate to the nucleus and participate the induction of apoptosis there [46, 16]. Therefore, p73 function in our experimental setting was examined. After GO6976 treatment, cell lysates from HPNE cells with or without overexpressing V12C40, and from MIA or DU145 cells were prepared to test the expression of phor-p73 (Figure 4A). The phosphorylated form of p73 was detected in HPNE/V12C40, MIA or DU145 cells only after the co-suppression of PKC α and β. Subsequently, the subcellular localization of p73 in our experimental setting was analyzed (Figure 4B). The cytosol and nuclear fractions were isolated from HPNE or NPNE/V12C40 cells with or without GO6976 treatment and immunoblotted with anti-p73 antibody. This tumor suppressor was detected in the cytosol fractions of untreated HPNE and HPNE/V12C40 cells, which was absent in the nuclear fractions. After GO6976 treatment, p73 was disappeared in the cytosolic fraction of HPME/V12C40 cells, and appeared in its nuclear fraction. The similar pattern of the nuclear translocation of p73 was observed in GO6976-treated MIA cells (Figure 4C). The data implicated that p73 was activated and perhaps played an indispensable role in this apoptotic process.
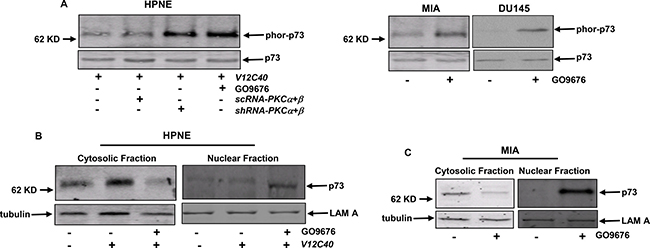
Figure 4: p73 is phosphorylated and translocated to the nucleus in response to GO6976 treatment. (A) HPNE cells ectopically expressing V12C40 as well as pancreatic cancer MIA or DU145 cells were either co-infected with shRNA-PKC α/β or treated with GO6976. Subsequently, lysates were immunoprecipitated with anti-p73 antibody and then immunoblotted with the antibody recognizing the phosphorylated p73. The evenly loadings of the samples were normalized by re-probing the blot with anti-p73 antibody. (B and C) After the treatments, the cytosolic or nuclear fractions were isolated and immunoblotted for p73 expression. The evenly loadings were normalized by tubulin or Lam A.
To further test if both the upregulation of ROS and induction of apoptosis occurred in our experimental setting were p73-dependent, the level of ROS (Figure 5A) and onset of apoptosis (Figure 5B) were analyzed. Again, a slight increase of ROS was revealed in untreated HPNE/V12C40 cells. The knockdown of p73 did not affect the upregulation of ROS mediated by GO6976 treatment. The similar results were obtained from GO6976-treated pancreatic or prostate cancer cells after p73 was knockdown. Subsequently, the induction of apoptosis in the cells was analyzed after knockdown of p73. In the absence of p73, HPNE/V12C40, MIA and DU145 cells were no longer sensitive to GO6976 treatment for the induction of apoptosis. The data suggested that the perturbation of ROS in our experimental setting occurred upstream of p73 and, the induction of apoptosis was dependent upon p73.
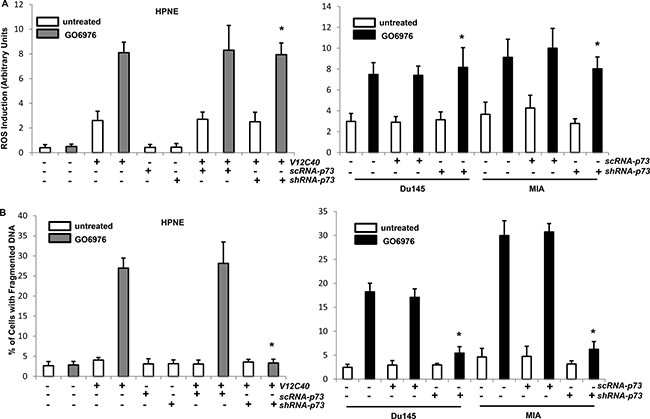
Figure 5: Induction of ROS and apoptosis in the absence of p73. (A) After infected with sc or shRNA-p73, cells were treated with NAC or KP-372-1 prior to GO6976 treatment, and the levels of ROS were then measured. The error bars are SD from 5 independent experiments (n = 5, ρ < 0.05). (B) After the treatments, DNA fragmentation assay was conducted to detect the induction of apoptosis in the cells. The error bars are SD from 5 independent experiments (n = 5, ρ < 0.01).
UPR was activated after the co-suppression of PKC α and β
The ER plays an important role in sensing cellular insults, persistent of which often activates the unfolded protein response (UPR) [36–38]. PERK is one of UPR sensors and activated or phosphorylated during persistent ER stress. To test whether the UPR was activated in our experimental setting, the expression of the phosphorylated form of PERK in the cells after the treatment of GO6976 was analyzed (Figure 6A). The phosphorylated PERK was detected in GO6976-treated HPNE/V12C40 or MIA cells, which was blocked by the addition of NAC or infection of shRNA-p73. PERK was not phosphorylated in untreated cells, as expected. GADD153 is an apoptotic factor and often induced in UPR-mediated apoptosis [36–38]. Immunoblotting analysis was performed to test GADD153 expression (Figure 6B). GADD153 was almost undetectable in untreated cells. After treated with GO6976, this apoptotic factor was induced in HPNE/V12C40 and MIA cells, which was suppressed by the addition of NAC or by knockdown of p73. It appeared that apoptosis triggered by the co-suppression of PKC α and β was the consequence of UPR activation, in which ROS and p73 were required.
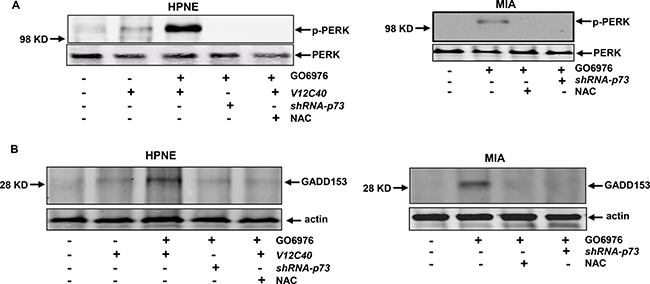
Figure 6: ER stress and UPR activation were triggered in the cells in response to GO6976 treatment. (A) In the presence or absence of p73, the phosphorylated PERK in HPNE, HPNE/V12C40 or MIA cells was analyzed by immunoblotting. (B) GADD153 expression was analyzed after the treatments as described above.
Induction of apoptosis occurs in xenografted pancreatic tumors
To further confirm the results from the in vitro experiments, xenograft assay was performed (Figure 7). After the inoculation of human pancreatic cancer MIA or Panc1 cells without or with stably expressing scRNA-p73 or shRNA-p73 into nude mice, respectively, GO6976 (30 mg/kg) was injected into the mice intraperitoneally, which was given every 3 days. One week after the inoculation when the tumors became detectable, the diameters of the tumors were measure every week for consecutive 4 weeks (Figure 7A). The xenografted tumors were formed in untreated mice inoculated with MIA or Panc1 cells. In comparison, the xenografted MIA or Panc1 tumors expressing scRNA-p73 in the mice grew very slow after received routine GO6976 injection. After the knockdown of p73, GO6976 injection could not block the tumor growth in the mice. The pictures of the tumors with or without the knockdown of p73 were taken (Figure 7B). The slides mounted with the tumor samples were stained with TUNEL reagent (Figure 7C). TUNEL staining was negative in untreated or GO6976-injected tumors after p73 was knockdown. However, the samples from GO6976-injected tumor cells transfected with scRNA-p73 were strongly stained with TUNEL reagent. The xenograft assay was also conducted with DU145 cells with or without the knockdown of p73. The similar data were obtained (data not shown). The in vivo data corroborated well with those obtained from the in vitro experiments. The data further suggested that the co-suppression of PKC α and β sensitized cancer cells harboring oncogenic ras, which is dependent upon p73.
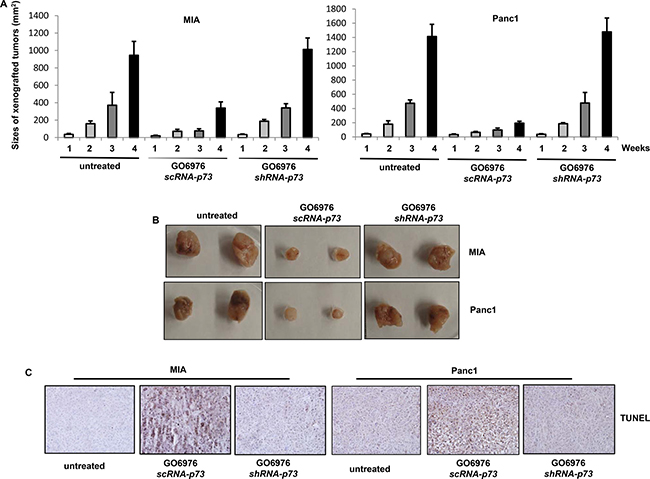
Figure 7: Induction of apoptosis in the xenografted tumors. (A) Cells were inoculated subcutaneously into the nude mice. GO6976 was injected peritoneally after the inoculation and subsequently administrated every 3 days. One week later, the diameters of the tumors were measured and then every week for consecutive 4 weeks. The error bars represent the SD (n = 4, *p < 0.05). (B) After the tumors were dissected from the mice, the pictures of the tumors were taken. (C) The slides mounted with the tumor samples were stained with TUNEL reagent.
DISCUSSION
Mutated ras or aberrant ras effectors are often present in various types of human malignancies. For example, in more than 90% of pancreatic cancer patients, mutated K-ras is detected, and active Akt are often seen in refractory prostate cancer among other types of cancers. [47]. Lack of effective treatments for these devastating diseases is the big challenge. Therefore, the objective of our current study was to search for new targets, which might help developing effective therapeutic strategies against cancers harboring aberrant ras. Using the ras loop mutant constructs, we demonstrated that PI3K/Akt signaling appeared functioning downstream of mutant Ras and was able to activate cell death program after the co-suppression of PKC α and β. In this apoptotic process, ROS was highly increased, resulted in ER stress and UPR activation for the induction of apoptosis. Furthermore, p73 was phosphorylated and translocated from the cytosol to the nucleus. However, p73 appeared downstream of ROS and played an important role in the initiation of apoptosis. The animal experiments corroborated well with the data from the in vitro studies. The study suggested that mutated ras, together with loss of PKC α and β, are incompatible for the viability of pancreatic or refractory prostate cancer cells. It also indicated that tumor suppressor p73 is a key element for triggering apoptosis in the cancer cells.
The mutational activation of ras is one of the key events during the initiation and development of various types of human malignancies, especially pancreatic cancer [1–5]. In the process of the malignant transformation, oncogenic Ras interacts and further switches on various downstream effector pathways, which triggers phosphorylation chain reactions and activates transcriptional factors. Despite the essential involvement in cell growth and differentiation, hyper-active Ras was shown to be able to induce apoptosis when some of its effector or supporting factors are suppressed [13–17]. For example, the treatment of PKC inhibitors is able to sensitize transformed cells ectopically expressing oncogenic ras or human cancers harboring mutant K-ras to apoptosis [48, 49]. In current study, we further demonstrated the underlying mechanisms by which the co-suppression of PKC α and β appeared essential for the viability of pancreatic cancer cells harboring mutated K-ras or prostate cancer cells expressing active Akt. The data suggested that these two PKC isoforms coped with aberrant Ras or Akt to keep the homeostasis in the cancer cells. Once these two isoforms were knocked out or inhibited, the oncogenes could not maintain malignant metabolic needs and fatal crises occurred. Thus, PKC α and β isoforms seem potentially therapeutic targets for developing new strategies to treat cancers harboring aberrant ras or Akt.
Levels of ROS are often altered in response to mitogenic stimuli. Persistent increases of ROS trigger the structural changes of the cytoskeleton, which often promote cell transformation or tumorigenesis [19–22]. Upon Epithelial growth factor (EGF) stimulation, ROS production was increased in the cells overexpressing oncogenic ras, which caused cancer progression [50, 51]. Oncogenic stresses induced by myc or ras oncogene often perturbed the state of intracellular redox, which led to chromosomal aberrations and disruption of genetic integrity for tumor promotion. Also, high increases of ROS were shown to play an obligatory role in the induction of apoptosis [14, 52]. For example, in TNFα-induced cell death, NF-κB and JNK cooperated to mobilize ROS signaling and further trigger caspase cascade [14, 52]. Ectopic expression of oncogenic ras in murine fibroblasts activated cell death program via inducing ER stress and UPR [14, 15]. In this study, we demonstrated that the concurrent inhibition of PKC α and β caused a significant increase of ROS that disrupted the equilibrium of the redox state in cancer cells harboring mutant K-ras or active Akt. The study indicated the important roles of PKC α and β in maintaining the homeostasis of these cancer cells.
PKC family consists of multiple serine/threonine kinase isoforms, some of which are structurally distinct or functionally diverse. The roles of some of PKC isoforms in the regulation of cell growth or death are rather controversial, depending upon cell types or cellular contexts. For example, PKC α, β, δ possessed dual roles in the regulation of cancer development or programmed cell death [16, 17, 48], which reflect the complexity of these isozymes. Cross-talks among PKC isoforms or with other intracellular signal transducers were often occurred in different subcellular compartments, which guided cells to participate different cellular activities. The interconnection of PKC and Ras signaling pathways was demonstrated in lymphocytes [53]. Upon mitogenic stimulations, the SH2 binding sites of PKC in T lymphocytes were shown to be phosphorylated and then recruited other signal transducers (such as Ras) via Grb2/SOS for forming the complexes, resulting in the activation of Ras signaling [53]. Using GO6976 that specifically inhibits PKC α and β isoforms or shRNAs-PKC α/β, we showed that the pancreatic cancer cells harboring mutant K-ras or prostate cancer cells expressing active Akt are being efficiently sensitized to apoptosis, which again suggested the cooperation of these pathways.
As a p53 family member, p73 shares the structural and functional homologue with p53 [54, 55]. This tumor suppressor plays an important role in DNA damage- or stress-induced apoptosis. The nuclear c-Abl was shown to be activated by genotoxic stress and further phosphorylated p73 for the induction of apoptosis [46, 55]. In this process, active p73 translocated to the nucleus, and interacted with c-Abl to initiate caspase cascade. In response to ionizing radiation or cisplatin treatment, p73 played a crucial role in initiating the apoptotic signaling [56, 57]. In our current experimental setting, p73 was phosphorylated and translocated from the cytosol to the nucleus for the induction of apoptosis. However, p73 activity is dispensable for the upregulation of ROS, but not for triggering UPR activation. The underlying mechanisms by which p73 elicits the sensors of UPR remain to be further investigated.
In summary, mutations in K-ras or Akt are uniquely detected in many types of human cancers. Thus, there is an eminent need for identifying intracellular targets to develop new strategies to treat these cancers. Our study demonstrated an apoptotic signaling pathway, in which mu-K-Ras utilized its downstream PI3K/Akt pathway to induce apoptosis in the absence of PKC α and β. In this apoptotic process, ROS was upregulated, accompanied with the activation of UPR and p73. The results indicated that PKC PKC α and β are important factors for the viability of cancer cells harboring mutant K-Ras or active Akt. Therefore, our investigation provides further information about the underlying mechanisms of apoptosis elicited by mutant K-Ras. Such knowledge may be exploited therapeutically for treating malignancies, especially pancreatic cancer or refractory prostate cancer.
MATERIALS AND METHODS
Cells and reagents
Human pancreatic epithelial HPNE, its stable mu-Kras transfectant HPNE/Kras, pancreatic cancer MIA, Panc1 and prostate cancer DU145 cells were purchased from ATCC (Manassas, VA) and the details of the phenotypes of the cells were provided by ATCC. The cells were frozen and kept in a -1500 C freezer after purchasing so that the authentications of the cells are preserved. HPNE cells were cultured in M3 medium containing 10% heat-inactivated fetal bovine serum and epithelial growth factor (EGF), penicillin and streptomycin (Invitrogen, CA). HPNE/Kras cells were maintained in the same M3 medium with 150 ug/ml of purimycin to keep the transfection pressure. Other cell lines were grown in Dulbecco’s Modified Eagles’s medium supplemented with 10% new born calf serum (Atlanta Biologicals, Flowery Branch, GA) and the antibiotic. Antibodies were purchased from BD (San Jose, CA), Santa Cruz Biotechnology (Santa Cruz, CA), Cell Signaling Technology (Dancers, MA) or Abcam (Cambridge, MA). The shRNAs were purchased from Origene (Rockville, MD).
Active Ras pull down and detection assay
Cell lysates were collected and assayed by the active Ras pull-down and detection kit (Thermo Scientific, Waltham, MA). Briefly, the GTP-form of Ras was pulled down by a GST-fusion protein with the Ras-binding domain (RBD) of Raf attached to glutathione agarose. The pull-down complexes were washed and separated on a 10% SDS-PAGE gel and immunoblotted with an anti-pan-Ras antibody.
Flow cytometry analysis
DNA fragmentation data were collected by a Muse Cell Analyzer, and analyzed by the Muse software program (BD Biosciences, Franklin Lakes, NJ). Following treatments, cells were harvested and fixed in 70% cold ethanol. Afterwards, cells were stained with 0.1 μg/ml propidium iodide containing 1.5ng/ml RNase. Fragmented DNA contents of cells were then analyzed using a flow cytometer.
Immunoblotting analysis
Cell lysates were extracted. After DNAs were removed, lysates were run on SDS-PAGE gels. Subsequently, gels were transferred to nitrocellulouses, blocked in TBS containing 5% non-fat milk and probed with corresponding antibodies. Membranes were then visualized by Odyssey infrared imaging system (Li-COR Biosciences, Lincoln, NE).
ROS analysis
Treated or untreated cells were washed with ice-cold PBS and resuspended in 5 μg/ml of 2′, 7′-dichlorodihydrofluorescein diacetate (DCF) (Thermo Scientific, MA). The samples were incubated for 10 min at room temperature and analyzed immediately.
Preparation of the cytosolic, cytoplasma membrane and nuclear fractions
Cells were suspended in lysis buffer (200 mM Sucrose, 20mM HEPES, 10 mM KCl, 1.5 M MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, Leupeptin, aprotinin at pH 7.4). The lysates then passed through a 25 gauge needle 15 times with syringes. Nuclear pellets were obtained by centrifugation. The remaining supernatants were further centrifuged at 8000 rpm and pellets were removed. The membrane and cytosolic fractions were separated by centrifuging the collected supernatants at 40,000 rpm. Subsequently, the supernatant cytosolic fractions were separated from the pellet plasma membrane fractions.
Xenograft assay
Cells (1 × 106) in 100 μl of PBS were inoculated into each BalB/c nude mouse. One group of mice (4 mice/group) was injected peritoneally with GO6976 (30 mg/kg) right after the inoculation and subsequently administrated the inhibitor every 3 days. The sizes of the tumors were measured weekly and plotted. After the mice were sacrificed, the tumors were isolated and slides mounted with tumor samples were stained with TUNEL staining. All the animal experiments were carried out according to the guidelines of the Animal Care and Use Committees of the Institute.
Statistical analysis
Statistical analysis was performed using a two-tailed Student’s t test for comparison of two groups or a one-way analysis of variance for comparison of more than two groups followed by Tukey’s multiple comparison tests. Tumor-free probabilities were estimated using Kaplan-Meier method and were compared among groups. Standard deviations are displayed in the figures. A p value < 0.05 was considered significant.
ACKNOWLEDGMENTS AND FUNDING
The authors thank Dr. T. Zhu (Sichuan University, China) for providing the reagents and constructs. This study is supported by the research funds to C. Chen (NIH R01CA153354 and R01CA100498).
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
1. Khosravi-Far R, Der CJ. The Ras signal transduction pathway. Cancer Metastasis Rev. 1994; 13:67–89.
2. Katz ME, McCormick F. Signal transduction from multiple Ras effectors. Curr Opin Genet Dev. 1997; 7:75–79.
3. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003; 3:11–22.
4. Cox AD, Der CJ. The dark side of Ras: regulation of apoptosis. Oncogene. 2003; 22:8999–9006.
5. McCormick F. Signaling networks that cause cancer. Trends Cell Biol. 1999; 9:M53–56.
6. Downward J. Ras signaling and apoptosis. Curr Opin Genet Dev. 1998; 8:49–54.
7. Jung JW, Cho SD, Ahn NS, Yang SR, Park JS. Ras/MAP kinase pathways are involved in Ras specific apoptosis induced by sodium butyrate. Cancer Lett. 2005; 225:199–206.
8. Guo J, Luo LY, Huang Y, Sunkavalli RG, Chen C. PI3K synergizes with loss of PKC to elicit apoptosis via UPR. J Cell Biochem. 2009; 107:76–83.
9. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO. 2006; 7:880–885.
10. Nogueira V, Park Y, Chen C, Xu P, Chen X, Tonic I, Unterman T, Hay N. Akt Determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008; 14:458–470.
11. Sawyer CL. Finding and drugging the vulnerabilities of Ras-dependent cancers. Cell. 2009; 137:796–768.
12. Singh A, Settleman J. Oncogenic K-ras ‘addiction’ and synthetic lethality. Cell Cycle. 2009; 8:2676–2678.
13. Liou JS, Chen JS, Faller DV. Characterization of p21Ras-mediated apoptosis induced by protein kinase C inhibition and application to human tumor cell lines. J Cell Physiol. 2004; 198:277–294.
14. Xia S, Forman LW, Faller DV. Protein kinase C delta is required for survival of cells expressing activated Ras. J Biol Chem. 2012; 282:13199–13210.
15. Guo J, Zhu T, Luo LY, Huang Y, Sunkavalli RG, Chen C. PI3K acts in synerby with loss of PKC to elicit apoptosis via the UPR. J Cell Bio. 2009; 107:76–85.
16. Zhu T, Tsuji T, Chen C. Roles of PKC isoforms in the induction of apoptosis elicited by aberrant Ras. 2010; 29:1050–1061.
17. Zhou X, Shen L, Yi B, Chen C. Regulation of the viability of Nf1 deficient cells by PKC isoforms. Oncotarget. 2014; 5:10709–10717. doi: 10.18632/oncotarget.2531.
18. Downward J. Cancer: a tumor gene’s fatal flaws. Nature. 2009; 462:44–45.
19. Suzuki YJ, Ford GD. Redox regulation of signal transduction in cardiac and smooth muscle. J Mol Cell Cardiol. 1999; 31:345–351.
20. Sen CK. Cellular thiols and redox-regulated signal transduction. Curr Top Cell Regul. 2000; 36:1–9.
21. Abid MR, Kachra Z, Spokes K, Aird WC. NADPH oxidase activity is required for endothelial cell proliferation and migration. FEBS Lett. 2000; 486:252–261.
22. Chen BT, Avshalumov MV, Rice ME. H2O2 is a novel endogenous modulator of synaptic dopamine release. J. Neurophysiol. 2001; 85:2468–2474.
23. Poulsen HE, Jensen BR, Weimann A. Antioxidants, DNA damage and gene expression. Free Radic Res. 2000; 33:S33–38.
24. Lassègue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003; 285:R277–289.
25. Guo J, Zhu T, Luo LY, Huang Y, Sunkavalli RG, Chen C. 2009 PI3K synergizes with loss of PKC to elicit apoptosis. J Cell Biochem 106:76–85.
26. Blackstone NW, Green DR. The evolution of a mechanism of cell suicide. Bioessays. 1999; 21:84–91.
27. Ueda S, Masutani H, Nakamura H, Tanaka T, Ueno M, Yodoi J. Redox control of cell death. Antioxid Redox Signal. 2002; 4:405–411.
28. Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009; 47:1239–1243.
29. Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep. 1997; 17:3–8.
30. Fridovich I. Superoxide anion radical (O2), superoxide dismutases, and related matters. J Biol Chem. 1997; 272:18515–18522.
31. Sundaresan M, Yu ZX, Ferrans V, Irani K, Finkel T. Requirement for generation of H2O2 for plateletderived growth factor signal transduction. Science. 1995; 270:296–305.
32. Bae YS, Sung JY, Kim OS. Platelet-derived growth factor-induced H2O2 production requires the activation of phosphatidylinositol 3-kinase. J Biol Chem. 2000; 275:10527–10534.
33. DeYulia GJ, Carcamo JM, Borquez-Ojeda O. Hydrogen peroxide generated extracellularly by receptor-ligand interaction facilitates cell signaling. Proc Natl Acad. Sci USA. 2005; 102:5044–5051.
34. Polyak K, Xia Y, Zweier J, Kinzler K, Vogelstein B. A model for p53 apoptosis. Nature. 1997; 389:300–303.
35. Li T, Chen A, Yu C.Activation of topoisomerase II-mediated excision of chromosomal DNA loops during oxidative stress. Genes Dev. 1999; 13:1553–1560.
36. Lee AS. Coordinated regulation of a set of genes by glucose and calcium ionophores in mammalian cells. 1987; Trends Biochem Sci; 12:20.27.
37. Shiu RP, Pouyssegur J, Pastan I. Glucose depletion accounts for the induction of twotrans formation-sensitive membrane proteins in Rous Sarcoma virus-transformed chick embryo fibroblasts. Proc Natl Acad Sci USA. 1997; 74:3840–3845.
38. Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001; 26:504–509.
39. Wooden SK, Lee AS. Comparison of the genomic organizations of the rat grp78 and hsc73 gene and their evolutionary implications. 1992; DNA Seq 3:41–49.
40. Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001; 26:504–509.
41. Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004; 14:20–27.
42. Kaufman RJ. 2002 Orchestrating the unfolded protein response in health and disease. J Clin Invest 110: 1389.
43. Zhang K, Kaufman RJ. Unfolding the toxicity of cholesterol. 2003; Nat Cell Bio; l 5: 769–774.
44. Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005; 74:739–743.
45. Ron D. Cell biology. Stressed cells cope with protein overload. Science. 2006; 313:52–58.
46. Alsafadi S, Tourpin S, Bessoltane N, Salome-Desnoulez S, vassal G, Andre F, Ahomadegbe JC. Nuclear localization of the caspase 3-cleaved form of p73 in anoikis. Oncotarget. 2015; 7:12331–12343. doi: 10.18632/oncotarget.6329.
47. Conley-LaComb MK, Saliganan A, Kandagatla P, Chen YQ, Cher ML, Chinni SR. PTEN loss mediated Akt activation promotes prostate tumor growth and metastasis via CXCL12/CXCR4 signaling. Mole Cancer. 2013; 12:85–91.
48. Zhu T, Chen L, Du W, Tsuji T, Chen C. Synthetic lethality induced by loss of PKC delta and mutated Ras. Genes Cancer. 2010; 1:142–51. doi: 10.1177/1947601909360989.
49. Shen L, Kim SH, Chen C. Sensitization of human pancreatic cancer cells harboring mutated K-ras to apoptosis. 2012; PloS One 7: e40435.
50. Irani K, Goldschmidt-Clemont PJ. Ras, superoxide and signal transduction. 1998; Biochem Pharmacol 55: 1339–1346.
51. Liou JS, Chen C, Chen JS, Faller DV. Oncogenic Ras mediates apoptosis in response to protein kinase C inhibition through the generation of reactive oxygen species. J Biol Chem. 2000; 275:39001–39011.
52. Chen CY, Juo P, Liou J, Yu Q, Blenis J, Faller DV. The recruitment of Fas-associated death domain/caspase-8 in Ras-induced apoptosis. Cell Growth Differ. 2001; 12:297–306.
53. Kawakami Y, Kitaura J, Yao L, McHenry RW, Kawakami Y, Newtori AC. A Ras activation pathway dependent on syk phosphorylation of protein kinase C. Proc Nat Ace Sci USA. 2003; 100:9470–9476.
54. Jost CA, Marin MC, Kaelin WG. p73 is a human p53-related protein that can induce apoptosis. Nature. 1997; 389:191–197.
55. Kulesz-Martin M. Melanocyte and keratinocyte Carcinogenesis: p53 family protein activities and intersecting mRNA expression profiles. J Investig Dermatol Symp Proc. 2005; 10:142–148.
56. Ren J, Datta R, Shioya H, Li Y. p73β is regulated by protein kinase Cδ catalytic fragment generated in the apoptotic response to DNA damage. J Biol Chem. 2002; 277:33758–33762.
57. Yang A, Kaghad M, Caput D, McKeon F. On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet. 2002; 18:90–96.