
INTRODUCTION
Prolactin Receptor (PRLR) belongs to the class I cytokine receptor superfamily. The long form of PRLR has an extracellular domain, a single transmembrane domain and intracellular cytoplasmic domain which is required for signal transduction. Prolactin (PRL) hormone binds to PRLR with high affinity and mediates its actions predominantly through JAK-2/STAT5 signaling pathway and other pathways that involve MAPK and the participation of JAK2/c-SRC family kinases/focal adhesion kinase via phosphatidylinositol 3-kinase [1–3]. PRLR is well known to play an important role in the physiology of the human breast and in the etiology, progression breast cancer [4–6]. Also, substantial clinical and etiological evidence indicates that estrogen exposure promotes the development, progression and invasion of breast cancer. Its effects are mediated via estrogen receptors through a host of estrogen responsive genes [7–9], and through signaling pathways generate through non-genomic pathways [10]. ER-independent effects resulting from direct genotoxic insult of its metabolites have been also recognized [11]. An important connection has been established between ERα and PRLR where both liganded and un-liganded ERα were found to participate in PRLR regulation [12, 13].
Our previous studies demonstrated upregulation of transcription/expression of the PRLR gene by estradiol through its preferential utilized promoter PIII promoter (hPIII/hE13), which is one of six promoters of the hPRLR gene with their corresponding alternative non-coding exons 1. Among these, in addition of the generic promoter 1/exon1 (hPIII/hE13) which is also present in rat and mouse, the other five are human specific (promoters1-5/exon1-5, hEN1-5) [14–16]. Estradiol (E2) through activation of the hPIII promoter lacking an estrogen responsive element induce increases of PRLR non-coding exon-1 hE13 mRNA transcripts, PRLR mRNA and protein. The hPIII promoter contains functional Sp1 and C/EBP sites that bind Sp1/Sp3 and C/EBPβ, respectively. Non-DNA bound ERα activated by E2 associates in a complex with Sp1 and C/EBPβ bound to their cognate elements. These in turn recruit coactivators and through epigenetic changes favor the recruitment of TFIIB, and Pol II for transcriptional induced expression of PRLR gene by E2 in MCF-7 breast cancer cells [12, 17].
In related studies we demonstrated that ERα preexist as homodimers even in absence of E2, and that a conformation change in ERα induced by E2 has an important role in its increased interaction with C/EBPβ and Sp1 [18].
Our recent studies revealed upregulation of the PRLR induced by endogenous and exogenos prolactin (PRL) through its receptor in the absence of estradiol in MCF-7 and T47D-cells where unliganded-ERα, JAK2/STAT5, mitogen-activated protein kinase (MAPK) and PI3K pathways were found to have essential roles. Phospho- ERα associates with the C/EBPβ-SP1 as complex at the PRLR hPIII promoter. JAK2- activated STAT5a and b (phospho-forms) associate as tetramer of homodimers or heterodimers with a functional GAS element in the non-coding exon 1 (hE13) corresponding to the hPIII promoter [13]. This exon was found in our previous studies to be required for the transcriptional activity of the hPIII promoter (Figure 1A, left) [15, 16]. Other recent studies have indicated a role for paracrine EGF via EGFR/ERBB1 independent of estrogen and prolactin in the transcriptional activation of PRLR gene expression. Unliganded ERα and STAT5b, which are both phosphorylated by EGF-induced activation of EGFR/ERRB1 through separate independent mechanisms, are both required in the PRLR activation of gene expression by EGF [19]. Of major relevance were our findings whereby interaction of ERα with STAT5 at the promoter was found to be required for association of phospho-ERα to the complex with consequent induction of the PRLR gene by PRL or EGF. These findings were indicative of STAT5 as stabilizing factor on unliganded ERα association to the complex at the PIII promoter.
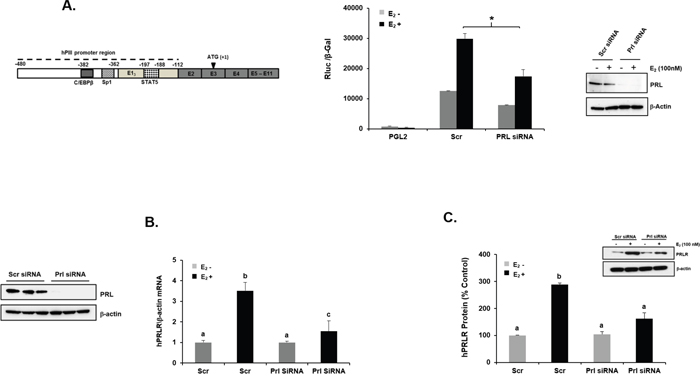
Figure 1: E2 induced upregulation of PRLR gene transcription/expression inhibited by knock-down of endogenous PRL. (A) Left- Schematic representation of PRLR gene with the generic promoter hPIII (indicated in dotted line -480/-112) which includes the non-coding exon-1 (hE13) required for promoter activity [15, 16] and also the DNA sites of relevant transcription factors within hPIII are indicated. In addition the common non-coding exon 2 and coding exons 3-11 are shown. Right- Effect of E2 (100 nM) on PRLR promoter activity in cells transfected with PGL2 construct (control) or wild type hPIII/hE13 and with/without Scrambled (Scr) or PRL siRNAs. Results presented are relative luciferase activities (Rluc) normalized to the activities of co-transfected β-galactosidase. Asterisks (*) indicate statistically significant reduction in PRL siRNA treated group induced by E2 compared to Scr siRNA treated group induced by E2 (Student t-test; P < 0.01). Relative mRNA (B) and protein expression (C) levels of PRLR normalized by endogenous β-actin upon E2 induction in MCF-7 cells transfected with either Scr or PRL siRNAs. Left- Western blot of PRL protein expression in Scr siRNA and PRL siRNA transfected samples. Different superscript letters indicate significant differences (Tukey’s multiple comparison test, P < 0.05). (C) Right- Western blot PRLR protein expression in Scr siRNA and PRL siRNA transfected samples (right). Results in this figure and below are reported as the mean ± SE of three independent experiments.
Since PRLR and EGFR are expressed in breast tumors and STAT5 was found essential for unliganded ERα dimer association with the complex at the PRLR promoter [13, 19], we envisioned commonalities in the mechanism that participate in the E2/ERα induction of PRLR gene transcription/expression to that observed for PRL and EGF in MCF-7 cells. Thus, it was of much interest to apply our current knowledge to further studies on the intrinsic mechanism of upregulation of PRLR by E2/ERα. Phosphorylation of ERα is effectively induced by E2 at S118 in the AF1 domain via cyclin dependent kinase CDK7/cyclin H complex [20] and JAK2 derived signal transduction pathways, MAPK and PI3K, induced by endogenous PRL via PRLR could also contribute to ERα phosphorylation. Moreover, since other aspects of the PRL/PRLR/JAK2/STAT5 are of relevance in transcriptional activation of PRLR we proceeded to investigate their role in the E2/ERα upregulation of the PRLR. In this study we demonstrated the requirement of the participation of PRL/PRLR in transcriptional upregulation/expression of the PRLR induced by E2/ERα in breast cancer cells.
RESULTS
PRL is required for E2 induced PRLR transcription/ expression
In previous studies we determined that endogenous PRL which is present in breast cancer cells contributes to basal levels of PRLR expression in MCF-7 and T47D cells and these were magnified by addition of exogenous PRL. Also, in early studies we demonstrated that E2/ERα induces upregulation of PRLR transcription. We initiated studies to determine whether endogenous PRL action in breast cancer cells is required for E2 induced increase in PRLR gene expression. Cells cultured in the absence of E2 transfected with PRL siRNA showed marked depletion of endogenous PRL levels compared to control cells transfected with scrambled siRNA. Control cells (Scrambled siRNA) upon E2 stimulation showed significant increases in PRLR promoter (hPIII generic) activity (Figure 1A), PRLR mRNA (Figure 1B) and protein levels (Figure 1C) by E2 over basal. In contrast, minimal or non-significant increases of these parameter induced by E2 were observed in cells where endogenous PRL was depleted by PRL siRNA, (Figure 1). These findings indicate that endogenous PRL is required for the increases in PRLR induced by E2 in breast cancer cells.
Role of STAT5 in the stimulation of PRLR by E2/ERα
Subsequent studies were directed to determine the mechanism of the PRL participation for the increase in PRLR induced by E2. In cells transfected with hPIII WT PRLR promoter E2 significantly stimulated the promoter activity while in cells transfected with hPIII with mutated STAT5 element, residing in non-coding exon-1 of the PRLR promoter, the activation by E2 was minimally present (Figure 2A). This indicated that PRL/PRLR through activation JAK2 and Stat5 is required for the E2/ERα stimulus of PRLR. Activated STAT5 induced by PRL independent of E2 associates with STAT5 DNA element to stimulate PRLR transcription [13]. In the case of the PRLR increase through E2 stimulation the findings indicate the requisite participation of PRL in STAT5 activation. Since in previous studies PRL-induced STAT5 activation in absence of E2 and ERα was found to interact with STAT5 in Re-ChIP assay [13], we initially investigated whether STAT5a or STAT5b associated with ERα. Immunoprecipitates of cells cultured in presence or absence of E2 with STAT5a and STAT5b antibodies revealed that only STAT5a interacts with ERα. Furthermore an association of phospho ERα at S118 with STAT5a was demonstrated (Figure 2B). In cultures where STAT5a or STAT5b was significantly depleted by their corresponding siRNAs (Figure 2C, right panel), E2 stimulation of PRLR mRNA expression was completely abolished in STAT5a depleted cultures while no effects were observed in the case of cells with depleted STAT5b (Figure 2C).
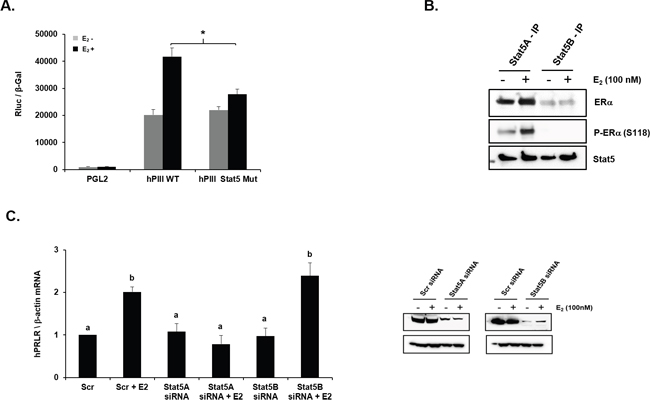
Figure 2: Role of STAT5 in E2-induced promoter activity and mRNA expression of PRLR. (A) Effect of E2 on PRLR promoter activity of cells transfected with pGL2 vector (basal) or hPIII construct (WT) or hPIII construct with mutation in STAT5 binding site. Asterisk (*) indicate the statistically significant reduction in E2-induced cells transfected with hPIII construct with mutation in STAT5 binding site when compared with E2 induced cells transfected hPIII construct (WT) (Student t-test; P < 0.01). (B) Immuno-precipitation analysis showing interaction between ERα/pERα and STAT5a in MCF-7 cells treated in presence or absence of E2. (C) Left- Expression of PRLR mRNA in cells transfected with coding region of STAT5a siRNA or STAT5b siRNA or Scr siRNA. Right- Western blot showing knock-down of STAT5a and STAT5b by siRNA. Different superscript letters indicate significant differences (Tukey’s multiple comparison test, P < 0.05).
We previously demonstrated that ERα associates in a DNA independent manner to C/EBPβ/Sp1 which in turn associate at their DNA sites at the hPIII promoter [18] and this was markedly enhanced by E2 treatment of cells with complex formation of E2/ERα.-C/EBPβ/Sp1 [12, 18] Studies on the association of these transcription factors to the hPIII promoter revealed that in cells with knock-down of endogenous PRL by specific siRNA (Left, Figure 3 Above), recruitment of ERα-induced by E2 was significantly decreased compared to controls with scrambled siRNA (Figure 3A) Also, the increased recruitment of C/EBPβ to the promoter induced by E2 in control cells was completely abolished by PRL knock-down (Figure 3B). Sp1 is constitutive associated to the hPIII promoter and E2 as in previous studies did not induce recruitment. However, it is apparent that in cells with PRL depleted Sp1 recruitment was markedly reduced and no significant differences were observed in presence or absence of E2 (Figure 3C). Recruitment of STAT5a, presumably induced by endogenous PRL/PRLR, in control cells was not influenced by E2 treatment. Consequently in cells with PRL knock-down STAT5a was significantly reduced and no differences were observed in presence or absence of estradiol (Figure 3D). The protein expression of these transcription factors were not influenced by the depletion of endogenous PRL (Figure 3E).
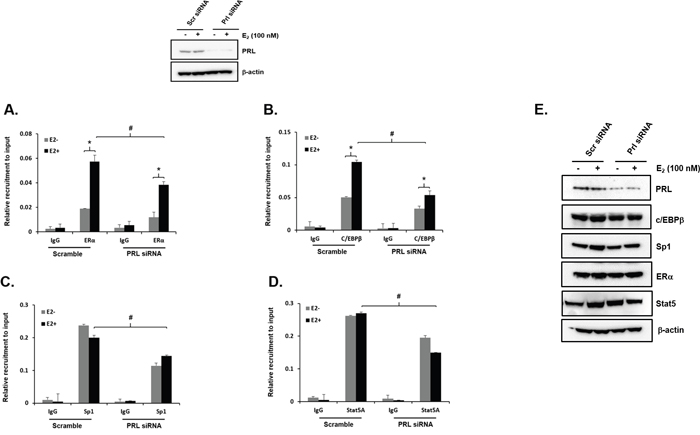
Figure 3: Effect of endogenous PRL knockdown on Recruitment of ERα, Sp1, C/EBPβ and STAT5a on to the PRLR promoter. Above- Western blot of MCF-7 Cells transfected with Scr siRNA or PRL siRNA for 48 h subsequently incubated with E2 or ethanol (control) after serum starvation for 2 days showing depletion of endogenous PRL by siRNA vs Scr siRNA for A-D shown below. ChIP assay was performed using these cells to observe the recruitment of ERα (A), C/EBPβ (B), Sp1 (C) and STAT5a (D) onto the PRLR promoter. Asterisks (*) indicate statistically significant changes between E2 untreated and treated groups (Student-t test; P < 0.001). Hash (#) indicate statistically significant changes between Scr and PRL siRNA transfected groups E (Student-t test; P < 0.05). Western blot showing depletion of endogenous PRL by siRNA versus Scr siRNA, and unchanged levels of ERα, C/EBPβ, Sp1 and STAT5a expression in PRL depleted versus control cells.
Role of CDK7 in the E2/ERα induced PRLR transcription
Subsequent studies in MCF7 cells demonstrated phosphorylation of ERα at Ser 118 in a ligand-dependent manner by CDK7 [20] inhibited by the specific covalent CDK7 inhibitor THZ1 [21], while the AG490 (JAK2 inhibitor) or MEK inhibitor had no effect (Figure 4A and 4B). Further studies demonstrated that the CDK7 inhibitor completely abrogated the stimulation of hPIII PRLR promoter activity by E2 (Figure 4C). Moreover, THZ1 treatment of MCF7 cells caused complete inhibition of E2-induced recruitment of ERα to the hPIII promoter versus basal control (Figure 4D). These findings indicate that inhibition of E2-induced ERα (S118) phosphorylation by the CDK7 inhibitor contributed to the near complete inhibition of E2-induced PRLR promoter activation.
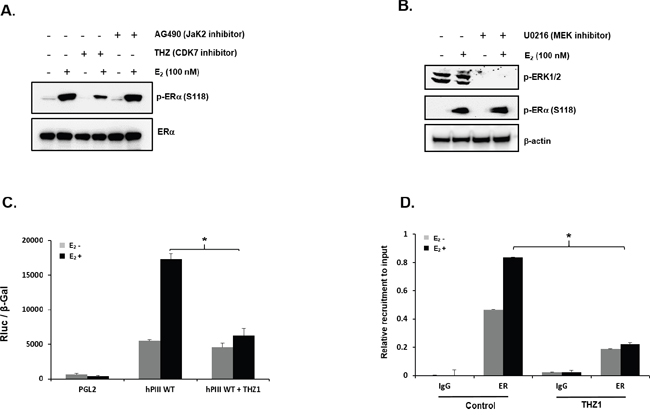
Figure 4: Role of CDK-7 in E2 induced promoter activity. (A) Representative Western blot showing the phosphorylation status of ERα at serine 118 (S118) in MCF-7 cells in controls and after treatment with the CDK7 inhibitor, THZ1 (100 nM), or AG-490 (100 μM) for 3 h prior to E2 treatment for 30 min. Endogenous ERα and β-actin are used as loading controls. (B) Western blot showing effect of U0126 (10 μM) on phosphorylation status of ERα at S118 and ERK1/2 in cells cultured in presence or absence of E2 for 30 min following pre-incubation with or without inhibitor. β-actin is used as loading control. (C) Effect of CDK-7 inhibitor on E2 induced upregulation of hPIII transcriptional activity. MCF-7 cells transfected with PGL2 vector (control) or with hPIII promoter construct were pre-incubated with or without THZ1 (100 nM) inhibitor for 3 h prior to addition of E2 and further incubated for 18 h. Results presented are relative luciferase activities (RLuc) normalized to the activities of co-transfected β-galactosidase. Asterisks (*) indicate statistically significant changes between E2 untreated and treated groups (Student t-test; P < 0.001). (D) Effect of THZ1 (100 nM) treatment of cells as in (C) on E2-induced recruitment of ERα to the complex at the promoter assessed by ChIP assay.
Role of endogenous PRL and CDK7 in E2 induced cell migration
To assess whether endogenous PRL and CDK7 have a role in E2-induced cell migration we performed wound healing/scratch assays in MCF-7 cells exposed to THZ1 inhibitor (Figure 5A) or PRL siRNA (Figure 6A), before E2 treatment in serum starved conditions. In untransfected (control) or scrambled (Scr) siRNA transfected cells, percentage wound gap was significantly narrower under E2 treatment than untreated controls. THZ1 treatments (Figure 5A) and knockdown of endogenous PRL by PRL siRNA (Figure 6A) resulted in increased wound gap in the cells with or without E2 induction. Similarly, the inhibitory role of the CDK7 inhibitor on E2-induced cell migration was demonstrated by Transwell assays (Figure 5B). Also, significant reduction of E2 induced cell migration was observed when endogenous PRL was knock-down by siRNA (Figure 6B). These results indicate that both endogenous PRL and CDK7 are involved in E2-induced cell migration in MCF-7 cells. The findings derived from migration studies are of much relevance since these resemble the essential role of PRL and CDK7 in PRLR upregulation induced by E2/ERα (Figures 1-5), and PRLR is require to mediate the actions of PRL.
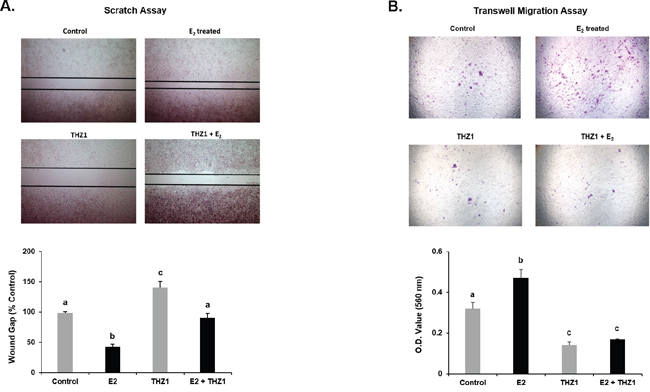
Figure 5: Effect of CDK-7 treatment on E2 induced MCF-7 cell migration by Scratch (A) and Trans-well (B) assays. (A) MCF-7 cells pre-incubated with THZ1 (100 nM) for 3 h were subjected to scratch/wound healing assay to assess the E2 induced cell migration. After 48 h photographs were taken and wound width was calculated using ImageJ software. Representative images showing the wound width in control and treated groups. Graph showing the wound gap (percentage of control) in different groups. Different superscript letters indicate statistical significance between experimental groups (Tukey’s multiple comparison test, P < 0.05). (B) Effect of THZ1 (100 nM) on migration induced by E2 assessed by Transwell assay following pre-treatment of MCF7 cells with THZ1 for three hours (see methods). Below Relative absorbance of lyzed cells detected on migrated membranes. Different superscript letters indicate statistical significance between experimental groups (Tukey’s multiple comparison test, P < 0.05). Data represents the Mean ± SE from three independent experiments.
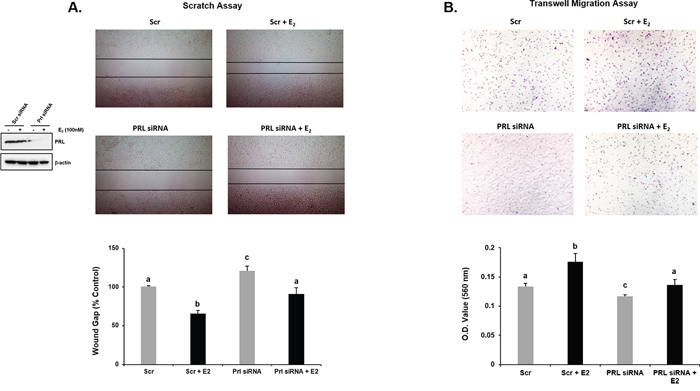
Figure 6: Effect of PRL knockdown on E2 induced MCF-7 cell migration by Scratch (A) and Trans-well (B) assays. (A) Left siRNA and control treated with Scr siRNA were assessed for E2- induced cell migration. MCF-7 cells transfected with Scr or PRL siRNA were subjected to scratch/wound healing assay to assess the E2 induced cell migration. After 48 h photographs were taken and wound width was calculated using ImageJ software. Representative images showing the wound width in control and treated groups. Graph showing the wound gap (percentage of control). Different superscript letters indicate statistical significance between experimental groups (Tukey’s multiple comparison test, P < 0.05). (B) Effect of PRL siRNA knock-down on migration induced by E2 assessed by Transwell assay. Below Relative absorbance of corresponding to lyzed cells present on migrate membranes. Different superscript letters indicate statistical significance between experimental groups (Tukey’s multiple comparison test, P < 0.05).
DISCUSSION
In this study we have analyzed the intrinsic mechanism of the PRLR upregulation induced by Estradiol via its ERα receptor previously recognized in our laboratory [12, 18]. We were prompted by our subsequent findings on the modalities of upregulation of the PRLR receptor by un-liganded ERα [13], and its independent regulation by Epidermal Growth through its receptor EGF/ERBB1 [19]. The finding of a functional GAS site in non-coding exon-1 [13] which is contained in the hPIII of the PRLR participating in both aspects of regulation provided fertile ground to expand our knowledge on the mechanism of E2/ERα.
In this study we demonstrate the essential role of endogenous PRL in the upregulation PRLR induced by E2/ERα with a requisite participation of STAT5a induced by PRL via PRLR. Phosphorylated STAT5a which associates with its functional element at hPIII interacts with non-DNA bound E2/ERα which in turn associates in a complex to Sp1 and C/EBPβ bound to their cognate DNA sites at the PRLR hPIII promoter. E2 favors ERα phosphorylation at S118 by CDK7 kinase, (Figure 4A) and greatly increase the recruitment of E2/ERα to the PRLR promoter over basal unliganded ERα and its association with pSTAT5a (Figure 2B). Phosphorylation of ERα at S118 is necessary for its association to the complex and its interaction with STAT5a. Inhibition by the specific CDK7 inhibitor, THZ1 markedly reduced the E2-induced ERα phosphorylation at S118, while the JAK2 inhibitor AG490 or MEK inhibitor U0219 which inhibits downstream JAK2 induced pathways [13] known to phosphorylate unliganded ERα at S118 and S167 had no effect (Figure 4A and 4B). THZ1 effectively abolished E2 induced ERα recruitment to the PRLR hPIII promoter (Figure 4D) and consequently caused a marked reduction of E2-induced PRLR transcription to control levels (Figure 4C). This clearly indicated the role of pERα (S118) and the essential requirement of CDK7 in the transcriptional upregulation of the PRLR receptor induced by E2 (Figure 4C).
Endogenous prolactin is essential for activation of pSTAT5a which its presence at the DNA site is required for the recruitment and stability of the complex. This has been established in ChIP studies where PRL was knock-down by siRNA and recruitment of STAT5a was effectively reduced at its site in the promoter. In this instance a major reduction was observed on ERα and C/EBPβ induced by E2. In the absence of E2 significant reductions on ERα recruitment to hPIII (basal) (Figure 3A) was also observed in this study as in our previous studies on the unliganded ERα [13] with PRL knock-down, which are comparable to the basal values in this study (Figure 3A). This led us to propose a role for endogenous PRL/PRLR/STAT5 and of STAT5a interaction in the stability of the complex. This proposal has been demonstrated in this study where E2 recruitment of ERα to the complex was significantly reduced with PRL knock-down. In addition, basal and E2 stimulated C/EBPβ were similarly reduced (Figure 3A-3C). The efficiency of C/EBPβ recruitment to its DNA site was greatly enhanced when ERα/Sp1 complexes were preformed and DNA-bound Sp1 was the preferred interacting partner of ERα [18]. However, the reduction of Sp1 recruitment was not expected since it appeared to be associated to the complex constitutively and not shown to be induced by E2 in previous [12, 18] and this study Figure 3C. The absence of stabilization of the complex by reduction of STAT5a/ERα interaction could reduce the recruitment C/EBPβ and Sp1. In this regard studies where C/EBPβ was depleted with siRNA in cells treated in presence or absence of E2 a marked reduction of Sp1 association to its DNA site was observed when compared to control. Thus, the association of ERα to Sp1 seems to be important for recruitment of C/EBPβ to its site and in turn C/EBPβ and -Sp1 associations secure the stable recruitment of Sp1 and the overall complex formation (Figure 7).
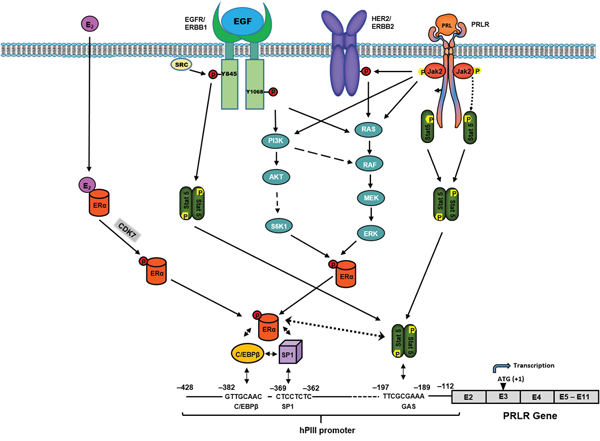
Figure 7: Mechanisms of the upregulation of hPRLR induced by E2/ERα revealed in this study and others defined in our previous studies [13, 19] Requirement of association of pERα (S118) induced by E2 via phosphorylation by CDK7 (non-DNA bound) and complex formation with C/EBP/Sp1 bound to their respective DNA elements at the hPIII promoter. PRL/PRLR is essential for transcriptional activation of PRLR through activation of STAT5 which associates its site at the hPIII promoter and stabilizes the complex by association with ERα, a requirement for recruitment of coactivators, TFIIB and Pol II [12]. Inhibition of pERα (S118) by the CDK7 inhibitor causes abolition complex formation and of transcription/expression. The use of this inhibitor could provide an alternative efficacious adjuvant therapy to prevent inductive and progression effects of E2/ERα in breast cancer. Also, presented schematically are other previously described modalities on the up-regulation of PRLR via PRL/PRLR/JAK2/STAT5 per se, or through HER2 [13] transactivation by JAK2. In addition, are shown EGF functional effects via its cognate receptor ERBB1, independent of PRL/PRLR and E2 [19], on the activation of intrinsic receptor tyrosine kinase and signal transduction pathways participating in STAT5 and ERα phosphorylation/activation, respectively, -essential for PRLR gene transcription through hPIII promoter.
The mechanism of upregulation of PRLR by E2/ERα defined in this study displays commonalties and specific differences with those described for unliganded ERα and EGF/EGFR [13, 19]. Although mechanistically all require STAT5 association with hPIII the specific STAT5 class participation which associates to hPIII reveal differences, STAT5a and b or specifically STAT5a, in both cases where involvement of prolactin was required and STAT5b in the case of EGF/EGFR participation. This may be related in part to specific requirements for their phosphorylation which is a requisite for their recruitment to its site at hPIII and/or presumably for their interaction with ERα. Also, differences in modality of phosphorylation of ERα have been indicated above and summarized in Table 1.
Table 1: Comparative summary of mechanisms of upregulation of PRLR induced by E2, PRL and EGF through liganded and un-liganded pERα and STAT5
Phosphorylation |
E2 |
PRL* |
EGF** |
|---|---|---|---|
ERα |
Cdk 7 (S118) |
1) PRLR/JAK2/PI3K/MEK/ERK (S118; S167) |
1) ERBB1 1068,1086 RAS/RAF/MEK/ERK (S118) |
STAT5 |
PRL/PRLR/JAK2 |
PRL/PRLR/JAK2 |
EGF/ERBB1Y845 c-SRC |
*independent of E2; ** independent of E2 and PRL
ERα Serine (S) (phospho-position)
Recent studies using kinase inhibitors and gene editing demonstrated that triple-negative breast cancer cells are transcriptional dependent on CDK7. A cluster of genes in these cells were found to be exquisitely sensitive to the CDK7 inhibitor THZ1 [22]. In this regard, it is of interest that the inhibition of CDK7 which participates in the phosphorylation of ERα at S118 require for E2 induced upregulation of PRLR, also effectively inhibited the well-known induction of cell migration induced by E2 (Figures 5 and 6). Prolactin as E2 through similar mechanism has been reported to promote cell migration [23–25]. This was further established in this study by knock-down of endogenous PRL which resulted in inhibition of E2-induced cell migration. This indicated a dependence on PRL/PRLR on E2-induced cell migration and their mutual link to CDK7 participation (Figure 6). Targeting CDK7 kinase, which is known to regulate both transcription and the cell cycle and ERα phosphorylation with the THZ1 inhibitor was found to effectively inhibit the transcription of the PRLR and its contribution to cell migration induced E2/ERα in breast cancer cells. THZ1 treatment could provide an additional avenue singly or in combination with other inhibitory approaches targeting receptor function (PRLR [26], ERα [13]), HER2 [27], ERBB1 [19]) and/or signaling pathways (MAPK, PI3K, c-SRC [13, 19], ERBB1Tyrosine kinase, [19]) to effectively ablate transcription of PRLR and its contribution to breast cancer.
MATERIALS AND METHODS
Reagents and antibodies
Phenol-red free RPMI 1640 media and RPMI 1640-GlutaMAX were purchased from ThermoFisher Scientific. Charcoal/Dextran treated Fetal Bovine Serum (FBS) was obtained from Atlanta Biologicals. STAT5a, STAT5b, STAT5, ERα, β-actin antibodies and AG-490 (JAK2 inhibitor) were obtained from Santa Cruz Biotechnology. Phospho (p)-ERK1/2, and U1026 (MEK1/2 inhibitor) were from Cell Signaling and the CDK7 inhibitor, THZ1 hydrochloride from Med Chem Express. pERα (Ser118) antibody was obtained from EMD Millipore. PRLR antibody were obtained from Sigma-Aldrich. Human PRL antibody were obtained from National Hormone and Peptide Program, Harbor-UCLA Med. Ctr., Torrance, CA 90502.
Cell culture and reporter gene assay
The MCF-7A2 (MCF-7) ER positive breast cancer cells (gift from E. Berleth, C. Roswell Park Cancer Institute, New York, NY) were maintained in RPMI 1640-GlutaMAX supplemented with 10% charcoal stripped FBS at 37°C in CO2 incubator. Cells were cultured in 24 well plates using phenol red-free RPMI media with 5% and 1% charcoal-treated FBS for 2 days at each serum concentration. The PRLR generic promoter (hPIII promoter/non-coding exon 1 [hE13]) with reporter pGL2 gene construct (bp −480/−112) containing C/EBPβ, Sp1 sites in hPIII and putative STAT5 binding site in hE13, and construct with the STAT5 site mutated, and pGL2 empty vector were used for transient transfection in MCF-7 cells using Lipofectamine 3000 reagent (ThermoFisher Scientific) as described previously [18]. Cells were treated with E2 (100 nM) in RPMI phenol red-free media for 18 h. To study the effect of CDK-7 inhibitor (THZ1) on the PRLR generic promoter activity, cells pre-incubated with THZ1 for 3 h prior treatment with 100 nM E2 and incubation for 24 h. Cells were harvested to the determine PRLR promoter activity using Brigh-Glo luciferase assay system (Promega). The E2 100 nM dose used in the present studies was derived from our previous work demonstrating that addition of E2 (1-100 nM) to the cultures caused dose dependent increases in PRLR promoter activity, mRNA and protein [12].
Cell-migration assays
Wound healing/scratch assay
MCF-7 cells (5 x 104 cells/well) were cultured in 6 well plates with phenol red free RPMI media containing 5% charcoal stripped FBS at 37°C in CO2 incubator. Then next day cells were transfected with 20 nM scrambled and PRL silencer select siRNAs using siPORTNeoFX reagent (Life Technologies) and grown in 1% charcoal stripped FBS till cells reach 90% confluent. Then cell monolayers were wounded by scratching with sterile 100 μL micropipette tips. Fresh medium containing 100 nM E2 was added every 12 h. After 48 h cells were photographed under phase contrast inverted microscope and cell migration was assessed by measuring migration distance (wound gap) using ImageJ Software from National Institutes of Health. To study the effect of THZ1 inhibitor on cell migration, cells were pre-incubated with the inhibitor for 3 h prior treatment with E2.
Transwell assay
MCF-7 cells were cultured and transfected as indicated above and incubated in presence or absence of THZ1 inhibitor for 3 h. Transwell assay was performed using 6.5 mm inserts with 8 μm pore size polycarbonate membrane inserts (Cell Biolabs, Inc). Subsequently, 0.5 X 106 cells in 300 μl of RPMI phenol red free were seeded on the top chamber and 500 μl RPMI containing 10% charcoal-treated FBS with or without E2 (100 nM) was applied to the lower chamber. Migration was assessed after incubation of culture plates at 37°C in a 5% CO2 incubator for 24-48 h. After removal of non-migrated cells from the upper chamber, membranes were fixed in methanol for 5 min. The cells that are migrated to the lower side of the membrane were stained with cell stain solution (crystal violet) provided in the kit for 5 min. The migrated cells were by visualized under a light microscopy and photograph at 5X magnification. Also, migrated cells were lyzed using 200 μl of extraction solution and the absorbance at 560 nm was recorded for indirect estimation of the number of migrated cells [28].
Western blot analysis
Whole cell lysates from MCF-7 cells cultured in serum starved conditions in presence and absence of E2 (100 nM) or treated with U0126 (10 μM) or THZ1 (100 nM) inhibitors were extracted using RIPA lysis buffer (Thermo Scientific, Rockford, IL) in presence of 1x protease and Phosphatase inhibitor cocktail (Thermo Scientific). The protein samples were run on NuPAGE 4 -12% Bis-Tris gradient gels and transferred to nitrocellulose membranes using iBlot 2 (ThermoFisher Scientific). Membranes were blocked with 5% skimmed milk powder in phosphate buffer saline and incubated with different primary antibodies or β-actin (loading control) and washed and incubated with secondary antibody conjugated with HRP (Pierce). Immunodetection was performed using super-signal chemiluminescence system (Pierce).
siRNA analysis
Silencer Select pre-designed validated siRNAs from Ambion (ThermoFisher Scientific) were used to knock-down the endogenous expression of PRL, STAT5a, STAT5b in MCF-7 cells. Scrambled siRNA was used as negative control. The MCF-7 cells were transfected with 20 nM siRNA using siPORTNeoFX or Lipofectamine RNAiMAX reagent (ThermoFisher Scientific) as described previously [18]. After 48 h of siRNA transfection cells were grown in 5% charcoal treated FBS for 1 day and 1% for additional 2 days. The cells were harvested for Chip assays and Western blots after treatment with E2 (100 nM) in serum free medium for 18 h. All the Silencer Select siRNA sequences used for RNAi interference are shown in Table 2.
Table 2: List of validated siRNAs used for gene knockdown and their sequence information
Gene |
Sequence (Sense) |
|---|---|
PRL |
GCGAAUUCGAUAAACGGUATT |
STAT5a |
AUGGAUAUGUGAAACCACATT |
STAT5b |
CACCCGCAAUGAUUACAGUTT |
Scrambled siRNA |
AATTCTCCGAACGTGTCACGT |
Chromatin immunoprecipitation (ChIP) assay
MAGnify™ Chromatin Immunoprecipitation system from Invitrogen was used to perform ChIP assays according to the manufacturer's protocol as described previously [18]. The relative binding of Sp1, C/EBPβ and STAT5 proteins to their respective putative DNA binding sites on PRLR promoter was quantitatively evaluated by qRT-PCR of the precipitated DNA and input DNA using SYBER Green FAST Master Mix in an ABI 7500 Fast Real-Time PCR system. The primers used for amplification of the PRLR promoter sequence that spans the STAT5 site and Sp1 and C/EBPβ sites are 5’GCATGCTGAAGAAAATCACTGTTTTGCC3’ (forward) and 5’ TGCACGAGGACATGAAGCTCCA 3’ (reverse).
Statistical analysis
The significance of the differences among groups were determined by multiple Tukey’s multiple-comparison test (one-way ANOVA) and significance of the differences between E2 treated versus control were determined by Student’s t-test using the Prism software program (GraphPad Software, Inc, San Diego, California)
ACKNOWLEDGMENTS
This work was supported by the NIH Intramural Research Program through the Eunice Kennedy Shriver National Institute for Child Health and Human Development.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Finidori J, Kelly PA. Cytokine receptor signaling through two novel families of transducer molecules: Janus kinases, and signal transducers and activators of transcription. J Endocrinol. 1995; 147:11-23.
2. Qazi AM, Tsai-Morris CH, Dufau ML. Ligand-independent homo- and heterodimerization of human prolactin receptor variants: inhibitory action of the short forms by heterodimerization. Mol Endocrinol. 2006; 20:1912-1923.
3. Aksamitiene E, Achanta S, Kolch W, Kholodenko BN, Hoek JB, Kiyatkin A. Prolactin-stimulated activation of ERK1/2 mitogen-activated protein kinases is controlled by PI3-kinase/Rac/PAK signaling pathway in breast cancer cells. Cell Signal. 2011; 23:1794-1805.
4. Touraine P, Martini JF, Zafrani B, Durand JC, Labaille F, Malet C, Nicolas A, Trivin C, Postel-Vinay MC, Kuttenn F. Increased expression of prolactin receptor gene assessed by quantitative polymerase chain reaction in human breast tumors versus normal breast tissues. J Clin Endocrinol Metab. 1998; 83:667-674.
5. Meng J, Tsai-Morris CH, Dufau ML. Human prolactin receptor variants in breast cancer: low ratio of short forms to the long-form human prolactin receptor associated with mammary carcinoma. Cancer Res. 2004; 64:5677-5682.
6. Wennbo H, Tornell J. The role of prolactin and growth hormone in breast cancer. Oncogene. 2000; 19:1072-1076.
7. Howell A. Clinical evidence for the involvement of oestrogen in the development and progression of breast cancer. Proceedings of the Royal Society of Edinburgh. Section B. Biological Sciences. 1989; 95:49-57.
8. Spink BC, Bennett JA, Pentecost BT, Lostritto N, Englert NA, Benn GK, Goodenough AK, Turesky RJ, Spink DC. Long-term estrogen exposure promotes carcinogen bioactivation, induces persistent changes in gene expression, and enhances the tumorigenicity of MCF-7 human breast cancer cells. Toxicol Appl Pharmacol. 2009; 240:355-366.
9. Saha Roy S, Vadlamudi RK. Role of estrogen receptor signaling in breast cancer metastasis. Int J Breast Cancer. 2012:654698.
10. Giretti MS, Fu XD, De Rosa G, Sarotto I, Baldacci C, Garibaldi S, Mannella P, Biglia N, Sismondi P, Genazzani AR, Simoncini T. Extra-nuclear signalling of estrogen receptor to breast cancer cytoskeletal remodelling, migration and invasion. PLoS One. 2008; 21;3:e2238.
11. Yue W, Yager JD, Wang JP, Jupe ER, Santen RJ. Estrogen receptor-dependent and independent mechanisms of breast cancer carcinogenesis. Steroids. 2013; 78:161-70.
12. Dong J, Tsai-Morris CH, Dufau ML. A novel estradiol/estrogen receptor alpha-dependent transcriptional mechanism controls expression of the human prolactin receptor. J Biol Chem. 2006; 281:18825-18836.
13. Kavarthapu R, Tsai Morris CH, Dufau ML. Prolactin induces upregulation of its cognate receptor in breast cancer cells via transcriptional activation of its generic promoter by cross-talk between ERα and STAT5. Oncotarget. 2014; 5:9079-9091. doi: 10.18632/oncotarget.2376.
14. Hu ZZ, Zhuang L, Meng J, Dufau ML. Transcriptional regulation of the generic promoter III of the rat prolactin receptor gene by C/EBPbeta and Sp1. J Biol Chem. 1998; 273:26225-35.
15. Hu ZZ, Zhuang L, Meng J, Leondires M, Dufau ML. The human prolactin receptor gene structure and alternative promoter utilization: the generic promoter hPIII and a novel human promoter hP(N). J Clin Endocrinol Metab. 1999; 84:1153-6.
16. Hu ZZ, Zhuang L, Meng J, Tsai-Morris CH, Dufau ML. Complex 5' genomic structure of the human prolactin receptor: multiple alternative exons 1 and promoter utilization. Endocrinology. 2002; 143:2139-42.
17. Leondires MP, Hu ZZ, Dong J, Tsai-Morris CH, Dufau ML. Estradiol stimulates expression of two human prolactin receptor isoforms with alternative exons-1 in T47D breast cancer cells. J Steroid Biochem Mol Biol. 2002; 82:263-8.
18. Kang JH, Tsai-Morris CH, Dufau ML. Complex formation and interactions between transcription factors essential for human prolactin receptor gene transcription. Mol Cell Biol. 2011; 31:3208-3222.
19. Kavarthapu R, Dufau ML. Role of EGF/ERBB1 in the transcriptional regulation of the prolactin receptor independent of estrogen and prolactin in breast cancer cells. Oncotarget. 2016; 7:65602-65613. doi: 10.18632/oncotarget.11579.
20. Chen D, Washbrook E, Sarwar N, Bates GJ, Pace PE, Thirunuvakkarasu V, Taylor J, Epstein RJ, Fuller-Pace FV, Egly JM, Coombes RC, Ali S. Phosphorylation of human estrogen receptor alpha at serine 118 by two distinct signal transduction pathways revealed by phosphorylation-specific antisera. Oncogene. 2002; 21:4921-4931.
21. Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, Jenkins CE, Hannett NM, McMillin D, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014; 511:616-620.
22. Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, Yuzugullu H, Von T, Li H, Lin Z, Stover DG, Lim E, Wang ZC, et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell. 2015; 163:174-186.
23. Ho JY, Chang FW, Huang FS, Liu JM, Liu YP, Chen SP, Liu YL, Cheng KC, Yu CP, Hsu RJ. Estrogen Enhances the Cell Viability and Motility of Breast Cancer Cells through the ERα-ΔNp63-Integrin β4 Signaling Pathway. PLoS One. 2016;11:e0148301.
24. Sun Y, Wang Y, Fan C, Gao P, Wang X, Wei G, Wei J. Estrogen promotes stemness and invasiveness of ER-positive breast cancer cells through Gli1 activation. Mol Cancer. 2014; 13:137. doi: 10.1186/1476-4598-13-137.
25. da Silva PL, do Amaral VC, Gabrielli V, Montt Guevara MM, Mannella P, Baracat EC, Soares-Jr JM, Simoncini T. Prolactin Promotes Breast Cancer Cell Migration through Actin Cytoskeleton Remodelling. Front Endocrinol (Lausanne). 2015; 6:186. doi: 10.3389/fendo.2015.00186.
26. Damiano JS, Wasserman E. Molecular pathways: blockade of the PRLR signaling pathway as a novel antihormonal approach for the treatment of breast and prostate cancer. Clin Cancer Res. 2013; 19:1644-50.
27. Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2011; 9:16-32.
28. Kramer N, Walzl A, Unger C, Rosner M, Krupitza G, Hengstschläger M, Dolznig H. In vitro cell migration and invasion assays. Mutat Res. 2013; 752:10-24.