
Introduction
The introduction of targeted therapy has been accompanied by an intensive search for biomarkers to select patients for treatment. The identification of companion diagnostics may improve treatment outcomes and cost-effectiveness of increasingly expensive oncolytic drugs. Several powerful biomarker-drug combinations have been introduced in the clinic, such as crizotinib in ALK mutant lung cancer and vemurafenib in BRAF V600E mutant melanoma [1, 2]. For these treatments, evident tumor regression can be observed in selected populations. However, for drugs that induce disease stabilization, such as everolimus, it is more difficult to determine treatment benefit. Everolimus is an orally administered drug with proven efficacy in advanced clear cell renal cell carcinoma, neuroendocrine tumors and breast cancer [3-6]. Everolimus inhibits the mammalian Target of Rapamycin (mTOR) pathway and its downstream substrates, S6K and 4EBP1, which promote cell growth, proliferation and survival [7]. mTOR can be activated by upstream pathways such as the MAPK pathway and the AKT/PI3K pathway [7]. Previous studies have described several genetic aberrations that might be predictive for response to mTOR inhibition such as mutations in or loss of PIK3CA, PTEN, TSC and KRAS [8-22].
These genetic aberrations in the mTOR pathway and its interconnected pathways are present across many different tumor types (COSMIC database). It is therefore reasonable to believe that patients with other tumor types harboring the proper molecular profile might also benefit from treatment [21, 23]. Unfortunately, despite extensive knowledge on the mechanism of action of everolimus, no tissue broad biomarkers have yet been identified and clinically validated.
To address this issue, we revised genomic profiles and drug sensitivity of 835 cell lines to generate hypothesis about potential biomarkers, and conducted a prospective biomarker identification study for everolimus. This study was conducted by the Center for Personalized Cancer Treatment (www.cpct.nl), a large consortium of hospitals in the Netherlands devoted to personalized medicine.
Results
Exploration of cell line data
The GDSC1000 cell line data from the Sanger Institute was used to search for potential markers for treatment sensitivity. As everolimus was not screened, we used the rapamycin analog temsirolimus as a proxy. IC50 values for temsirolimus were available for 835 cell lines, and sensitivity differed significantly between tumor types (p < 0.001; ANOVA). Specifically, non-small cell lung cancer (NSCLC), neuroblastoma, pancreatic and colon tumor cell lines were in general more resistant than e.g. kidney and bladder tumors (Supplementary Figure 1). After selecting only solid tumors and correcting for tissue of origin, the elastic net analysis identified a small number of genetic aberrations that could be associated with response: PTEN mutations, FGFR2 mutations and CDKN2A loss were associated with increased sensitivity (Table 1). Gains in CCNE1 and ERCC5, as well as mutations in RB1, HGF, SOX9 and CIC were associated with temsirolimus resistance (Table 1). The strongest effect was seen for CCNE1 gain and FGFR2 mutations. Gain of CCNE1 was observed in 48 cell lines including breast (10/42) and NSCLC (11/100). Only within breast cancer cell lines, was CCNE1 gain alone also associated with temsirolimus resistance (P = 0.010; one-tailed t-test). FGFR2 mutations were only observed in eight cell lines, distributed over seven tumor types.
Next, we focused on genes in the mTOR pathway or genes previously reported in association with sensitivity to mTOR inhibition. In our model including all tumor types, both PIK3CA and PTEN mutations were associated with increased sensitivity (P = 0.041 and P = 0.016, respectively; Wald test) (Table 1). PIK3CA mutations were most common in breast, NSCLC, ovarian, stomach, colorectal and aerodigestive tract tumor cell lines. PTEN mutations were also frequently observed in endometrial tumors. Within those subtypes, PIK3CA mutations were only associated with temsirolimus sensitivity in tumor cells from the aerodigestive tract (P = 0.014; one-tailed t-test) and cervical tumor cell lines (P = 0.023), but no such association could be seen for PIK3CA mutation status in breast cancer (P = 0.411), and an opposite effect was observed in endometrial cancer (P = 0.053). PTEN mutations were significantly associated with response in ovarian (P = 0.0211) and endometrial tumor cells (P = 0.031), but not in breast cancer or colorectal cancer cell lines (P = 0.278 and P = 0.423, respectively).
Table 1: Cell line data
genetic aberration |
Estimate |
Std Error |
t-value |
Pr(>|t|) |
sign level |
FGFR2_mut |
-2,014 |
0,505 |
-3,989 |
0,000 |
*** |
CCNE1 gain |
0,897 |
0,242 |
3,714 |
0,000 |
*** |
PTEN_mut |
-0,536 |
0,192 |
-2,788 |
0,005 |
** |
CDKN2A loss |
-0,307 |
0,116 |
-2,645 |
0,008 |
** |
RB1_mut |
0,486 |
0,198 |
2,447 |
0,015 |
* |
CIC_mut |
1,324 |
0,550 |
2,408 |
0,016 |
* |
gain_cnaPANCAN384_,ERCC5,ING1,IRS2,TFDP1, |
0,523 |
0,223 |
2,346 |
0,019 |
* |
SRGAP3 loss |
0,328 |
0,141 |
2,334 |
0,020 |
* |
loss_cnaPANCAN216 |
0,724 |
0,313 |
2,313 |
0,021 |
* |
HGF_mut |
1,408 |
0,700 |
2,011 |
0,045 |
* |
SOX9_mut |
1,048 |
0,544 |
1,927 |
0,054 |
. |
genetic aberration |
Estimate |
Std Error |
t-value |
Pr(>|t|) |
sign level |
PTEN_mut |
-0,457 |
0,189 |
-2,422 |
0,016 |
* |
PIK3CA_mut |
-0,384 |
0,188 |
-2,044 |
0,041 |
* |
gain_cnaPANCAN164_,KRAS, |
0,285 |
0,171 |
1,660 |
0,097 |
. |
gain_cnaPANCAN395_,AKT1,HSP90AA1,PPP2R5C, |
0,411 |
0,316 |
1,298 |
0,195 |
|
loss_cnaPANCAN44_,BMPR1A,FAS,PTEN, |
-0,208 |
0,183 |
-1,137 |
0,256 |
|
KRAS_mut |
-0,193 |
0,195 |
-0,985 |
0,325 |
|
TSC1_mut |
0,307 |
0,490 |
0,627 |
0,531 |
|
gain_cnaPANCAN129_,MET, |
0,092 |
0,195 |
0,470 |
0,639 |
|
EGFR_mut |
-0,037 |
0,340 |
-0,110 |
0,912 |
|
gain_cnaPANCAN301_,CDK12,ERBB2,MED24, |
-0,018 |
0,246 |
-0,071 |
0,943 |
|
gain_cnaPANCAN124_,EGFR, |
0,010 |
0,180 |
0,055 |
0,956 |
|
Legend: This table illustrates genetic aberrations that could potentially predict sensitivity to mTOR inhibition, based on an in vitro drug screen with temsirolimus. Part A of the table demonstrates the relation between genetic aberrations and sensitivity to temsirolimus, corrected for tumor type and excluding blood cell tumors. Part B shows similar data, but is analyzed per gene and specifically directed at genetic aberrations previously associated with sensitivity to mTOR inhibition.
Patient baseline data
A total of 73 patients were included in the study. Seventy-one (97%) patients underwent a tumor biopsy according to protocol. Fifty-nine patients (81%) started treatment with everolimus, of whom 43 (59%) were evaluable for efficacy according to TTP ratio and 51 (70%) for efficacy according to RECIST. Tumor material sufficient for sequencing analysis was obtained in 37 patients (51%) of the TTP cohort and 43 patients (59%) of the RECIST cohort (Figure 1). Nine biopsies were insufficient for sequencing due to a low or absent tumor percentage. Six biopsies were insufficient due to a low DNA yield. We obtained sequence data of 1,977 genes for 38 patients, and whole exome sequencing data for five patients. The sequencing data reached an average coverage of 159x. All 43 patients were also sequenced on the IonTorrent (panel of 50 genes and custom-made primers for mTOR pathway related genes) to validate the mutations. Baseline characteristics of the patients are described in Table 2.
Table 2: Baseline characteristics
Demographic or Clinical Characteristic |
No. of patients |
% |
No. of patients |
43 |
|
Sex Male |
26 |
60.5 |
Age, years Mean Range |
60 31 – 79 |
|
WHO PS 0 1 2 Missing |
14 26 1 2 |
32.6 60.5 2.3 4.7 |
Primary tumor Colorectal NET Esophageal Breast NSCLC Ovarian Renal cell Sarcoma Cervical Head and Neck Bladder Mesothelioma Thyroid Thymoma Gastric Pancreatic Melanoma Unknown origin |
12 7 4 3 2 2 2 1 1 1 1 1 1 1 1 1 1 1 |
27.9 16.3 9.3 7.0 4.7 4.7 4.7 2.3 2.3 2.3 2.3 2.3 2.3 2.3 2.3 2.3 2.3 2.3 |
No. of previous treatments 1 2 3 >3 |
9 5 4 25 |
20.9 11.6 9.3 58.2 |
Biopsy characteristics Tumor percentage - Median - SD DNA yield (ng) - Median - SD |
60 23 1440 2123 |
|
Legend: This table contains the baseline characteristics of all patients of whom both molecular and clinical response data was available. DNA yield is depicted in nanogram.
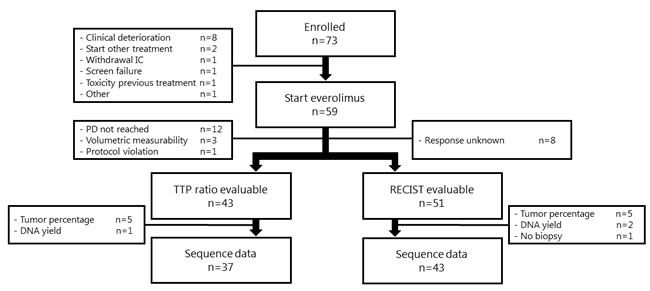
Figure 1 : Evaluability of patients. This figure illustrates the evaluability of patients for the biomarker analyses. A single patient can be evaluable according to both RECIST and TTP ratio. Abbreviations: IC, Informed Consent; PD, Progressive Disease.
Tumor mutation and copy number data
We detected on average sixteen somatic mutations per patient in the 1,977 gene set (range zero to eighty-six). The most frequently mutated genes included TP53 (N = 24), APC (N = 9), KRAS (N = 9) and PIK3CA (N = 7). In addition, we detected copy number gains and amplification of several well-known oncogenes such as ERBB2 (N = 6), PIK3CA (N = 4), CCND1 (N = 3), MYC (N = 3), EGFR (N = 2), MET (N = 2), MDM2 (N = 2) and KRAS (N = 1), and amplification of TERT in 5 samples. Losses were detected of SMAD4 and CDKN2A (both N = 8), TP53 (N = 5), APC (N = 5), PTEN, VHL and RB1 (all N = 4), and specifically TSC1 (N = 3) and TSC2 (N = 1).
Genomic variations and treatment response in patients
When exploring the cell line data, several hypotheses were generated with regard to the correlation between genomic aberrations and treatment response. The first step was to evaluate if these hypotheses could be tested in our patient data set. The rest of the paragraph is focused on TTP ratio assessment, because only one patient had a RECIST response, and because PFS is a longitudinal endpoint similar to TTP ratio, but without the correction for individual tumor growth rate.
In vitro data suggested increased resistance to mTOR inhibitors in the presence of a gain of CCNE1 or mutation in RB1. In our patient data however, all patients with a gain of CCNE1 (N = 2) or mutation in RB1 (N = 2) had clinical benefit from treatment (defined as TTP ratio response) (Table 3, Supplementary Table 1). Mutations in FGFR2, PTEN and loss of CDKN2A were associated with increased sensitivity to mTOR inhibition in vitro. In our patient data set, there was only one patient with a loss of FGFR2, this patient had a favorable outcome in terms of TTP ratio. Loss of CDKN2A (N = 7) was not correlated with TTP ratio response (either as a binary outcome or as a continuous outcome). Five patients had either a mutation or copy number loss of PTEN. Despite the fact that it was only possible to generate a TTP ratio for three of these patients (which classified them as responders) the other two patients also had clinical signs of a treatment effect: in one patient, central necrosis of all target lesions was observed at first response evaluation whereas the other patient had a PFS of 90 weeks, with a TTP1 period of 23 weeks. This patient was not evaluable for TTP ratio analysis, due to loss of volumetric measurability. When categorizing these five patients as responders, there was a significant correlation between treatment response and PTEN status (P = 0.046; one-tailed Fisher’s exact test). It should be noted however that these PTEN aberrations often coincided with other mTOR pathway related mutations (Supplementary Table 1). PIK3CA was also associated with increased sensitivity to mTOR inhibition in vitro. However, we could not find an association in our patient data (seven responders versus three non-responders).
Table 3: Genetic aberrations and response
Gene |
Clinical benefit |
Statistics |
|
|
Yes |
No |
p value |
KRAS |
5 |
5 |
.327 |
PIK3CA |
7 |
3 |
.377 |
MAPK |
5 |
4 |
.623 |
CDKN2A |
5 |
2 |
.326 |
PTEN |
5 |
0 |
.046* |
ERBB2 |
3 |
2 |
.625 |
TSC1 |
2 |
2 |
NA |
AKT |
2 |
0 |
NA |
CCNE1 |
2 |
0 |
NA |
RB1 |
2 |
0 |
NA |
TSC2 |
1 |
0 |
NA |
MTOR |
1 |
0 |
NA |
FGFR2 |
1 |
0 |
NA |
Legend: This table contains the number of patients that have, or have not experienced clinical benefit from treatment, stratified per afflicted gene.
Using an unbiased, overall analysis, no other somatic mutations or copy number alterations showed a significant correlation with response. Similarly, combining genetic aberrations or comparing somatic mutations on the pathway level did not yield significant results. To evaluate if genetic aberrations had a downstream effect by activating respectively the mTOR or MAPK pathway, we evaluated pS6 and pERK status. However, when incorporating pS6 and pERK status in previously mentioned analyses, we were still not able to predict clinical benefit, nor were pS6 and pERK predictive for response as single markers.
When focusing on mTOR (or interconnected) pathway related genes, we observed an equal distribution of responders and non-responders in KRAS mutated patients. This equal distribution was also observed in patients with other MAPK mutated genes. More directly upstream of MTOR are TSC1 and TSC2. One breast cancer patient harbored a missense TSC1 mutation and indeed responded to everolimus. Four tumor samples showed copy number loss or a mutation of TSC1 and one tumor showed loss of TSC2; these events were evenly divided over responders and non-responders.
Pre- and post-treatment comparison
Nine patients underwent a post-treatment biopsy procedure, of which four biopsies were of sufficient quality for DNA sequencing. Two of these patients had a TTP ratio response. In patient #1 (breast cancer), no resistance mechanisms were detected. Patient #2’s tumor initially harbored a very focal, high level amplification of the MET proto-oncogene (Figure 2). During treatment this amplification was clearly reduced, while a second high level gain on chromosome 7 appeared, i.e. affecting BRAF.
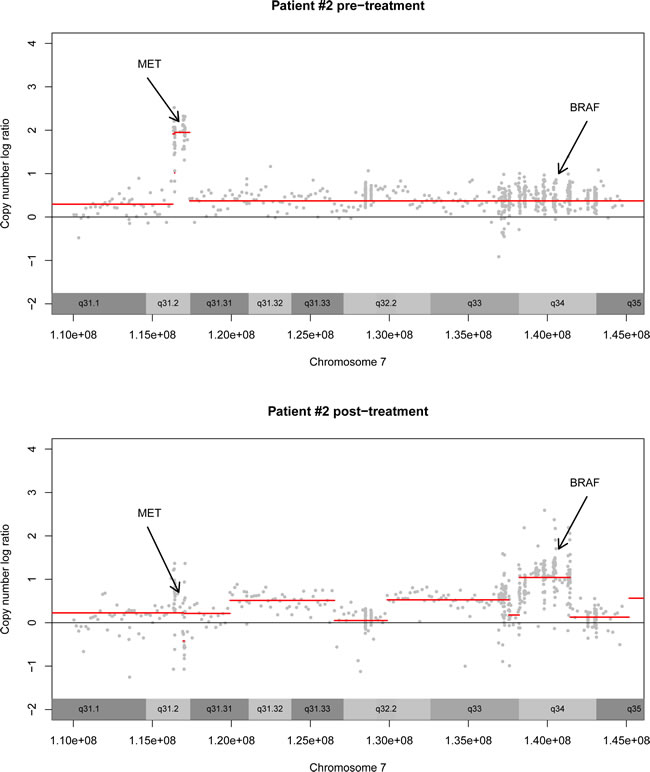
Figure 2 : Pre-post treatment biopsy. This figure demonstrates the copy number profile of chromosome 7 in patient #2 pre-treatment and post-treatment. Pre-treatment, there is an amplification of MET. This amplification is not present in the post-treatment biopsy. Instead, there is an amplification of BRAF wild-type.
Discussion
In this study, we found that copy number loss or mutation of PTEN was associated with treatment benefit of everolimus, suggesting that PTEN status could be a predictive biomarker for benefit from treatment. PTEN was frequently speculated to be a marker of interest, however, most clinical biomarker studies did not find a significant correlation with response [8, 9, 11, 12, 14, 17, 18]. This could be a result of the method used to determine PTEN status, as many studies used immunohistochemistry instead of DNA sequencing. Janku et al. were one of the few to combine immunohistochemistry and DNA sequencing in their biomarker study and had similar findings to our study [14]. Furthermore, this was the first study to employ an intra-patient control to determine treatment benefit. These findings should be further confirmed in other trial designs, such as basket studies.
Although currently published (pre-)clinical data report contradictory results, KRAS and PIK3CA mutation status have previously been associated with respectively resistance and sensitivity to mTOR inhibition in particular tumor types. In our dataset, we did not observe an association between mutations in either of these genes and treatment response. However, the sample size of this study is insufficient to make any statements regarding the absence of such an association.
Interestingly, a new amplification of wild-type BRAF was identified in a post-treatment biopsy, suggestive of a potential mechanism of resistance to mTOR inhibition. This tumor had a pre-treatment MET amplification. Both MET and BRAF can activate the MAPK-signaling pathway, but while MET functions upstream of mTOR, BRAF is just downstream of mTOR/Akt so activation of MAPK at this level circumvents the possible effect of mTOR inhibition. This data illustrates, that although post-treatment biopsies are difficult to acquire, they do provide hypothesis-generating information.
While this study yielded interesting findings and the data produced will be released to large sequencing databases to facilitate data sharing in further biomarker discovery efforts, there is an important side note. This study was drafted and implemented five years ago, when next generation sequencing technology had only just found its way to research centers and hospitals worldwide. The unprecedented wealth of genetic information fueled faith and optimism to identify markers for response and select patients for treatment. The past years have revealed that the implementation of genomics-based personalized medicine is not as simple as initially thought [24]. Complicating factors are amongst others varying degrees of tumor type dependence for the efficacy of biomarker-drug combinations, discrepancies between in vitro and in-patient findings, and a lower than expected incidence of actionable mutations [24]. In our study, the discrepancies between in vitro and in-patient findings can also be a result of the use of different mTOR inhibitors. Negative results for the first large genotype-matched drug trial (SHIVA), where context, i.c. tumor type, dependence was not taken into account, have also raised concerns [24, 25]. Another major hurdle is, that for many targeted agents, there are no established biomarkers. To identify single (or combinations of) molecular alterations that can predict treatment outcome, other study designs with more homogeneous patient groups (basket trials) or large cohorts of patients ( > 1000) are necessary [26, 27]. The latter can only be achieved by world-wide collaborations and sharing of data [28]. National- and worldwide sequencing initiatives such as the CPCT or project GENIE (by the American Association of Cancer Research) have been established to facilitate these efforts [28]. And whereas many of these efforts mainly focus on genomics-based analyses, we should aim to incorporate other types of analyses such as transcriptomics or proteomics.
To conclude, this study identified an association between PTEN status and treatment benefit from everolimus, identifying PTEN status as a potential biomarker for everolimus therapy. BRAF wildtype amplification could be a potential mechanism of resistance.
materials and Methods
Cell line data
Genetic profiles and drug sensitivity measurements of cell lines treated with the mTOR inhibitor temsirolimus were analyzed for potential biomarkers for treatment. This dataset (GDSC1000 v17a) was downloaded from http://cancerrxgene.org/gdsc1000/Pharmacogenomic_interactions.html [29].
Patients
The CPCT-03 study was an open-label, single arm, biomarker study. The primary objective was to identify genetic predictors for response to mTOR inhibition by everolimus. Patients with advanced solid malignancies without regular treatment options were eligible for inclusion. Inclusion and exclusion criteria, as well as detailed information on everolimus treatment, safety assessments and study design have been described previously [30]. The protocol was approved by the Institutional Review Board of The Netherlands Cancer Institute and complied with the Declaration of Helsinki, Dutch law and Good Clinical Practice guidelines. All patients provided written informed consent prior to study-related procedures. Patients were accrued at the Netherlands Cancer Institute, UMC Utrecht Cancer Center, and Erasmus MC Cancer Institute. The study was registered on ClinicalTrials.gov (NCT01566279).
Clinical efficacy assessments
Efficacy was measured according to three endpoints, TTP ratio, Response Rate (RR) and Progression-free survival (PFS). The TTP ratio uses an intra-patient control to correct for natural tumor growth rate and has been described previously by Cirkel et al. [30].
Tumor biopsy
After inclusion, all patients underwent a pre-treatment histological tumor biopsy of a metastatic lesion. A post-treatment tumor biopsy was optional. Biopsies were snap-frozen and stored at -80°C. Safety and feasibility of the CPCT ‘biopsy pipeline’ has been described by Bins et al. [31]. Blood samples (10mL) were collected in K2EDTA tubes, as a reference to determine somatic mutations.
Evaluability
Patients evaluable according to either RECIST or TTP ratio were evaluable for biomarker analyses in case of an adequate tumor biopsy (tumor percentage ≥30% and DNA yield ≥500ng).
DNA sequencing
Histological assessment to confirm the presence of tumor cells and mark regions with high tumor cellularity for macro-dissection was performed by a pathologist (S.W.). DNA was extracted from whole blood and macro-dissected tumor sections. Barcoded libraries were generated as previously described and enriched for a “Cancer mini-genome” of 1,977 cancer genes, based on Vermaat et al. and Hoogstraat et al. [32-34]. Enriched libraries were sequenced to an average coverage of 150x on a SOLiD 5500xl instrument according to manufacturer’s protocol. Whole exome sequencing was performed for six patients using the NextSeq 500 v2 as our sequencing facility switched platforms. Somatic mutations were validated with the Ion Ampliseq Cancer Panel or custom-made primers for mTOR-related genes. Mapping, variant calling and annotation was done as previously described [33]. Sam tools mpileup was used to ensure the absence or presence of a variant in a given sample [35]. Copy number profiles were generated using CNVkit [36]. Detailed information on sequencing methods and bioinformatics pipelines can be reviewed in online-only supplementary materials.
Immunohistochemistry
In order to determine activation of mTOR and interconnected pathways, all available biopsies (N = 33) were stained for phospho-S6 and phospho-ERK. Phospho-S6 is a marker for activation of mTOR, pERK is a marker for MAPK pathway activation. Slides were scored for intensity (0-3) and percentage of positive tumor cells by a pathologist blinded for treatment outcome.
Statistical analyses
No formal sample size calculation was performed due to an unknown expected RR of a heterogeneous group of tumors with unknown frequencies of genetic aberrations that might be predictive for response. The study was open for accrual of 60 evaluable patients or 15 evaluable TTP ratio responders. R (version 3.2.1) was used for downstream analyses of mutations and copy number variation, and to detect associations between genetic variation, tumor type and treatment response. All genetic aberrations (copy number gain, loss or mutation) were encoded as binary variables, where 0 = absence and 1 = presence of the mutation. On the cell line data, elastic net feature selection was performed using the R-package glmnet [37]. We used linear models to assess the significance of the presence of (multiple) genetic aberrations while correcting for tumor type and ANOVA to determine the effect of tissue type on treatment response. Univariate analyses of single genes within specified tumor types were done using one- or two-tailed t-tests, depending on context. If previous data or literature had already provided an indication of the direction of the effect, a one-tailed test was used. In our patient data, we tested associations between single variables and response using Fisher’s exact test and associations between multiple variables and outcome were modeled using logistic regression. We assessed pathway enrichment of genetic variation in responders and non-responders as described previously [38]. Briefly, we used the Kyoto Encyclopedia of Genes and Genomes (KEGG) to define pathways. A pathway was considered to be affected if at least one of its genes was found mutated. We performed the Fisher’s exact test to correlate pathway activation and treatment response.
Abbreviations
4EBP1 - 4E-Binding Protein
APC - Adenomatous Polyposis Coli
AKT - Protein kinase B
ALK - Anaplastic Lymphoma Kinase
ANOVA - Analysis of Variance
BRAF - B-Raf Proto-Oncogene, Serine/Threonine Kinase
CDKN2A - Cyclin-Dependent Kinase Inhibitor 2A
CCND1 - Cyclin-D1
CCNE1 - Cyclin-E1
CIC - Capicua transcriptional repressor
COSMIC - Catalogue Of Somatic Mutations In Cancer
CPCT - Center for Personalized Cancer Treatment
DNA - Deoxyribonucleic acidEGFR - Epidermal Growth Factor Receptor
ERBB2 - Erb-B2 Receptor Tyrosine Kinase 2
ERCC5 - Excision Repair Cross Complement group 5
FGFR2 - Fibroblast Growth Factor Receptor-2
HGF - Hepatocyte Growth Factor
IC50 - Inhibitory Concentration Of 50%
KEGG - Kyoto Encyclopedia of Genes and Genomes
KRAS - Kirsten rat sarcoma viral oncogene homolog
MAPK - Mitogen-Activated Protein Kinases
MDM2 - Mouse double minute 2 homolog
MET - proto-oncogene MET
mTOR - mammalian Target of Rapamycin
MYC - Avian Myelocytomatosis Viral Oncogene Homolog
NSCLC - Non-Small Cell Lung Cancer
pERK - phospho Extracellular Regulated Kinase
PFS - Progression-Free Survival
PI3K/PIK3CA - Phosphoinositide-3-kinase
pS6 - phosphorylated S6 Ribosomal Protein
PTEN - Phosphatase and tensin homolog
SOX9 - SRY-Box 9
RB1 - Retinoblastoma 1
RECIST - Response Evaluation Criteria In Solid Tumors
RR - Response Rate
TERT - Telomerase Reverse Transcriptase
TP53 - Tumor Protein p53
TSC - Tuberous Sclerosis Complex
TTP - Time To Progression
VHL - Von Hippel-Lindau.
Author contributions
Study design: G.A.C., M.P.L., E.E.V.
Study conduct: F.W., G.A.C., S.B., C.G.M.G.-v.H., S.O., S.M.W., M.v.S., W.B.V., N.J.M.B., N.S., M.J.d.J, M.H.G.L., J.H.S., S.S., M.P.L., E.E.V.
Data analysis: F.W., G.A.C., M.H., E.v.W., H.M.H., L.F.A.W., E.P.J.G.C., M.P.L., E.E.V.
Study supervision: M.P.L., E.E.V.
Writing of the manuscript: F.W., G.A.C., M.H., S.B., E.v.W., S.M.W., M.v.S., W.B.V., N.J.M.B., H.M.H., N.S., M.J.d.J, M.H.G.L., L.F.A.W., E.P.J.G.C., J.H.S., S.S., M.P.L., E.E.V.
Acknowledgments
We would like to thank the Core Facility - Molecular Pathology & Biobanking of the Netherlands Cancer Institute, and especially Ingrid Hofland and Dennis Peters, for immunohistochemical stainings.
Conflicts of interest
The authors report no conflict of interest.
Funding
The Center for Personalized Cancer Treatment is financially supported by:
Dutch Cancer Society, Amsterdam, The Netherlands (grant HUBR 2011-4880)
NutsOhra Foundation, Amsterdam, The Netherlands (project 1102-062)
Barcode for Life, Utrecht, The Netherlands (www.barcodeforlife.nl)
Friends of the UMC Utrecht Foundation, Utrecht, The Netherlands (www.vriendenumcutrecht.nl)
Everolimus was provided by Novartis.
References
1. Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, Riely GJ, Solomon B, Ou SH, Kim DW, Salgia R, Fidias P, Engelman JA, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012; 13: 1011-1019.
2. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363: 809-819.
3. Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012; 366: 520-529.
4. Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, Tomassetti P, Pavel ME, Hoosen S, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011; 364: 514-523.
5. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grunwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008; 372: 449-456.
6. Yao JC, Fazio N, Singh S, Buzzoni R, Carnaghi C, Wolin E, Tomasek J, Raderer M, Lahner H, Voi M, Pacaud LB, Rouyrre N, Sachs C, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016; 387: 968-977.
7. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009; 8: 627-644.
8. Andre F, Hurvitz S, Fasolo A, Tseng LM, Jerusalem G, Wilks S, O’Regan R, Isaacs C, Toi M, Burris H, He W, Robinson D, Riester M, et al. Molecular Alterations and Everolimus Efficacy in Human Epidermal Growth Factor Receptor 2-Overexpressing Metastatic Breast Cancers: Combined Exploratory Biomarker Analysis From BOLERO-1 and BOLERO-3. J Clin Oncol. 2016; 34: 2115-24.
9. Di Nicolantonio F, Arena S, Tabernero J, Grosso S, Molinari F, Macarulla T, Russo M, Cancelliere C, Zecchin D, Mazzucchelli L, Sasazuki T, Shirasawa S, Geuna M, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest. 2010; 120: 2858-2866.
10. Grunwald V, Keilholz U, Boehm A, Guntinas-Lichius O, Hennemann B, Schmoll HJ, Ivanyi P, Abbas M, Lehmann U, Koch A, Karch A, Zorner A, Gauler TC. TEMHEAD: a single-arm multicentre phase II study of temsirolimus in platin- and cetuximab refractory recurrent and/or metastatic squamous cell carcinoma of the head and neck (SCCHN) of the German SCCHN Group (AIO). Ann Oncol. 2015; 26: 561-567.
11. Holsinger FC, Piha-Paul SA, Janku F, Hong DS, Atkins JT, Tsimberidou AM, Kurzrock R. Biomarker-directed therapy of squamous carcinomas of the head and neck: targeting PI3K/PTEN/mTOR pathway. J Clin Oncol. 2013; 31: e137-140.
12. Hortobagyi GN, Chen D, Piccart M, Rugo HS, Burris HA 3rd, Pritchard KI, Campone M, Noguchi S, Perez AT, Deleu I, Shtivelband M, Masuda N, Dakhil S, et al. Correlative Analysis of Genetic Alterations and Everolimus Benefit in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results From BOLERO-2. J Clin Oncol. 2016; 34: 419-426.
13. Ihle NT, Lemos R Jr, Wipf P, Yacoub A, Mitchell C, Siwak D, Mills GB, Dent P, Kirkpatrick DL, Powis G. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009; 69: 143-150.
14. Janku F, Hong DS, Fu S, Piha-Paul SA, Naing A, Falchook GS, Tsimberidou AM, Stepanek VM, Moulder SL, Lee JJ, Luthra R, Zinner RG, Broaddus RR, et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014; 6: 377-387.
15. Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, Hong DS, Naing A, Falchook GS, Moroney JW, Piha-Paul SA, Wheler JJ, Moulder SL, Fu S, et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther. 2011; 10: 558-565.
16. Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna I, Luthra R, Lee JJ, Lu KH, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012; 30: 777-782.
17. Mackay HJ, Eisenhauer EA, Kamel-Reid S, Tsao M, Clarke B, Karakasis K, Werner HM, Trovik J, Akslen LA, Salvesen HB, Tu D, Oza AM. Molecular determinants of outcome with mammalian target of rapamycin inhibition in endometrial cancer. Cancer. 2014; 120: 603-610.
18. Seront E, Rottey S, Sautois B, Kerger J, D’Hondt LA, Verschaeve V, Canon JL, Dopchie C, Vandenbulcke JM, Whenham N, Goeminne JC, Clausse M, Verhoeven D, et al. Phase II study of everolimus in patients with locally advanced or metastatic transitional cell carcinoma of the urothelial tract: clinical activity, molecular response, and biomarkers. Ann Oncol. 2012; 23: 2663-2670.
19. Tredan O, Treilleux I, Wang Q, Gane N, Pissaloux D, Bonnin N, Petit T, Cretin J, Bonichon-Lamichhane N, Priou F, Lavau-Denes S, Mari V, Freyer G, et al. Predicting everolimus treatment efficacy in patients with advanced endometrial carcinoma: a GINECO group study. Target Oncol. 2013; 8: 243-251.
20. Treilleux I, Arnedos M, Cropet C, Wang Q, Ferrero JM, Abadie-Lacourtoisie S, Levy C, Legouffe E, Lortholary A, Pujade-Lauraine E, Bourcier AV, Eymard JC, Spaeth D, et al. Translational studies within the TAMRAD randomized GINECO trial: evidence for mTORC1 activation marker as a predictive factor for everolimus efficacy in advanced breast cancer. Ann Oncol. 2015; 26: 120-125.
21. Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, Pirun M, Sander C, Socci ND, Ostrovnaya I, Viale A, Heguy A, Peng L, et al. Genome sequencing identifies a basis for everolimus sensitivity. Science. 2012; 338: 221.
22. Wagner AJ, Malinowska-Kolodziej I, Morgan JA, Qin W, Fletcher CD, Vena N, Ligon AH, Antonescu CR, Ramaiya NH, Demetri GD, Kwiatkowski DJ, Maki RG. Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol. 2010; 28: 835-840.
23. Si L, Xu X, Kong Y, Flaherty KT, Chi Z, Cui C, Sheng X, Li S, Dai J, Yu W, Guo J. Major response to everolimus in melanoma with acquired imatinib resistance. J Clin Oncol. 2012; 30: e37-40.
24. Voest EE and Bernards R. DNA-Guided Precision Medicine for Cancer: A Case of Irrational Exuberance? Cancer Discov. 2016; 6: 130-132.
25. Le Tourneau C, Delord JP, Goncalves A, Gavoille C, Dubot C, Isambert N, Campone M, Tredan O, Massiani MA, Mauborgne C, Armanet S, Servant N, Bieche I, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015; 16: 1324-1334.
26. Herbst RS, Gandara DR, Hirsch FR, Redman MW, LeBlanc M, Mack PC, Schwartz LH, Vokes E, Ramalingam SS, Bradley JD, Sparks D, Zhou Y, Miwa C, et al. Lung Master Protocol (Lung-MAP)-A Biomarker-Driven Protocol for Accelerating Development of Therapies for Squamous Cell Lung Cancer: SWOG S1400. Clin Cancer Res. 2015; 21: 1514-1524.
27. Andre F, Delaloge S, Soria JC. Biology-driven phase II trials: what is the optimal model for molecular selection? J Clin Oncol. 2011; 29: 1236-1238.
28. Siu LL, Lawler M, Haussler D, Knoppers BM, Lewin J, Vis DJ, Liao RG, Andre F, Banks I, Barrett JC, Caldas C, Camargo AA, Fitzgerald RC, et al. Facilitating a culture of responsible and effective sharing of cancer genome data. Nat Med. 2016; 22: 464-471.
29. Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Goncalves E, Barthorpe S, Lightfoot H, Cokelaer T, Greninger P, van Dyk E, et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell. 2016; 166: 740-54.
30. Cirkel GA, Weeber F, Bins S, Gadellaa-van Hooijdonk CG, van Werkhoven E, Willems SM, van Stralen M, Veldhuis WB, Ubink I, Steeghs N, de Jonge MJ, Langenberg MH, Schellens JH, et al. The Time To Progression Ratio: a new individualized volumetric parameter for early detection of clinical benefit of targeted therapies. Ann Oncol. 2016; 27: 1638-43.
31. Bins S, Cirkel GA, Gadellaa-van Hooijdonk CG, Weeber F, Nijman IJ, Bruggink AH, van Diest PJ, Willems SM, Veldhuis WB, van den Heuvel MM, de Knegt RJ, Koudijs MJ, van Werkhoven E, et al. Implementation of a Multicenter Biobanking Collaboration for Next-Generation Sequencing-Based Biomarker Discovery Based on Fresh Frozen Pretreatment Tumor Tissue Biopsies. Oncologist. 2016; 22: 33-40.
32. Harakalova M, Mokry M, Hrdlickova B, Renkens I, Duran K, van Roekel H, Lansu N, van Roosmalen M, de Bruijn E, Nijman IJ, Kloosterman WP, Cuppen E. Multiplexed array-based and in-solution genomic enrichment for flexible and cost-effective targeted next-generation sequencing. Nat Protoc. 2011; 6: 1870-1886.
33. Hoogstraat M, de Pagter MS, Cirkel GA, van Roosmalen MJ, Harkins TT, Duran K, Kreeftmeijer J, Renkens I, Witteveen PO, Lee CC, Nijman IJ, Guy T, van ‘t Slot R, et al. Genomic and transcriptomic plasticity in treatment-naive ovarian cancer. Genome Res. 2014; 24: 200-211.
34. Vermaat JS, Nijman IJ, Koudijs MJ, Gerritse FL, Scherer SJ, Mokry M, Roessingh WM, Lansu N, de Bruijn E, van Hillegersberg R, van Diest PJ, Cuppen E, Voest EE. Primary colorectal cancers and their subsequent hepatic metastases are genetically different: implications for selection of patients for targeted treatment. Clin Cancer Res. 2012; 18: 688-699.
35. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011; 27: 2987-2993.
36. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol. 2016; 12: e1004873.
37. Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw. 2010; 33: 1-22.
38. Lips EH, Michaut M, Hoogstraat M, Mulder L, Besselink NJ, Koudijs MJ, Cuppen E, Voest EE, Bernards R, Nederlof PM, Wesseling J, Rodenhuis S, Wessels LF, et al. Next generation sequencing of triple negative breast cancer to find predictors for chemotherapy response. Breast Cancer Res. 2015; 17: 134.