
INTRODUCTION
Gastrointestinal stromal tumors (GISTs) are common neoplasms that originate in the mesenchymal tissue of the gastrointestinal tract, and so-called micro GISTs are found in ~20-30% of adults [1-3]. Malignant, metastatic GISTs serve as a clinical paradigm for inhibiting activating driver mutations to confer a major clinical benefit to patients and a rational clinical model to evaluate new targeted therapies and identify molecular mechanisms of drug response and resistance [4]. However, the genetic events responsible for the clinical progression of this tumor type are poorly understood. GISTs are generally considered to emanate from the interstitial cells of Cajal (ICC), which are pacemaker cells regulating gut motility. ICCs originate from the same precursor cells as smooth muscle tissue, common intestinal mesenchymal precursor cells [5]. GISTs are most frequently found in the stomach, small intestine, colon, esophagus, and rectum [1, 6, 7]. Clinical GIST is a relatively rare cancer; the average annual incidence ranges from 11 to 19.6 per million individuals [4].
In the last few decades, several genetic events in the initiation and progression of GIST have been reported. Specifically, a large portion of the GISTs harbor oncogenic mutations in KIT or platelet-derived growth factor receptor-α (PDGFRA) [8-12], and some GISTs without KIT or PDGFRA mutations are called wild-type GISTs. This review provides an overview of inspiring developments in the oncology, pathology and pharmacology of GISTs. The focus is placed on the driver mutations and chromosomal alterations involved in GIST initiation and progression as well as the relationship between resistance to targeted therapeutics and secondary mutations.
KIT/PDGFRA GAIN-OF-FUNCTION MUTATIONS
KIT mutations
KIT, also known as CD117, is a member of the type III receptor tyrosine kinase family [8]. The family consists of PDGFRA, PDGFRB, FLT3 (Fms-like tyrosine kinase 3) and CSF1R (macrophage colony-stimulating-factor receptor) [13]. KIT protein is expressed in several cell types, including mast cells, melanocytes, hematopoietic progenitor cells, germ cells, and ICCs. Normally, KIT does not have kinase activity until it binds stem cell factor (SCF) and homodimerizes [14].
Moreover, 78.5% GISTs contain KIT mutations (Figure 1A), and these alterations contribute to the constitutive activation of the KIT oncoprotein. Therefore, mutant KIT is a crucial diagnostic marker and clinical therapeutic target for the treatment of GISTs [15].
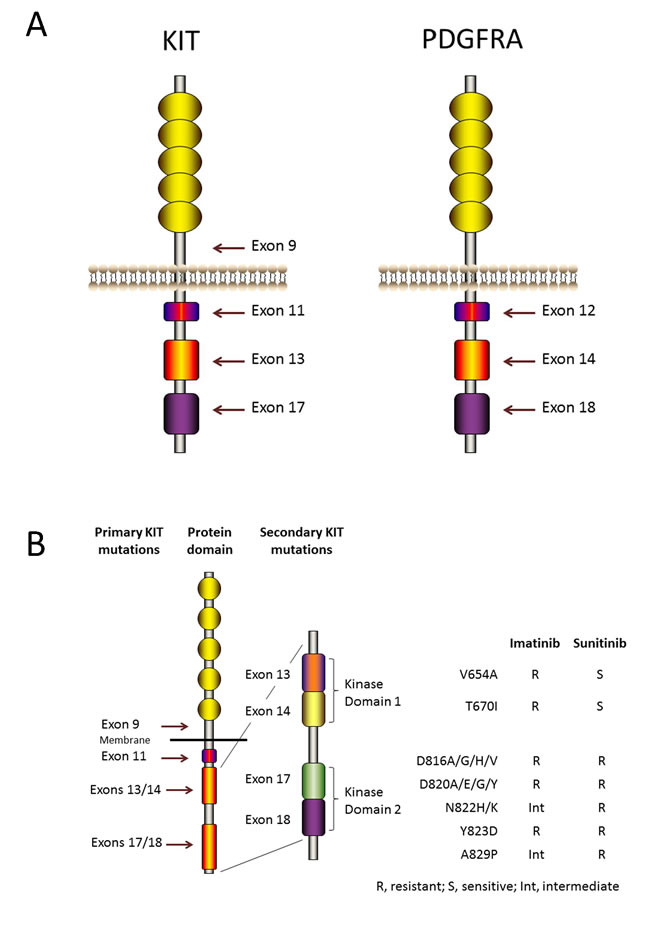
Figure 1: KIT and PDGFRA structure and mutations. A. KIT and PDGFRA are type III receptor tyrosine kinases and have the same topology: an extracellular ligand-binding domain that consists of five immunoglobulin-like repeats, a transmembrane sequence, a juxtamembrane domain and a cytoplasmic kinase domain. Mutations in the juxtamembrane domain, which is encoded by exon 11 of KIT or by exon 12 of PDGFRA, allow receptor dimerization without ligands and modulate the kinase activation loop from swinging to activating. Mutations in the activation loop, which is encoded by exon 17 of KIT or by exon 18 of PDGFRA, and mutations in the ATP-binding region, which is encoded by exon 13 of KIT or by exon 14 of PDGFRA, all stabilize the active conformation of KIT or PDGFRA. Mutations in the extracellular domain of KIT (encoded by exon 9) favor receptor dimerization. B. Secondary KIT mutations and drug sensitivities. Secondary mutations cluster in two regions of the KIT oncoprotein: the tyrosine kinase domain (TKD) 1 (encoded by exons 13 and 14) and the TKD 2 (encoded by exons 17 and 18).
Oncogenic KIT mutants result in spontaneous receptor homodimerization and kinase activation without SCF [16]. The most frequent mutations occur in exon 11 of KIT, which encodes the juxtamembrane domain. Specifically, 85% of KIT mutations are caused by mutations in exon 11, and these mutations change the juxtamenbrane structure and modulate the KIT protein activation loop from swinging to activating [17, 18]. In addition to exon 11 mutations, approximately one tenth of KIT mutations occur in exon 9, which encodes an extracellular domain. The conformations of these mutant KITs are similar to that of the extracellular domain bound with KIT ligand [19]. Besides, other uncommon mutations, such as mutations in the KIT activation loop and mutations in the ATP-binding pocket, encoded by exon 17 and exon 13, respectively, occur, too [20, 21]. These mutations all stabilize the active structure of KIT.
The KIT oncoprotein activation stimulates downstream signaling pathways (Figure 2), including the PI3K-AKT pathway, the MAPK pathway, and the STAT3 (signal transducer and activator of transcription 3) pathway [4, 22].
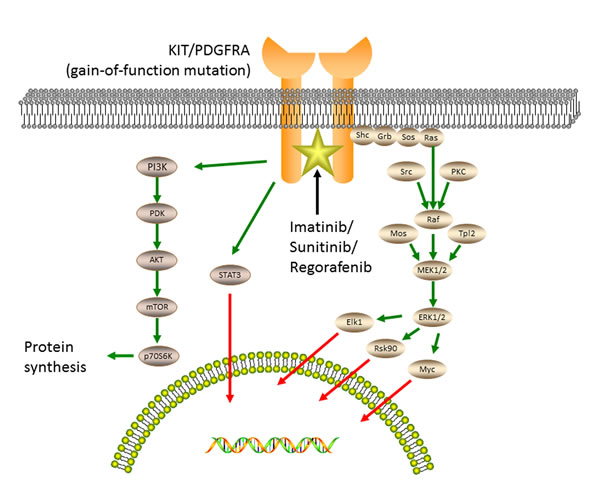
Figure 2: Oncogenic tyrosine kinase signaling and accessory pathways responsible for the pathogenesis of GISTs. The kinase activation of KIT or PDGFRA stimulates downstream signaling pathways, including the MAPK pathway, the PI3K-AKT pathway and the STAT3 pathway. Imatinib, sunitinib and regorafenib inhibit several kinases, including KIT and PDGFRA, by binding to the ATP-pocket and yield durable responses.
PDGFRA mutations
PDGFRA shares similar homologue with KIT. Approximately 5-8% GISTs contain a mutation in PDGFRA gene (Figure 1A). Immunoblots of tumor samples have shown that a minority of GISTs without KIT mutations exhibit high levels of PDGFRA phosphorylation [11]. Specifically, activated PDGFRA is detected in GISTs that harbor mutations in exons 12, 14 and 18, which encode the PDGFRA juxtamembrane domain, and the kinase domains, respectively. What’s more, mutant PDGFRA exhibits activated kinase activity without their ligand PDGFA. It shares the same downstream signaling pathways with those in KIT-mutant GISTs. Furthermore, heat shock protein 90 (HSP90) stabilizes mutant PDGFRA [23, 24].
In addition to these similarities, several pathological features distinguish PDGFRA-mutant GISTs from KIT-mutant GISTs, such as different whole genome expression profiles, a significantly higher morbidity in the stomach, variable KIT expression and a lower possibility for malignancy [9, 25, 26]. Nevertheless, the mechanisms for these distinctions remain unclear.
KIT/PDGFRA inhibitors
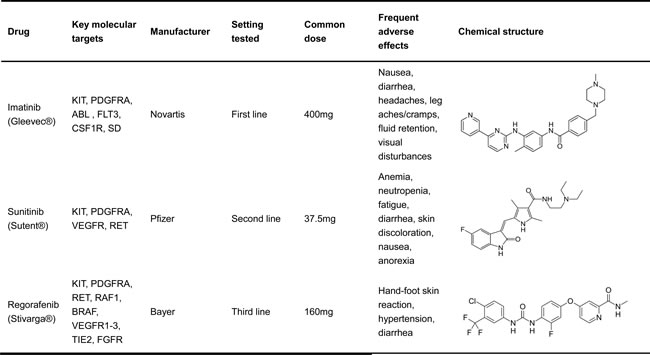
Because of these mutations in KIT and PDGFRA, tyrosine kinase inhibitor (TKI) therapy has been developed to treat GISTs. TKIs have revolutionized the treatment of GISTs in the last 15 years [27-30], and three TKIs have been approved to treat GIST (Table 1). Specifically, imatinib was approved as the standard first-line drug. It inhibits several kinases, including KIT and PDGFRA, by binding to the ATP-binding domain and induces dramatic disease control in about 85% of GISTs. In addition, it has shown excellent initial clinical responses in approximately 80% of GIST patients, with only 10-15% of patients exhibiting primary resistance [28, 29, 31]. More specifically, imatinib produces higher partial response rates in GISTs with the exon 11 KIT mutation than in GISTs with the exon 9 mutation or patients without a KIT or PDGFRA mutation. Conversely, patients with tumors harboring PDGFRA mutations (D842V) are strongly resistant to imatinib [32-34]. Therefore, mutational tests for KIT and PDGFRA should be considered for metastatic or advanced GIST treatment planning. In 2006, the median survival for patients being treated with imatinib was 4.8 years. Although imatinib shows remarkable initial success in the treatment of GISTs, its long-term efficacy is not as dramatic because many secondary mutations in KIT cause the tumors to become resistant [35]. Similar to imatinib, sunitinib inhibits KIT and PDGFRA activities by binding to the ATP-binding domain. However, the binding features of sunitinib differ from those of imatinib. Sunitinib can inhibit the vascular endothelial growth factor receptor (VEGFR) and the rearranged during transfect (RET) kinase as well. Thus, sunitinib was approved to combat imatinib resistance in 2006. In a clinical study, 312 patients were given with sunitinib for four weeks at a daily dose of 50 mg, and followed by a two-week break. The result showed that in the sunitinib and placebo arms, the median progression-free survival times were 6.3 and 1.5 months with a hazard ratio (HR) of 0.33. Compared with the placebo arm, overall survival was superior in the sunitinib group [36]. However, sunitinib likely caused significant side effects, including skin pigmentation disorders, fatigue and so on. Thus, the benefits of sunitinib therapy are conserved than those of first-line imatinib standard treatment [36]. However, the inherent heterogeneity of resistant mutations in GIST tumors precludes the universal efficacy of sunitinib [21]. Regorafenib is an oral kinase inhibitor that binds several kinases functioned in tumorigenesis, including KIT and PDGFRA [37-40]. The most frequent side effects were hand-foot skin reactions and hypertension.
Table 1: FDA approved TKIs for the treatment GISTs
CDKN2A/P53 DYSREGULATIONS
CDKN2A (cyclin-dependent kinase inhibitor 2A) is a tumor suppressor gene encoding the p16 and p14. p16 suppresses cyclin dependent kinases 4 and 6 (CDK4 and CDK6) and thus activates the retinoblastoma (Rb) family members, which inhibit cell cycle from the G1 to the S phase. p14 activates the p53 tumor suppressor. Furthermore, several studies indicated that CDKN2A significantly impacts the pathogenesis of GIST [41, 42], and most advanced GISTs harbor CDKN2A inactivation due to chromosome 9p21 deletion, which may be biallelic or combined with other mutations or promoter methylation [43, 44]. In 2015, the Food and Drug Administration (FDA) approved ibrance to treat (in combination with letrazole) breast cancer patients with human epidermal growth factor receptor 2 (HER2)(-) and estrogen receptor (ER) (+). Since ibrance is a selective inhibitor of CDK4 and CDK6, it may be efficacious to treat GISTs. Adverse reactions to ibrance include neutropenia, infections and pulmonary embolism.
p53 protein, encoded by the TP53 gene in humans, also acts as a tumor suppressor. It participates in many biological processes, including cell cycle, programmed cell death and DNA repair. Mutations in the TP53 gene frequently occur in neoplasms, including GISTs, and these mutations result in p53 protein inactivation. Most mutations in TP53 are centered in exons 4 to 8, which are also the most highly conserved exons in vertebrates. These mutations alter the central DNA-binding core domain. The TP53 protein binds to DNA and has the following three main functions: activating DNA repair mechanisms, holding the cell cycle at the G1/S regulation point to allow DNA repair and consequently arrest cell growth, and initiating apoptosis. These functions lead to cell stability, but when TP53 is mutated, the cell is allowed to grow and divide uncontrollably, leading to cancer. Another gene, MDM2, encodes a protein that negatively controls TP53 by degrading it, and MDM2 is induced by TP53 in a negative feedback loop. However, MDM2 is not induced by mutated TP53, and mutated TP53 consequently accumulates at very high concentrations. Some studies showed that impaired p53 expression is common in advanced GISTs, and p53 has a strong effect on the progression-free survival. Moreover, the accumulation of p53 protein correlated significantly with the mitotic rate and the risk of malignancy. Mutations in TP53 or reduced p53 expression are associated with an unfavorable prognosis [45, 46].
DYSTROPHIN INACTIVATION
Dystrophin tumor suppressor in GIST
Dystrophin is a rod-shaped structure protein, forming a protein complex providing a link between the cytoskeleton of a muscle fiber and the surrounding extracellular matrix (Figure 3A). In cancer, dystrophin suppresses many cell behaviors, such as cell migration, invasion, anchorage independence, in sarcomas with myogenic features. A recent study showed that the somatic DMD deletion is a common mechanism by which myogenic tumors progress to advanced sarcomas (Figure 3B). DMD is one of the largest human genes and consists of 79 coding exons that span 2.2 Mb of the genome, with multiple isoforms. DMD deletions target 427-kDa myogenic isoform, while the expression of a 71-kDa isoform, which is essential for cell viability, is preserved. Dystrophins are expressed highly in the ICC cells and low-risk GISTs, whereas dystrophin inactivation was identified in 96% of metastatic GISTs [47].
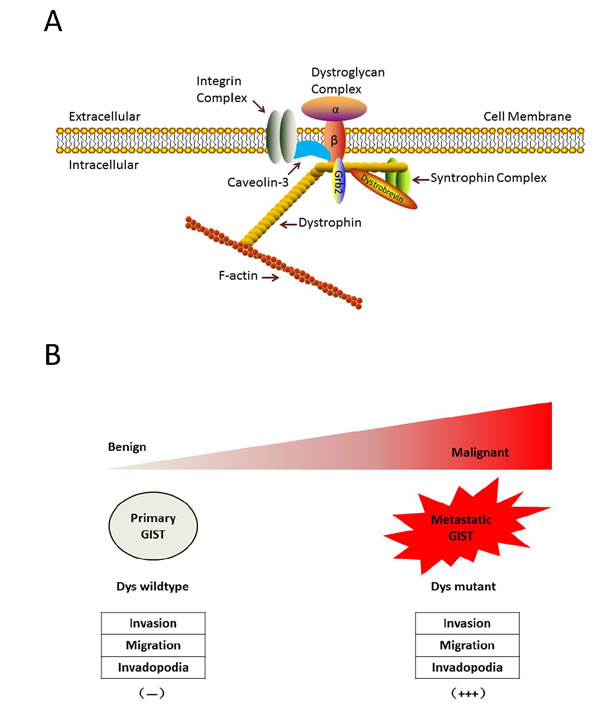
Figure 3: Dystrophin is a novel tumor suppressor that regulates GIST invasion, migration, anchorage-independent growth and invadopodia formation. A. Dystrophin structure. Dystrophin is a rod-shaped structure protein, forming a protein complex providing a link between the cytoskeleton of a muscle fiber and the surrounding extracellular matrix. B. Myogenic dystrophin 427kDa isoform was expressed strongly (and had no demonstrable genomic mutations) in low-risk primary GISTs, whereas dystrophin expression was undetectable in most metastatic GISTs, most of which had inactivating intragenic DMD mutations. Dystrophin inactivation increases cell invasion, migration, anchorage-independent growthand invadopodia formation.
DMD is an X-linked tumor suppressor gene, but the deletion rates do not differ between males and females. Specifically, copy number profiles demonstrated that the inactive X chromosome was intact in female GISTs with heterozygous DMD mutations. Both heterozygous and homozygous DMD mutations in females resulted in entire DMD knockout. We demonstrated that dystrophin dysregulation can be used as a prognostic factor in GISTs and as a novel target of attack for GIST therapy. This is a significant finding that impacts the direction of future research studies. The implication is that by detecting key genetic defects such as the lack of dystrophin in GIST patients, it may eventually become possible to predict those who will develop metastatic diseases. These studies determine whether loss of dystrophin expression identifies a high-risk group of patients, with higher frequency of subsequent metastases, and lower recurrence-free and overall survival, compared to patients whose primary tumors retain dystrophin expression. DMD deletions are enriched in myogenic cancers, and DMD inactivation selectively increases metastatic potential in GISTs. What’s more, the knockdown of the Dp71 isoform decreased cell viabilities, showing that 71-kDa isoform is required for cancer cell growth. Moreover, dystrophin interacts with dystrophin glycoprotein complex. The evidence of dystrophin tumor suppressor suggests that other components in dystrophin complex are likely to play key roles in GIST tumorgenesis [48, 49].
Targeting dystrophin deficiency
Some studies have attempted to correct the dystrophin gene reading frame by modulating pre-mRNA splicing with antisense oligonucleotides (AOs)-mediated exon skipping, which has shown success both in vitro and in vivo. Genetic testing can detect the precise character and DMD mutation site. DMD mutations in muscular dystrophy cluster in hotspot regions, which are mainly located on exons 45-53 and exons 2-20. Most patients with DMD would benefit from the creation of generally needed AOs. Specifically, up to 50% of these patients could benefit from AO cocktails to produce the skipping of the exons 45-53 [50-52].
Recent studies demonstrate that the functions of utrophin and dystrophin are equivalent in muscle
Dystrophin-deficient mdx mouse can be cured by utrophin therapy. Utrophin mRNA levels can be increased through its promoter manipulation. Similarly, SMT C1100 is invented to increase and maintain utrophin transcription. It induces increased utrophin mRNA and protein levels in human muscle cells and reduces dysrophin-deficient muscle pathology. Furthermore, it provides significant benefits to whole body strength and endurance. In theory, the development of small molecules (such as SMT C1100) can increase the level of utrophin to treat DMD. The most significant advantage of this approach is that it can treat all disease associated with DMD deficiency, irrespective of the dystrophin mutation [53].
Some studies show that the 70-kDa heat shock protein 1 (Hsp72) inducer and Wnt7a help to ameliorate muscular dystrophy. Intracellular Ca2+ is deregulated in dystrophic muscle fibers, which inducing inflammation. Inflammation contributes to dystrophic pathology via the pro-inflammatory cytokine TNF-α, which activates the NF-kB and JNK signaling pathways. Accordingly, dystrophin-deficient mdx mice treated with BGP-15, a Hsp72 activator, showed improved muscular dystrophy phenotype [54].
The non-canonical Wnt receptor Frizzled-7 is selectively expressed in satellite stem cells. Wnt7a protein induces the expansion of satellite stem cells, and its overexpression enhances muscle regeneration. In addition, Wnt7a activity results in muscle hypertrophy, and a preclinical study showed that Wnt7a can increase the specific force in dystrophic mice [55].
Dystrophin expression does not reduce the number of viable cells in GIST. More importantly, dystrophin deficiency could be detected in both TKI-sensitive and TKI-resistant GIST, suggesting the limited value of targeting dystrophin deficiency to overcome TKI resistance.
1p, 14q, 22q ALTERATIONS
Although gene mutations play a key role in GIST tumorigenesis, other molecular events are crucial in GIST progression [56], such as chromosomal alterations [57]. Among these chromosomal alterations, losses of chromosome arms 1p, 14q and 22q are the most commonly detected abnormities. According to research by Maria Debiec-Rychter et al., 11 tumors from 39 GIST samples harbored chromosome 1 aberrations. Specifically, one sample exhibited a loss of whole chromosome 1, one sample displayed a loss of 1q32-qter and the other seven tumors showed copy number losses in different bands of 1p [58, 59]. Furthermore, Julie Breiner’s research indicated that a deletion of the long and/or short arms of chromosome 1 is related to the malignancy of GIST [60]. Chromosome 1 copy number abnormities look like as a secondary change that is crucial in GIST tumorgenesis, and chromosome arm 14q changes were detected in both primary and recurrent tumors. About 67% of GIST samples exhibit either total chromosome 14 loss or a partial 14q loss. According to the copy number and loss of heterozygosity (LOH) studies, tumor suppressor genes, that promote GIST initiation, may locate in two bands of the chromosome 14. Specifically, loss of 14q11.2 involving the PARP2, APEX1 and NDRG2 and loss of 14q32 involving the SIVA, and the inactivation of these genes may be important in the pathogenesis of GIST. A deletion of the long arm of chromosome 22 is found in about half GISTs, and this loss is invariably related to chromosome 1 aberrations. Moreover, losses of 22q are related to a poor prognosis. What’s more, another tumor suppressor gene NF2 is located in 22q12. Copy number losses of chromosomes 14 and 22 were identified in low-risk, intermediate-risk and high-risk GISTs, showing that these losses may occur early in GIST progression [61, 62].
TKI RESISTANCE
TKI resistance mechanisms
GISTs are resistant to traditional chemotherapeutic regimes and radiation treatments because of the dose-limiting effects on nearby vital organs. Based on studies with long-term follow up, adverse events, including recurrence, metastases, and death, occur in up to 85% of patients who have undergone surgical resection. Prior to the development of highly specific TKIs, no treatment options existed for patients with malignant, non-operable GISTs. Targeted TKIs work by competitively binding to the ATP binding domain of the tyrosine kinase active site in protein products of oncogenes, such as KIT [28], ABL [27, 63], BCR-ABL [63], and PDGFRA [32]. In the presence of the drug, ATP cannot bind and activate the protein, and the protein is consequently sustained in an inactive conformation. While in the inactive state, the proteins cannot dimerize, and downstream enzymatic activity is ultimately blocked, which halts cell growth.
The tyrosine kinase inhibitor imatinib was originally developed to treat CML, a previously fatal disease. However, the five-year survival rate of this disease was 95% in 2008, and imatinib has extended the life expectancy of these patients from 3-5 years to 30 years [63, 64]. After being FDA approved for the treatment of CML, imatinib was approved as the first-line drug for metastatic and recurrent GIST. However, the majority of patients will develop secondary resistance within two years [65, 66]. Most secondary resistance has been established to be caused by a secondary mutation in KIT or PDGFRA, and these secondary mutations are in cis with the primary mutation without exception [21, 67-71]. In a Phase 2 study, imatinib was used to treat advanced GISTs. The results showed that two-thirds of patients with imatinib refractory had a secondary KIT mutation [72]. These mutations especially frequently occurred in exon 11-mutated GISTs, while wild-type GISTs were not the case. The secondary mutations cluster in the following 2 regions of the KIT kinase domain that were targeted by imatinib: the ATP-binding pocket, which is encoded by exons13 and 14 whose mutations directly hinder drug binding, and the activation loop, which is encoded by exons 17and 18 whose mutations make KIT oncoprotein more stable in the active conformation and interfere with TKI interation [67, 73-76]. Several secondary mutations are relatively common, including V654A [77], a point mutation found in KIT exon 13, which is involved with the KIT ATP-binding pocket, and D820A or various point mutations involving N882, which are located in exon 17 in the activation loop of the protein (Figure 1B). Sometimes, the so-called “gatekeeper” mutation, T670I, is also seen in exon 14, which, like V654A, is involved with the KIT ATP-binding pocket [78]. The secondary mutations V654A and T670I are intrinsically resistant to imatinib (Figure 1B). These secondary mutations are invariably found in cis with the primary mutation, but patients with primary resistance, progression within the first six months of treatment, to imatinib generally do not show secondary mutations. However, most patients who show secondary resistance (defined as progression after the first six months of treatment, typically within 2 years of treatment) will show secondary mutations. Primary mutations in exon 11 are more sensitive to TKI than those with exon 9 primary mutations; however, secondary resistance mutations are more frequent in patients with primary exon 11 mutations than in patients with primary exon 9 mutations. In terms of the effect of secondary mutations on the benefits of TKI treatment, patients with the KIT ATP-binding domain mutations (V654A and T670I) or with the activation loop mutations in exons 17 and 18 do worse on imatinib (Figure 1B). However, sunitinib effectively overcomes the ATP-binding pocket mutants, but not the activation loop mutants. Other drugs are proving to exert specific effects on different mutations in clinical trials. For example, sorafenib is showing remarkable clinical effects against the gatekeeper mutation T670I [79, 80]. Imatinib resistance was also identified in GISTs with PDGFRA mutations, and the major mutation is D842V mutation [74, 81]. Viable tumor cells are found in most patients who have had their tumors resected during imatinib therapy, suggesting that a certain level of drug resistance is inherent in most GISTs. Many secondary KIT mutations have been found at low frequencies, and mutations often exhibit inter-tumor or even intra-tumor heterogeneity in the same patient [69, 76, 82, 83]. This heterogeneity is unfavorable for the subsequent treatment of imatinib resistance.
Strategy to overcome resistance
Several approaches are available to combat imatinib resistance (Table 2). First, increasing the dose of imatinib is recommended, but the effect of this strategy is unfortunately limited. Presumably, imatinib resistance is caused by either inadequate drug concentrations , associated secondary KIT mutations or activation of signaling pathways that by pass KIT, and higher drug concentrations are thought to inhibit tumor growth in relative rather than absolute imatinib resistance [84]. In addition to the many new TKIs, including sunitinib and regorafenib, that are being developed, and other inhibitors that target GIST via mechanisms independent of KIT or PDGFRA are being developed [85-87].
Table 2: Novel therapies being tested for the treatment of GISTs
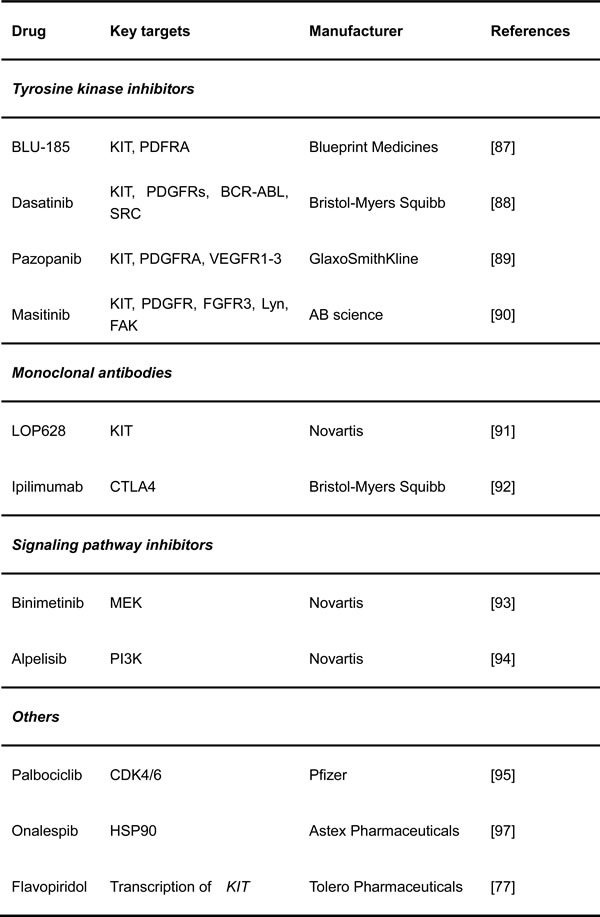
Tyrosine kinase inhibitors
BLU-285 (Blueprint Medicines, Cambridge, MA, USA) is an orally available agent that was developed to specifically target the mutant forms of KIT and PDGFRA. It potently and selectively inhibits exon 17-mutated KIT and the PDGFRA D842V mutant [88].
Dasatinib (Sprycel, Bristol-Myers Squibb, New York, NY, USA) is a BCR-ABL inhibitor approved as the first-line drug in patients with CML. The main targets of dasatinib are BCR-ABL, SRC, KIT, PDGFRs and several other tyrosine kinases. Combination with dasatinib plus imatinib or ipililumab has better efficiency for blockade of KIT signaling [89].
Pazopanib (Votrient, GlaxoSmithKline, Brentford, UK) is a broad-spectrum tyrosine kinase inhibitor that is efficacious against KIT, FGFR, PDGFR and VEGFR, as reported at the 2015 Annual Meeting of ASCO [90]
Masitinib (Masivet, AB Science, Paris, France) is an orally available TKI with more efficiency than imatinib against KIT exon 11-mutated and wild-type in GIST. It also effectively inhibits PDGFR and FGFR. The compound has already undergone several clinical trials to study its efficacy and safety and compare them to those of sunitinib. This study showed that masitinib prolonged survival and had a better safety profile than sunitinib, although the progression-free survival curves for these two drugs were similar. GIST patients with no evidence of disease after surgery but with a high potential of recurrence will be given with masitinib, and a phase 3 study is being planned to test masitinib in the adjuvant setting and evaluate its efficacy and safety in the first-line setting for GIST [91].
Monoclonal antibodies
LOP628 (Novartis, Cambridge, MA, USA) is a humanized monoclonal antibody targeting the KIT receptor and is linked to the cytotoxic agent maytansine, which inhibits the assembly of microtubules, molecules that are necessary for cell division. The mechanism of action of this “antibody-drug conjugate” requires two steps. First, the monoclonal antibody portion of LOP628 targets and binds to the cell surface receptor KIT that is expressed on all GIST cells. This process is followed by the internalization of the LOP628-KIT complex and binding of the maytansine portion of LOP628 to the tubulin molecules inside the cell, which inhibits cell division and tumor growth. The FDA has approved a similar concept to treat advanced breast cancer with HER2(+) using an anti-HER2 antibody (trastuzumab, Herceptin) conjugated to the cytotoxic maytansinoid DM1 (trastuzumab emtansine). This therapeutic concept has great potential, especially for the treatment of GIST, because most GISTs depend on the activation of KIT signaling, even when they are resistant to TKIs. LOP628 can only bind to KIT-expressing cells, but its activity is not inhibited by secondary resistance mutations.
Another monoclonal antibody is ipilimumab (BMS, New York, NY, USA) in combination with dasatinib. Ipilimumab binds cytotoxic T-lymphocyte-associated antigen-4 (CTLA4) directly, a protein receptor enriched on T cells that functions as an immune checkpoint, to downregulate the immune system. The inhibition of CTLA4 can re-activate the known-tumor activities of cytotoxic T cells, and augmenting the immune system with a CTLA4 inhibitor significantly enhanced the effect of imatinib treatment [92].
Signaling pathway inhibitors
Several trials are testing inhibitors to block downstream pathways of KIT in conjunction with imatinib. One prospective study is using the MEK inhibitor MEK162 (Binimetinib, Novartis, Cambridge, MA, USA), combining imatinib in advanced GISTs. This strategy is based on the discovery that MEK/MAPK signaling activates ETV1, a lineage-specific survival factor for GIST and its precursor, interstitial cells of Cajal. In preclinical studies, MEK162 synergistically destabilized ETV1 protein with imatinib and suppressed GIST formation and progression [93]. Another inhibitor, BYL-719 (Alpelisib, Novartis, Cambridge, MA, USA), is a selective inhibitor of the PI3K catalytic p110α subunit. Notably, neither compound significantly inhibited the downstream kinase mTOR, which is known to provide a negative feedback loop that reactivates the PI3K and the MAPK pathways [94].
Others
The efficacy and safety of Palbociclib (Ibrance, Pfizer, New York, NY, USA) for advanced GISTs who are refractory to imatinib and sunitinib are also being evaluated. PD-0332991 is an oral inhibitor of the CDK4 and CDK6, which are crucial promoters of cell division and often deregulated in cancer, including GIST. In fact, a publication by Bauer et al. showed that defects in the cell cycle are the most common aberrations in GIST after KIT/PDGFRA mutations.
AT13387 (Onalespib, Astex Pharmaceuticals, Pleasanton, CA, USA) is a KIT chaperone HSP90 inhibitor [23, 95], which has been shown to be effective against imatinib-resistant GIST [96].
Flavopiridol (Alvocidib, Tolero Pharmaceuticals, Lehi, UT, USA) can repress the transcription of KIT to downregulate the activity of pathways downstream of KIT.
The genotype of GISTs is very relevant to the response of the tumor to various drugs. Thus, more research is needed to identify mutations that endow resistance to a particular treatment and effective therapies for each unique genotype. As previously mentioned, imatinib-resistant tumors are highly heterogeneous. Thus, the use multiple agents to confront different secondary mutations is reasonable [97, 98]. Theoretically, a cocktail of inhibitors could suppress tumor growth by inhibiting multiple pathways. However, combining small-molecule drugs for synchronous treatment may be a challenge because most inhibitors can be metabolized through cytochrome P450 signaling pathways [99]. Therefore, the composition and effects of multiple agents should be explored further.
CONCLUSIONS
GISTs are common neoplasms, and so-called micro GISTs are found in ~20-30% of adults. Gain-of-function KIT or PDGFRA mutations are early genetic events (Figure 4), and most GISTs exhibit remarkable clinical responses to TKIs, including imatinib, sunitinib and regorafenib. The clinical success of TKIs in GIST serves as a paradigm for the molecularly targeted therapy of solid tumors. Unfortunately, even patients with dramatic initial clinical responses ultimately develop resistance to TKI therapy, resulting in disease progression. Genomic alterations contribute to tumorigenic progression in GIST, including cell cycle abnormalities due to CDKN2A or TP53 loss-of-function, dystrophin inactivation, 1p, 14q, 22q alterations and secondary KIT mutations (Figure 4). Further genomic screens pave the way to identify more drivers in GIST tumorgenesis that have a potential to be validated as a target, and novel insights into the genetic progression of GISTs are shedding new light on therapeutic innovations.
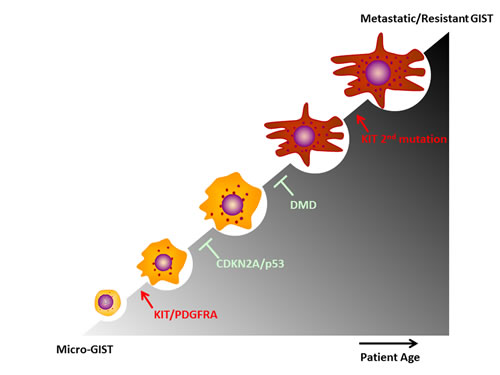
Figure 4: Major genomic alterations promote GIST progression.
Abbreviations
Aos = Antisense oligonucleotides; CDK4 and CDK6 = Cyclin dependent kinase 4 and 6; CDKN2A = Cyclin-dependent kinase inhibitor 2A; CML = Chronic myelogenous leukemia; CSF1R = Colony-stimulating-factor receptor; CTLA4 = Cytotoxic T-lymphocyte-associated antigen-4; ER = Estrogen receptor; FDA = Food and Drug Administration; FGFR = Fibroblast growth factor receptor; FLT3 = Fms-like tyrosine kinase 3; Fzd7 = Frizzled-7; GISTs = Gastrointestinal stromal tumors; HER2 = Human epidermal growth factor receptor 2; HR = Hazard ratio; HSP90 = Heat shock protein 90; ICC = Interstitial cells of Cajal; JNK = c-Jun N-terminal kinase; LMS = Leiomyosarcoma; LOH = Loss of heterozygosity; NF-κB = Nuclear factor kappa-light- chain-enhancer of activated B cells; PDGFRA and PDFGRB = Platelet-derived growth factor receptor α and β; PDN = Prednisolone; PR = Partial response; Rb = Retinoblastoma; RET = Rearranged during transfection; RMS = Rhabdomyosarcoma; SCF = Stem cell factor; SD = Stabilized disease; STAT3 = Signal transducer and activator of transcription 3; TKI = Tyrosine kinase inhibitor; TNF = Tumor necrosis factor; VEGFR = Vascular endothelial growth factor receptor.
ACKNOWLEDGMENTS
This work was supported by the 100-Talent Program of the Chinese Academy of Sciences (to Y.W.), the National Natural Science Foundation of China (81572642 to Y.W.), the Basic Research Project of Shanghai Science and Technology Commission (16JC1405600 to Y.W.), the Technology Foundation for Selected Overseas Chinese Scholar, the Ministry of Human Resources and Social Security, China (to Y.W.), the National Key R&D Program of China (2016YFC1302100), and the funds by the Chinese Academy of Sciences, Shanghai Changzheng Hospital and the Second Military Medical University.
CONFLICTS OF INTEREST
The authors declare that they have no conflict of interest.
REFERENCES
1. Muenst S, Thies S, Went P, Tornillo L, Bihl MP, Dirnhofer S. Frequency, phenotype, and genotype of minute gastrointestinal stromal tumors in the stomach: an autopsy study. Hum Pathol. 2011; 42:1849-1854.
2. Mazur MT, Clark HB. Gastric stromal tumors. Reappraisal of histogenesis. Am J Surg Pathol. 1983; 7:507-519.
3. Nishida T, Goto O, Raut CP, Yahagi N. Diagnostic and treatment strategy for small gastrointestinal stromal tumors. Cancer. 2016; 122:3110-3118.
4. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011; 11:865-878.
5. Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT)_ gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998; 152:11.
6. Kawanowa K, Sakuma Y, Sakurai S, Hishima T, Iwasaki Y, Saito K, Hosoya Y, Nakajima T, Funata N. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006; 37:1527-1535.
7. Abraham SC, Krasinskas AM, Hofstetter WL, Swisher SG, Wu TT. “Seedling” mesenchymal tumors (gastrointestinal stromal tumors and leiomyomas) are common incidental tumors of the esophagogastric junction. Am J Surg Pathol. 2007; 31:1629-1635.
8. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998; 279:577-580.
9. Subramanian S, West RB, Corless CL, Ou W, Rubin BP, Chu KM, Leung SY, Yuen ST, Zhu S, Hernandez-Boussard T, Montgomery K, Nielsen TO, Patel RM, et al. Gastrointestinal stromal tumors (GISTs) with KIT and PDGFRA mutations have distinct gene expression profiles. Oncogene. 2004; 23:7780-7790.
10. Yan L, Zou L, Zhao W, Wang Y, Liu B, Yao H, Yu H. Clinicopathological significance of c-KIT mutation in gastrointestinal stromal tumors: a systematic review and meta-analysis. Sci Rep. 2015; 5:13718.
11. Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD, Fletcher CD, Fletcher JA. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003; 299:708-710.
12. Taniguchi M, Nishida T, Hirota S, Isozaki K, Ito T, Nomura T, Matsuda H, Kitamura Y. Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res. 1999; 59:4297-4300.
13. Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988; 241:42-52.
14. Yuzawa S, Opatowsky Y, Zhang Z, Mandiyan V, Lax I, Schlessinger J. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell. 2007; 130:323-334.
15. Singer S, Rubin BP, Lux ML, Chen CJ, Demetri GD, Fletcher CD, Fletcher JA. Prognostic value of KIT mutation type, mitotic activity, and histologic subtype in gastrointestinal stromal tumors. J Clin Oncol. 2002; 20:3898-3905.
16. Tabone-Eglinger S, Subra F, El Sayadi H, Alberti L, Tabone E, Michot JP, Theou-Anton N, Lemoine A, Blay JY, Emile JF. KIT mutations induce intracellular retention and activation of an immature form of the KIT protein in gastrointestinal stromal tumors. Clin Cancer Res. 2008; 14:2285-2294.
17. Lasota J, Jasinski M, Sarlomo-Rikala M, Miettinen M. Mutations in exon 11 of c-kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol. 1999; 154:53-60.
18. Beghini A, Tibiletti MG, Roversi G, Chiaravalli AM, Serio G, Capella C, Larizza L. Germline mutation in the juxtamembrane domain of the kit gene in a family with gastrointestinal stromal tumors and urticaria pigmentosa. Cancer. 2001; 92:657-662.
19. Roskoski R, Jr. Structure and regulation of Kit protein-tyrosine kinase—the stem cell factor receptor. Biochem Biophys Res Commun. 2005; 338:1307-1315.
20. Lasota J, Corless CL, Heinrich MC, Debiec-Rychter M, Sciot R, Wardelmann E, Merkelbach-Bruse S, Schildhaus HU, Steigen SE, Stachura J, Wozniak A, Antonescu C, Daum O, et al. Clinicopathologic profile of gastrointestinal stromal tumors (GISTs) with primary KIT exon 13 or exon 17 mutations: a multicenter study on 54 cases. Mod Pathol. 2008; 21:476-484.
21. Kikuchi H, Miyazaki S, Setoguchi T, Hiramatsu Y, Ohta M, Kamiya K, Sakaguchi T, Konno H. Rapid relapse after resection of a sunitinib-resistant gastrointestinal stromal tumor harboring a secondary mutation in exon 13 of the c-KIT gene. Anticancer Res. 2012; 32:4105-4109.
22. Bauer S, Duensing A, Demetri GD, Fletcher JA. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene. 2007; 26:7560-7568.
23. Muhlenberg T, Zhang Y, Wagner AJ, Grabellus F, Bradner J, Taeger G, Lang H, Taguchi T, Schuler M, Fletcher JA, Bauer S. Inhibitors of deacetylases suppress oncogenic KIT signaling, acetylate HSP90, and induce apoptosis in gastrointestinal stromal tumors. Cancer Res. 2009; 69:6941-6950.
24. Liang L, Yan XE, Yin Y, Yun CH. Structural and biochemical studies of the PDGFRA kinase domain. Biochem Biophys Res Commun. 2016; 477:667-672.
25. Wasag B, Debiec-Rychter M, Pauwels P, Stul M, Vranckx H, Oosterom AV, Hagemeijer A, Sciot R. Differential expression of KIT/PDGFRA mutant isoforms in epithelioid and mixed variants of gastrointestinal stromal tumors depends predominantly on the tumor site. Mod Pathol. 2004; 17:889-894.
26. Perrone F, Tamborini E, Dagrada GP, Colombo F, Bonadiman L, Albertini V, Lagonigro MS, Gabanti E, Caramuta S, Greco A, Torre GD, Gronchi A, Pierotti MA, et al. 9p21 locus analysis in high-risk gastrointestinal stromal tumors characterized for c-kit and platelet-derived growth factor receptor alpha gene alterations. Cancer. 2005; 104:159-169.
27. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996; 2:561-566.
28. Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000; 96:925-932.
29. Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, Silberman S, Capdeville R, Dimitrijevic S, Druker B, Demetri GD. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001; 344:1052-1056.
30. Guerin A, Sasane M, Keir CH, Gauthier G, Macalalad AR, Wu EQ, Conley AP. Physician underestimation of the risk of gastrointestinal stromal tumor recurrence after resection. JAMA Oncol. 2015; 1:797-805.
31. Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002; 347:472-480.
32. Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, Shiraga S, Bainbridge T, Morich J, Heinrich MC. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005; 23:5357-5364.
33. Grimpen F, Yip D, McArthur G, Waring P, Goldstein D, Loughrey M, Beshay V, Chong G. Resistance to imatinib, low-grade FDG-avidity on PET, and acquired KIT exon 17 mutation in gastrointestinal stromal tumour. Lancet Oncol. 2005; 6:724-727.
34. Joensuu H, Martin-Broto J, Nishida T, Reichardt P, Schoffski P, Maki RG. Follow-up strategies for patients with gastrointestinal stromal tumour treated with or without adjuvant imatinib after surgery. Eur J Cancer. 2015; 51:1611-1617.
35. Chen LL, Trent JC, Wu EF, Fuller GN, Ramdas L, Zhang W, Raymond AK, Prieto VG, Oyedeji CO, Hunt KK, Pollock RE, Feig BW, Hayes KJ, et al. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res. 2004; 64:5913-5919.
36. Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006; 368:1329-1338.
37. Schroeder B, Li Z, Cranmer LD, Jones RL, Pollack SM. Targeting gastrointestinal stromal tumors: the role of regorafenib. Onco Targets Ther. 2016; 9:3009-3016.
38. Jakhetiya A, Garg PK, Prakash G, Sharma J, Pandey R, Pandey D. Targeted therapy of gastrointestinal stromal tumours. World J Gastrointest Surg. 2016; 8:345-352.
39. Rossi S, Sbaraglia M, Dell’Orto MC, Gasparotto D, Cacciatore M, Boscato E, Carraro V, Toffolatti L, Gallina G, Niero M, Pilozzi E, Mandolesi A, Sessa F, et al. Concomitant KIT/BRAF and PDGFRA/BRAF mutations are rare events in gastrointestinal stromal tumors. Oncotarget. 2016; 7:30109-30118. doi: 10.18632/oncotarget.8768.
40. George S, Wang Q, Heinrich MC, Corless CL, Zhu M, Butrynski JE, Morgan JA, Wagner AJ, Choy E, Tap WD, Yap JT, Van den Abbeele AD, Manola JB, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol. 2012; 30:2401-2407.
41. Schneider-Stock R, Boltze C, Lasota J, Miettinen M, Peters B, Pross M, Roessner A, Gunther T. High prognostic value of p16INK4 alterations in gastrointestinal stromal tumors. J Clin Oncol. 2003; 21:1688-1697.
42. Ricci R, Arena V, Castri F, Martini M, Maggiano N, Murazio M, Pacelli F, Potenza AE, Vecchio FM, Larocca LM. Role of p16/INK4a in gastrointestinal stromal tumor progression. Am J Clin Pathol. 2004; 122:35-43.
43. Schneider-Stock R, Boltze C, Lasota J, Peters B, Corless CL, Ruemmele P, Terracciano L, Pross M, Insabato L, Di Vizio D, Iesalnieks I, Dirnhofer S, Hartmann A, et al. Loss of p16 protein defines high-risk patients with gastrointestinal stromal tumors: a tissue microarray study. Clin Cancer Res. 2005; 11:638-645.
44. Cerski MR, Pereira F, Matte US, Oliveira FH, Crusius FL, Waengertner LE, Osvaldt A, Fornari F, Meurer L. Exon 11 mutations, Ki67, and p16(INK4A) as predictors of prognosis in patients with GIST. Pathol Res Pract. 2011; 207:701-706.
45. Feakins RM. The expression of p53 and bcl-2 in gastrointestinal stromal tumours is associated with anatomical site, and p53 expression is associated with grade and clinical outcome. Histopathology. 2005; 46:270-279.
46. Henze J, Muhlenberg T, Simon S, Grabellus F, Rubin B, Taeger G, Schuler M, Treckmann J, Debiec-Rychter M, Taguchi T, Fletcher JA, Bauer S. p53 modulation as a therapeutic strategy in gastrointestinal stromal tumors. PLoS One. 2012; 7:e37776.
47. Wang Y, Marino-Enriquez A, Bennett RR, Zhu M, Shen Y, Eilers G, Lee JC, Henze J, Fletcher BS, Gu Z, Fox EA, Antonescu CR, Fletcher CD, et al. Dystrophin is a tumor suppressor in human cancers with myogenic programs. Nat Genet. 2014; 46:601-606.
48. Adamo CM, Dai DF, Percival JM, Minami E, Willis MS, Patrucco E, Froehner SC, Beavo JA. Sildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2010; 107:19079-19083.
49. Wang Y, Fletcher JA. Cell cycle and dystrophin dysregulation in GIST. Cell Cycle. 2015; 14:2713-2714.
50. Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C, Guglieri M, Ashton E, Abbs S, Nihoyannopoulos P, Garralda ME, Rutherford M, McCulley C, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009; 8:918-928.
51. Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, Dickson G, Wood MJA, Wilton SD, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011; 378:595-605.
52. Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014; 345:1184-1188.
53. Tinsley JM, Fairclough RJ, Storer R, Wilkes FJ, Potter AC, Squire SE, Powell DS, Cozzoli A, Capogrosso RF, Lambert A, Wilson FX, Wren SP, De Luca A, Davies KE. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One. 2011; 6:e19189.
54. Gehrig SM, van der Poel C, Sayer TA, Schertzer JD, Henstridge DC, Church JE, Lamon S, Russell AP, Davies KE, Febbraio MA, Lynch GS. Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature. 2012; 484:394-398.
55. von Maltzahn J, Renaud JM, Parise G, Rudnicki MA. Wnt7a treatment ameliorates muscular dystrophy. Proc Natl Acad Sci U S A. 2012; 109:20614-20619.
56. El-Rifai W, Sarlomo-Rikala M, Andersson LC, Knuutila S, Miettinen M. DNA sequence copy number changes in gastrointestinal stromal tumors: tumor progression and prognostic significance. Cancer Res. 2000; 60:3899-3903.
57. Assamaki R, Sarlomo-Rikala M, Lopez-Guerrero JA, Lasota J, Andersson LC, Llombart-Bosch A, Miettinen M, Knuutila S. Array comparative genomic hybridization analysis of chromosomal imbalances and their target genes in gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2007; 46:564-576.
58. Debiec-Rychter M, Lasota J, Sarlomo-Rikala M, Kordek R, Miettinen M. Chromosomal aberrations in malignant gastrointestinal stromal tumors: correlation with c-KIT gene mutation. Cancer Genet Cytogenet. 2001; 128:24-30.
59. Debiec-Rychter M, Sciot R, Pauwels P, Schoenmakers E, Dal Cin P, Hagemeijer A. Molecular cytogenetic definition of three distinct chromosome arm 14q deletion intervals in gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2001; 32:26-32.
60. Breiner JA, Meis-Kindblom J, Kindblom LG, McComb E, Liu J, Nelson M, Bridge JA. Loss of 14q and 22q in gastrointestinal stromal tumors (pacemaker cell tumors). Cancer Genet Cytogenet. 2000; 120:111-116.
61. Yamamoto H, Tobo T, Nakamori M, Imamura M, Kojima A, Oda Y, Nakamura N, Takahira T, Yao T, Tsuneyoshi M. Neurofibromatosis type 1-related gastrointestinal stromal tumors: a special reference to loss of heterozygosity at 14q and 22q. J Cancer Res Clin Oncol. 2009; 135:791-798.
62. el-Rifai W, Sarlomo-Rikala M, Miettinen M, Knuutila S, Andersson LC. DNA copy number losses in chromosome 14: an early change in gastrointestinal stromal tumors. Cancer Res. 1996; 56:3230-3233.
63. Eide CA, Adrian LT, Tyner JW, Mac Partlin M, Anderson DJ, Wise SC, Smith BD, Petillo PA, Flynn DL, Deininger MW, O’Hare T, Druker BJ. The ABL switch control inhibitor DCC-2036 is active against the chronic myeloid leukemia mutant BCR-ABLT315I and exhibits a narrow resistance profile. Cancer Res. 2011; 71:3189-3195.
64. Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, Ryan CW, von Mehren M, Blanke CD, Rankin C, Benjamin RS, Bramwell VH, Demetri GD, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008; 26:5360-5367.
65. Lai S, Wang G, Cao X, Luo X, Wang G, Xia X, Hu J, Wang J. KIT over-expression by p55PIK-PI3K leads to imatinib-resistance in patients with gastrointestinal stromal tumors. Oncotarget. 2016; 7:1367-1379. doi: 10.18632/oncotarget.6011.
66. Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009; 15:7510-7518.
67. Wardelmann E, Merkelbach-Bruse S, Pauls K, Thomas N, Schildhaus HU, Heinicke T, Speidel N, Pietsch T, Buettner R, Pink D, Reichardt P, Hohenberger P. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006; 12:1743-1749.
68. Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, Town A, McKinley A, Ou WB, Fletcher JA, Fletcher CD, Huang X, Cohen DP, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008; 26:5352-5359.
69. Nishida T, Kanda T, Nishitani A, Takahashi T, Nakajima K, Ishikawa T, Hirota S. Secondary mutations in the kinase domain of the KIT gene are predominant in imatinib-resistant gastrointestinal stromal tumor. Cancer Sci. 2008; 99:799-804.
70. Wakai T, Kanda T, Hirota S, Ohashi A, Shirai Y, Hatakeyama K. Late resistance to imatinib therapy in a metastatic gastrointestinal stromal tumour is associated with a second KIT mutation. Br J Cancer. 2004; 90:2059-2061.
71. Mahadevan D, Cooke L, Riley C, Swart R, Simons B, Della Croce K, Wisner L, Iorio M, Shakalya K, Garewal H, Nagle R, Bearss D. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene. 2007; 26:3909-3919.
72. Antonescu CR, DeMatteo RP. CCR 20th anniversary commentary: a genetic mechanism of imatinib resistance in gastrointestinal stromal tumor-where are we a decade later? Clin Cancer Res. 2015; 21:3363-3365.
73. Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, Leversha MA, Jeffrey PD, Desantis D, Singer S, Brennan MF, Maki RG, DeMatteo RP. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005; 11:10.
74. Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, Eisenberg BL, von Mehren M, Fletcher CD, Sandau K, McDougall K, Ou WB, Chen CJ, Fletcher JA. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006; 24:4764-4774.
75. Agaram NP, Besmer P, Wong GC, Guo T, Socci ND, Maki RG, DeSantis D, Brennan MF, Singer S, DeMatteo RP, Antonescu CR. Pathologic and molecular heterogeneity in imatinib-stable or imatinib-responsive gastrointestinal stromal tumors. Clin Cancer Res. 2007; 13:170-181.
76. Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, Fletcher CD, Corless CL, Fletcher JA. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008; 216:64-74.
77. Roberts KG, Odell AF, Byrnes EM, Baleato RM, Griffith R, Lyons AB, Ashman LK. Resistance to c-KIT kinase inhibitors conferred by V654A mutation. Mol Cancer Ther. 2007; 6:1159-1166.
78. Serrano C, Wang Y, Marino-Enriquez A, Lee JC, Ravegnini G, Morgan JA, Bertagnolli MM, Beadling C, Demetri GD, Corless CL, Heinrich MC, Fletcher JA. KRAS and KIT gatekeeper mutations confer polyclonal primary imatinib resistance in GI stromal tumors: relevance of concomitant phosphatidylinositol 3-kinase/AKT dysregulation. J Clin Oncol. 2015; 33:e93-96.
79. Guo T, Agaram NP, Wong GC, Hom G, D’Adamo D, Maki RG, Schwartz GK, Veach D, Clarkson BD, Singer S, DeMatteo RP, Besmer P, Antonescu CR. Sorafenib inhibits the imatinib-resistant KITT670I gatekeeper mutation in gastrointestinal stromal tumor. Clin Cancer Res. 2007; 13:4874-4881.
80. Heinrich MC, Marino-Enriquez A, Presnell A, Donsky RS, Griffith DJ, McKinley A, Patterson J, Taguchi T, Liang CW, Fletcher JA. Sorafenib inhibits many kinase mutations associated with drug-resistant gastrointestinal stromal tumors. Mol Cancer Ther. 2012; 11:1770-1780.
81. Debiec-Rychter M, Cools J, Dumez H, Sciot R, Stul M, Mentens N, Vranckx H, Wasag B, Prenen H, Roesel J, Hagemeijer A, Van Oosterom A, Marynen P. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005; 128:270-279.
82. Loughrey MB, Waring PM, Dobrovic A, Demetri G, Kovalenko S, McArthur G. Polyclonal resistance in gastrointestinal stromal tumor treated with sequential kinase inhibitors. Clin Cancer Res. 2006; 12:6205-6206; author reply 6206-6207.
83. Wardelmann E, Thomas N, Merkelbach-Bruse S, Pauls K, Speidel N, Buttner R, Bihl H, Leutner CC, Heinicke T, Hohenberger P. Acquired resistance to imatinib in gastrointestinal stromal tumours caused by multiple KIT mutations. Lancet Oncol. 2005; 6:249-251.
84. Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, Issels R, van Oosterom A, Hogendoorn PCW, Van Glabbeke M, Bertulli R, Judson I. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004; 364:1127-1134.
85. Bauer S, Joensuu H. Emerging agents for the treatment of advanced, imatinib-resistant gastrointestinal stromal tumors: current status and future directions. Drugs. 2015; 75:1323-1334.
86. Cioffi A, Maki RG. GI stromal tumors: 15 years of lessons from a rare cancer. J Clin Oncol. 2015; 33:1849-1854.
87. Zhu JQ. Therapeutic targets in gastrointestinal stromal tumors. World J Transl Med. 2015; 4:25.
88. Evans EK, Hodous BL, Gardino AK, Davis A, Zhu J, Shutes A, Kim JL, Wilson KJ, Wilson D, Zhang Y, Chu T, Kohl NE, Kadambi V, et al. Abstract 791: BLU-285, the first selective inhibitor of PDGFR D842V and KIT exon 17 mutants. Cancer Res 2015; 75:791-791.
89. D’Angelo SP, Shoushtari AN, Keohan ML, Dickson MA, Gounder MM, Chi P, Loo JK, Gaffney L, Schneider L, Patel Z, Erinjeri JP, Bluth M, Sjoberg A, et al. Combined KIT and CTLA-4 blockade in patients with refractory GIST and other advanced sarcomas: a phase Ib study of dasatinib plus ipilimumab. Clin Cancer Res. 2016.
90. Ramaswamy A, Pande N, Shetty O, Shetty N, Gupta S, Ostwal V. Pazopanib in metastatic multiply treated progressive gastrointestinal stromal tumors: feasible and efficacious. J Gastrointest Oncol. 2016; 7:638-643.
91. Dubreuil P, Letard S, Ciufolini M, Gros L, Humbert M, Casteran N, Borge L, Hajem B, Lermet A, Sippl W, Voisset E, Arock M, Auclair C, et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS One. 2009; 4:e7258.
92. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, Sorenson EC, Popow R, Ariyan C, Rossi F, Besmer P, Guo T, Antonescu CR, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011; 17:1094-1100.
93. Ran L, Sirota I, Cao Z, Murphy D, Chen Y, Shukla S, Xie Y, Kaufmann MC, Gao D, Zhu S, Rossi F, Wongvipat J, Taguchi T, et al. Combined inhibition of MAP kinase and KIT signaling synergistically destabilizes ETV1 and suppresses GIST tumor growth. Cancer Discov. 2015; 5:304-315.
94. Massacesi C, Di Tomaso E, Urban P, Germa C, Quadt C, Trandafir L, Aimone P, Fretault N, Dharan B, Tavorath R, Hirawat S. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. Oncotargets Ther. 2016; 9:203-210.
95. Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 2006; 66:9153-9161.
96. Wagner AJ, Agulnik M, Heinrich MC, Mahadevan D, Riedel RF, von Mehren M, Trent J, Demetri GD, Corless CL, Yule M, Lyons JF, Oganesian A, Keer H. Dose-escalation study of a second-generation non-ansamycin HSP90 inhibitor, onalespib (AT13387), in combination with imatinib in patients with metastatic gastrointestinal stromal tumour. Eur J Cancer. 2016; 61:94-101.
97. Rutkowski P, Hompes D. Combined therapy of gastrointestinal stromal tumors. Surg Oncol Clin N Am. 2016; 25:735-759.
98. Saponara M, Pantaleo MA, Nannini M, Biasco G. Treatments for gastrointestinal stromal tumors that are resistant to standard therapies. Future Oncol. 2014; 10:2045-2059.
99. Schoffski P, Reichardt P, Blay JY, Dumez H, Morgan JA, Ray-Coquard I, Hollaender N, Jappe A, Demetri GD. A phase I-II study of everolimus (RAD001) in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors. Ann Oncol. 2010; 21:1990-1998.