
Introduction
Suicide gene therapy for cancer treatment, also referred to as gene-directed enzyme pro-drug therapy (GDEPT), aims at selectively targeting cancer cells as an alternative or complementary adjuvant to classical chemotherapies [1]. GDEPT is based on introducing into tumour cells a viral or a bacterial “suicide” gene encoding an enzyme able to activate a non- or mildly toxic pro-drug leading to tumour cell death. The suicide gene has to be selectively active in tumour cells and the pro-drug is delivered either by local or systemic administration and is only activated if the cells express the suicide gene. The most extensively studied suicide gene system is the combination of Herpes Simplex Virus Thymidine Kinase (HSV-TK) and the pro-drug Ganciclovir (GCV). HSV-TK has a high affinity for GCV and catalyses GCV mono-phosphorylation that is further converted into the di- and triphosphate derivatives by cellular kinases. DNA polymerase then incorporates GCV-triphosphate into replicating DNA leading to cell death by polymerase inhibition [2]. In a clinical setting, HSV-TK and GCV were first used to treat ovarian cancer and central nervous system malignancies via in situ transduction (for review see [3]). To be effective GDEPT approaches have to specifically target tumour cells.
The Long Interspersed Element-1 (LINE-1 or L1) retrotransposons make up 17% of the human genome [4]. The vast majority of L1s (approximately 500,000 L1 copies) are no longer mobile due to rearrangements, point mutations, or 5′-truncations [5]. Only around 100 members of the L1-Ta and pre-Ta subfamilies remain transposition-competent [6, 7] and are responsible for the bulk of on-going retro-transposition in humans (reviewed in [8-10]). L1 mobilization primarily occurs via target primed reverse transcription (TPRT), a process catalysed in cis by two proteins, ORF1p and ORF2p, translated from the bi-cistronic 6 kb L1 mRNA. The L1 ORF2p comprises endonuclease (EN) and reverse transcriptase (RT) activities essential for L1 retro-transposition and are also responsible for trans-mobilization of Alu and SVA retro-transposons [11-13]. Because of the potential harmful impact of L1 element mobility on genome integrity, their expression is held in check through a variety of genome defence mechanisms [14, 15] for reviews [16, 17]. However, L1 RNA expression has been shown in several adult tissues [18]. In contrast, expression of L1 ORF1/2 protein is not found in normal adult somatic cells, but is seen in many human cancers, including breast cancer [19], human bladder carcinoma, colon carcinoma, melanoma, and fibrosarcoma [20] that exhibit high levels of both L1 RNA and ORF1 protein [8, 9, 21-28]. Similarly, somatic insertions of L1 elements have been described in many cancers indicating that the expressed ORF machinery is functional. There exists little evidence for the presence of functional L1 ORF machinery in normal somatic cells, with only one report of weak ORF1 expression in normal esophagus and where somatic L1 insertions may take place early in the development of Barrett’s esopahgus disease [29].
Alu elements are the most abundant Short INterspersed Elements (SINEs), with over one million copies in the human genome [30]. Alu repeats compose greater than 10 % of the mass of the human genome. Full-length Alu elements are approximately 300 bp in length [4, 31]. Alu elements have no open reading frames, but use L1 ORF1p and ORF2p, for their mobility [11, 32]. Of the multiple Alu subfamilies, almost all of the recently integrated Alu elements within the human genome belong to one of several closely related “young” Alu subfamilies: Y, Yc1, Yc2, Ya5, Ya5a2, Ya8, Yb8, and Yb9 with the majority being Ya5 and Yb8 subfamily members [33-36].
It has been shown that endogenously expressed L1 ORF1/2p machinery can support exogenously expressed Alu retroposition [18, 37]. Here we take advantage of the selective expression of L1 ORF1/2 in many cancer cells to specifically express the HSV-TK suicide gene using an expression construct whose genomic integration is mediated by an Alu element. Treatment of HSV-TK-expressing cells with GCV efficiently blocks tumour cell proliferation and spheroid growth. Here we describe for the first time a strategy based on the tumour-specific L1 ORF1/2 expression as means of integrating a suicide gene and eliminating cancer cells that represents a new complement in the treatment of cancer.
Results
Designing and optimising a plasmid to integrate and express HSV-TK selectively in L1 ORF1/2 expressing cells using an Alu element
In order to establish a novel plasmid system to express a suicide gene selectively in cancer cells we designed a vector that is derived from a reporter plasmid used to detect Alu retro-transposition with the neoTet reporter gene that becomes functional only after a cycle of transcription, reverse transcription and integration (kindly provided by Dr T Heidmann [11]). The neoTet reporter gene with its own promoter is encoded on the negative strand and is rendered inactive by the presence of an autocatalytic Tetrahymena (Tet) intron that has to be spliced out of the transcribed RNA. This Tetrahymena group I intron can auto-splice and is thus independent from the spliceosome pathway [38]. We replaced the neomycin selection cassette by the HSV-TK gene that was human codon optimized and fused to bright monomer GFP [39] to monitor its expression (Figure 1A). Several different constructs were made to obtain maximal efficiency (Figure 1B). Two different “young” Alu elements, AluYa5 or AluYa8 [33] with internal Pol III promoters were inserted downstream of the enhancer of the Pol III-transcribed Alu-like 7SL RNA gene used in the original plasmid. As the efficiency of Tet self-splicing is dependent on the 5’ splice site delineated by the P1 helix formed by base pairing between the internal guide sequence (IGS) of the intron and the last six nucleotides of the 5’ exon [38], we selected two different positions (1 and 2) to introduce the Tet intron into the HSV-TK gene. These two positions differ by the HSV-TK-IGS pairing at P1 and P10 helixes (Supplemental Figure 1). We also optimised the poly(A) tail length since it has an essential role in L1 and Alu retro-transposition [37, 40-43]. Accordingly, we tried two poly(A) tails of 50 and 100 bases having the RNA polymerase III (Pol III) terminator signal very close in order to maximize activity [37]. To increase Pol II polymerase transcription of the integrated HSV-TK, we compared the CMV and SV40 promoters and we introduced an intron with splice donor and acceptors sites (Figure 1B).
Upon transfection of this vector, the insert is transcribed by Pol III to generate a transcript where the Tet intron is auto-spliced, the transcript is reverse transcribed and then reintegrated into the host genome via the action of the L1 ORF1/2 machinery and the Alu sequence after which Pol II generates a transcript encoding both HSV-TK and GFP (Figure 1A).
In total seven different vectors were generated and their efficiency was evaluated by transfection into HEK293-T cells that express the L1 ORF1p (see below), followed by measurement of the GFP signal. Quantification of the number of GFP expressing transfected cells showed that construct A5b comprising the AluYa8 element, the CMV promoter and a 100 base poly(A) tail was the most efficient (Figure 1C).
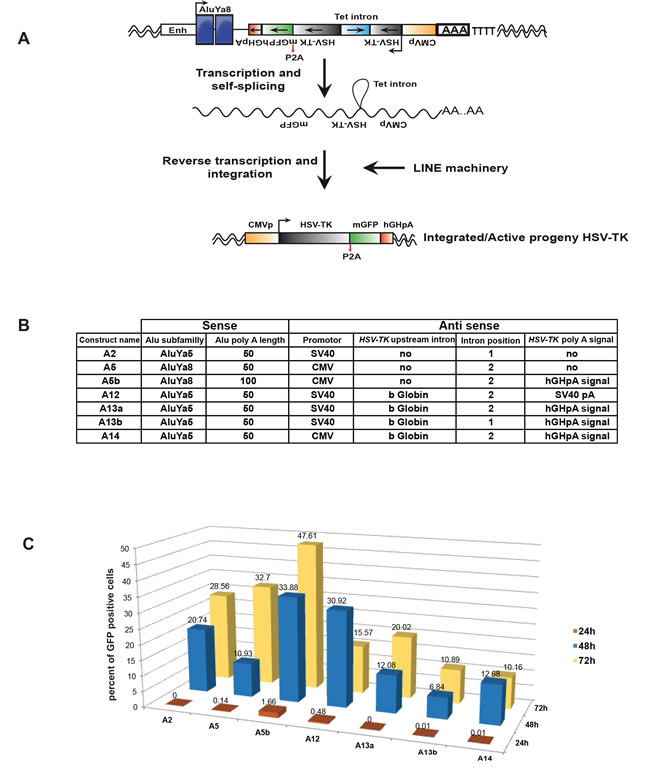
Figure 1: Design and optimisation of vectors. A. Schematic representation of vector design showing the AluYa8 element flanked with the HSV-TK gene (grey boxes) followed by a pA2 cleavage site and GFP (green box). Both HSV-TK and GFP are on the negative strand with the CMV promoter (yellow box). The HSV-TK coding sequence is interrupted by the presence of an autocatalytic Tetrahymena (Tet) intron (light blue box), which can be spliced out of the transposition RNA intermediate (middle). The predicted structure of the resulting de novo Alu insertion following splicing and reverse transcription is shown (bottom). B. Table summarizing the differences between the constructs. hGHpa is human growth hormone poly A. C. Histograms showing the percentage of DAPI stained cells with GFP signal for each construct. 5X104 HEK293T/17 cells were grown in a 12-well plate and transfected with 1µg DNA using FuGENE® (Promega) (DNA/FuGENE ratio 1/6). GFP signal was measured at 24, 48 and 72 hours post transfection using CellInsight, (Cellomics, Thermofisher) and the data were analysed using HCS Studio.
Selective targeting of L1 ORF 1/2 expressing cancer cells
We first screened several tumour cell lines for expression of L1 ORF1/2. Immunoblots with a novel ORF1p antibody showed potent ORF1p expression in HEK293-T cells, MCF-7 breast cancer cells, NCI-H1975 non-small cell lung cancer cells, and HT-29 colorectal adenocarcinoma cells (Figure 2A, note that no reliable and accessible ORF2p antibodies exist so its expression could not be tested in this way). In contrast, no ORF1p was detected in primary IMR-90 and WI-38 non-transformed lung fibroblasts that were used as negative control lines. As an additional control, we transfected a construct (A1) containing a Pol III termination signal within the HSV-TK gene resulting in a shortened transcript that encodes neither an intact HSV-TK, nor a poly(A) signal.
We transfected these cells twice within a 48-hour interval with the A1 and A5b constructs, added GCV 12 hours after the second transfection and counted the living cells immediately (as the base point (Day 0) as well as after 24 and 48 hours. In control A1 transfected MCF-7 cells, GCV addition slowed cell growth such that the number of cells increased only by 50% relative to day 0, whereas they increased by more than 250% in absence of GCV showing a previously reported low level of non-specific toxicity of the drug [44] (Figure 2B). In contrast, GCV treatment of the A5b-transfected cells led to a potent reduction in cell numbers to less than 50% of the starting population. Similar potent reductions in the numbers of viable cells were seen in A5b transfected and GCV treated HT-29 and NCI-H1975 cells. Importantly, no comparable reduction in cell numbers was seen using the IMR-90 and WI-38 cells that did not express L1 ORF1p (Figure 2A and 2B). These data demonstrate that transfection with A5b and GCV treatment can selectively eliminate L1 ORF1p-expressing tumour cells.
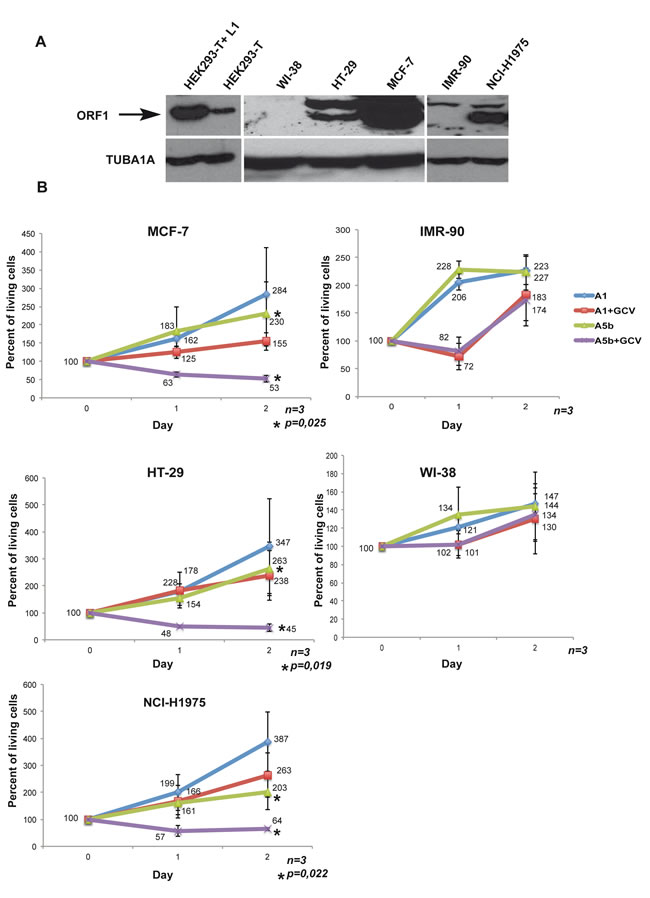
Figure 2: Selective targeting the growth of L1 ORF1/2 expressing cancer cells. A. Immunoblots showing recombinant ORF1p expression in HEK293-T cells transfected with an L1 expression vector and endogenous ORF1 protein in HEK293-T cells, primary WI-38 and IMR-90 fibroblasts and the HT-29, MCF-7 and NCI-H1975 tumour cells. TUBA1A is used as a loading control. B. All indicated cell lines were seeded into a 6-well plate transfected with 4 µg per dish of the A5b or A1 vectors. Transfection was carried out using Lipofectamine 2000® (DNA/lipofectamine 1/3) for HT-29 and FuGENE® (DNA/FuGENE 1/6) for the other lines. After 48 hours, cells were re-transfected and 24 hours later treated with GCV (Sigma-Aldrich) at 10 µg/ml. Living cells were counted at the time of GCV addition (Day 0 =100%), and then at day 1 and day 2. All experiments were performed in triplicate and the data are expressed as mean ± s.d. with a pvalue determined by Student’s t-test.
To demonstrate that HSV-TK integration and subsequent GCV-induced cell death were really mediated by the enzymatic activities encoded by the L1 ORF1/2 proteins and thus that L1 expression was the major determinant of sensitivity to our suicide gene approach, we expressed exogenous wild-type ORF1/2 or a version mutated in the reverse transcription (RT) function of ORF2 (D702A) [45] in transformed cells that expressed endogenous L1 ORF1/2 or primary WI-38 cells that did not express endogenous ORF1/2. These constructs or a GFP vector as a negative control were transfected in HeLa or HEK293T transformed cells that we chose as they expressed only low endogenous levels of ORF1 and in primary WI-38 fibroblasts that do not express ORF1 (Figure 3A). After transfection, strong expression of ORF1 was seen in the HeLa or HEK293T cells, while it accumulated to lower, but readily detectable levels in WI-38 cells (Figure 3A). Notably the mutant protein accumulated to lower levels than wild-type in each case.
Cells were co-transfected with these vectors together with the A5b vector with or without GCV treatment. In the absence of any exogenous L1 ORF1/2 the HeLa and HEK293T cells were specifically sensitive to GCV as seen above with the other transformed L1-expressing cells (Figure 3B). Expression of wild-type, but not mutant ORF1/2 led to a mild increase in the proliferation of these cells and also in the sensitivity to GCV treatment such that the overall effect was stronger than with the GFP or mutant ORF1/2 transfected cells (Figure 3B). Strikingly, expression of wild-type ORF1/2 sensitised the WI-38 cells to the effect of GCV. These cells that were normally insensitive to GCV after A5b transfection became sensitive in the presence of wild-type but not mutant ORF1/2.
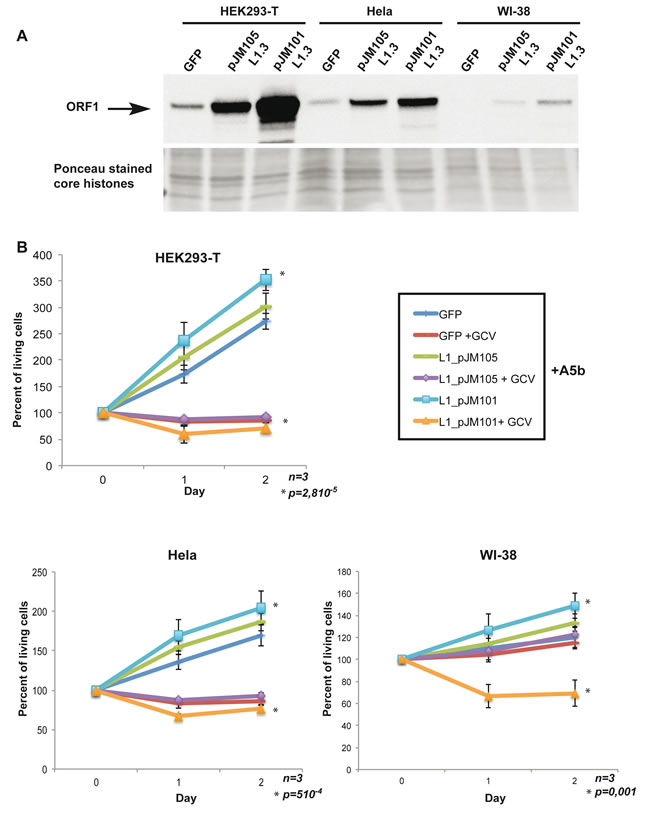
Figure 3: Expression of exogenous wild-type L1 ORF1/2 is necessary and sufficient to confer sensitivity to GCV. A. Immunoblots showing recombinant ORF1p expression in HEK293-T, HeLa and primary WI-38 cells transfected with a control GFP expression vector or expression vector expressing wild-type ORF1/2p (pJM101L1.3) or wild type ORF1p and a mutated version of ORF2p (D702A) (pJM105L1.3) [45]. Ponceau staining is used as a loading control. B. All indicated cell lines were seeded into a 24-well plate transfected with 0.8 µg per well of A5b vector, 24 hours later cells were transfected by triplicate with 0.8 µg/well of GFP, pJM105 and pJM101vectors, 24 hours later GCV (Sigma-Aldrich) were added at 10 µg/ml. Transfection was carried out using FuGENE® (Promega) (DNA/FuGENE 1/6) for HEK293-T cells, X-tremeGENE9® (Sigma-Aldrich) (DNA/X-tremeGENE9 1/4) and Lipofectamine 3000® (Invitrogen) (DNA/lipofectamine 1/3) for WI-38. Living cells were counted as described above. All experiments were performed in triplicate and the data are expressed as mean ± s.d. with a pvalue determined by Student’s t-test.
These data show that expression of exogenous wild-type, but not mutant ORF1/2, in transformed cells expressing endogenous ORF1/2 enhanced their sensitivity to GCV treatment after transfection with the A5b vector. More importantly, expression of wild-type, but not mutant L1 ORF1/2, in WI-38 cells conferred sensitivity to GCV after A5b transfection. Thus, we show that expression of wild-type L1 ORF1/2 is necessary and sufficient to render primary cells sensitive to targeting by the A5b vector and that sensitivity required the L1 encoded RT activity.
Growth of cancer cells as 3D-spheroids can be used to assess their tumour initiating capacity (for reviews see [46, 47]). To test the ability of our strategy to block spheroid growth, cells were transfected as described above as monolayers before culture as 3D spheroids. Alternatively, we introduced the A5b and A1 constructs into Adeno-Associated Virus vectors (AAVs) that were then used to infect cells seeded as monolayers before culture as 3D spheres. Cells were subsequently cultured under non-adherent conditions in presence or absence of GCV and their growth was observed over time. The NCI-H1975, WI-38 and IMR-90 cells did not readily form 3D spheroids in these experiments and these experiments were therefore restricted to the MCF-7 and HT-29 cell lines with the appropriate A1 transfected/infected cells as negative controls.
MCF-7 spheroids were grown in agarose-coated 96-well plates under conditions where a single spheroid grows in each well. In the absence of GCV, A5b or A1 infected MCF-7 cells formed large spheroids that grew from the agarose surface. GCV treatment of the A1 infected cells led to a reduction in spheroid size, whereas treatment of the A5b infected cells led to a potent inhibition of spheroid growth by 14 days (Figure 4A). Spheroids were re-infected with the AAVs and allowed to grow for a further 14 days. Large spheroids persisted in control A1 infected cells with or without GCV treatment and in the A5b infected cells in absence of GCV. In contrast, spheroid growth was completely abolished in the GCV-treated A5b infected cells (Figure 4B).
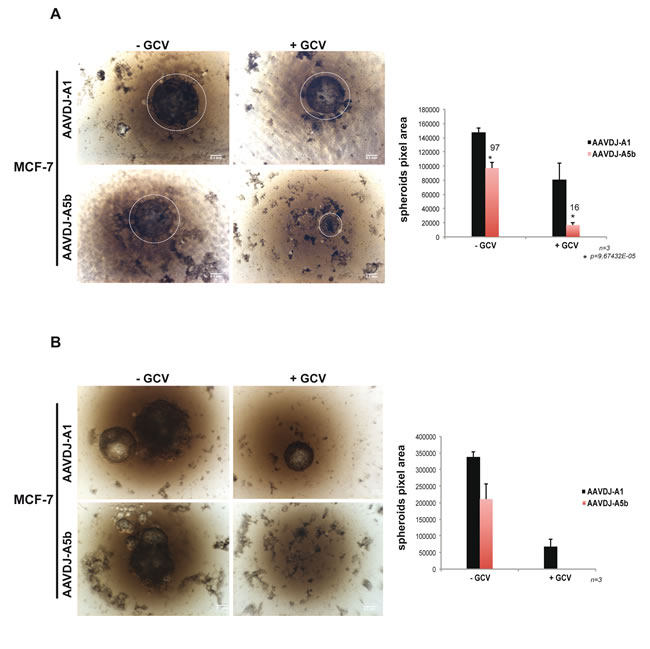
Figure 4: Targeting spheroid growth of MCF-7 cells. A.-B. MCF-7 cells were infected with recombinant A5b and A1-containing AAVs. The A5b and A1 inserts were cloned in pAAVDJ (Cell Biolabs). After two weeks of GCV treatment and double infection (see materiel and methods), spheroid growth was analysed using a bright field Macroscope (Leica M420) (Panel A). The pixel area (right) of each spheroid was determined using Image j (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997-2016). Fresh medium was added 1 week after reinfection and spheroids were grown for a further 7 days and imaged a second time (Panel B). All experiments were performed in triplicate and the data are expressed as mean ± s.d. with a pvalue determined by Student’s t-test. Scale bars. 100 µm.
In a parallel approach HT-29 cells were transfected and cultured in bacterial petri dishes where they formed multiple free-floating spheroids. After 7 days, the number and size of the spheroids derived from the A1 transfected cells was mildly reduced (30%) by GCV treatment, however this effect was much more potent in the spheroids derived from the A5b transfected cells with an 80% reduction in their number with respect to the absence of GCV (Figure 5A). HT-29 cells were also infected with the AAVs and grown on agarose-coated wells. After 14 days of culture, HT-29 cells formed large aggregated spheroid masses (Figure 5B). GCV treatment reduced spheroid size by 65% in A5b infected cells, compared to 10% in the A1 cells. Additionally HT-29 cells were also grown in agarose-coated 96 well plates under conditions were a single spheroid is produced per well. After 1 month, spheroid growth was completely abolished in the A5b infected cells (Figure 5C). As with the MCF-7 cells, after two serial AAV infections over a one month period, spheroid growth was completely inhibited only in cells infected with the A5b AAV and GCV treatment. Thus, spheroid growth of both of these cell lines can be completely abrogated using a combination of A5b-expressed HSV-TK and GCV.
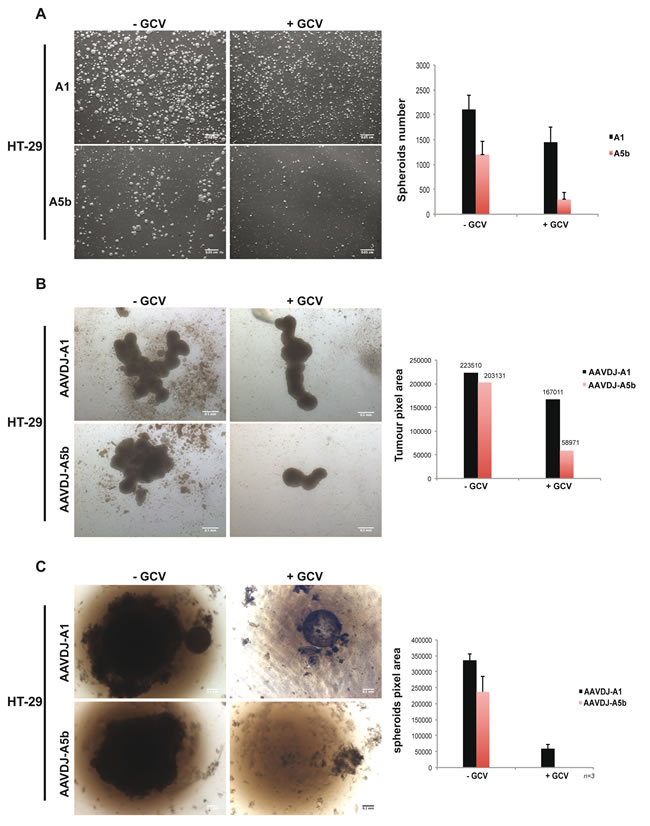
Figure 5: Targeting spheroid growth of HT-29 cells. A. HT-29 cells were transfected twice as described above and 48 hours post transfection, 106 cells were cultured in bacterial petri dishes, with or without GCV. One-week later, spheroids were imaged and counted. Scale bar; 50 µm. B. 105 HT-29 cells were transferred onto agarose-coated 12-well plates and infected at 107 MOI and GCV was added after 4 hours. Two weeks later spheroids were imaged and measured. Scale bar; 50 µm. C. Similar to MCF-7 cells above, HT-29 cells were grown as single spheroids in a 96 well plate with 2 cycles of infection and spheroids were imaged and measured after 1 month of growth. Scale bar; 50 µm.
Discussion
Here we describe a novel strategy for efficient and selective expression of the HSV-TK suicide gene in cancer cells based on their L1 ORF1/2p expression. We show that transfection with Alu-driven vectors or infection with the corresponding AAVs renders the tumour cells sensitive to efficient GCV mediated growth inhibition both as monolayers and spheroid cultures. We show that L1 ORF1/2 expression conferred GCV sensitivity to primary WI-38 and that this sensitivity was dependent on ORF2 RT-activity. These observations highlight the specificity of our strategy for selectively targeting cancer cells based on their reactivation of L1 expression and expression of functional ORF1/2 machinery.
Based on our data we suggest a new selective and non-toxic method for cancer treatment. Given the clinical experience of AAVs already used for human gene therapy [48], our strategy has potential to be adapted for use as an adjuvant to targeted inhibitor or immunotherapy for human cancers. Furthermore, the strategy could be further adapted by replacing the HSV-TK gene with other suicide genes or genes that mark cancer cells for elimination by the immune system.
Materials and Methods
Vectors
pJM101L1.3 and pJM105L1.3 have been described elsewhere [45].
A2, A5, A5b, A12, A13a, A13b and A14 were designed in silico using SnapGene software (from GSL Biotech; available at snapgene.com) and were synthetized in Genscript®. Sequences and maps are available upon request.
ORF1 antibody
Monoclonal anti-ORF1 was prepared as following; ORF1 protein fused at its N-terminus to 6XHistidine was expressed in E.coli and purified using Ni-NTA agarose (Qiagen). Mice were injected with purified recombinant ORF1 and monoclonal antibody production and screening were performed as previously described [49].
Cell lines
MCF-7 cells were grown in DMEM (1g/l glucose) supplemented with 10% foetal calf serum, insulin 0.6 µg/ml and gentamicin 40 µg/ml. HT-29 cells were grown in DMEM (4,5g/l glucose) supplemented with 10% foetal calf serum and gentamicin 40 µg/ml. NCI-H1975 cells were grown in RPMI (2,5g/l glucose), HEPES 10mM supplemented with 10% foetal calf serum, Na Pyruvate 1mM and gentamicin 40 µg/ml. WI-38 cells (purchased from the ATCC repository in 2015) were grown in MEM (Life Technologies) supplemented with 10% foetal calf serum, AANE 0,1 mM and gentamicin 40 µg/ml. IMR-90 cells (purchased from the ATCC repository in 2015) were grown in MEM w/Earle’s salts (Invitrogen) supplemented with 15% foetal calf serum, AANE 0,1 mM and gentamicin 40 µg/ml. MCF-7, HT-29 and NCI-H1975 cells were authentified by LGC July 2015. All cell lines are mycoplasma free (Venorgem mycoplasma PCR detection kit). HEK293T cells used for AAV production were grown in DMEM (4,5g/l Glutamax-I) supplemented with 10% foetal calf serum and gentamicin 40 µg/ml.
Immunostaining
HEK293T cells were seeded on coverslips in a cell culture dish 96 hours after transfection with A5b or after co-transfection with HSV-TK and GFP expression vectors as positive controls. Cells were fixed for 10 minutes with 4% paraformaldehyde, washed with PBS and permeabilized with 1% Triton X-100 in PBS for 15 minutes, followed by washing with PBS and incubation with polyclonal anti-HSV-TK (1:2000 dilution) anti-serum at 4°C overnight. Next day, cells were washed with PBS and incubated with Alexa Fluor 555 conjugated-anti-rabbit IgG (1: 500 dilution; Invitrogen) for 1 hour, and nuclei stained with DAPI (Invitrogen, Life Technologies) for 2 minutes. The slides were embedded in Vectashield and fluorescence was captured by confocal microscopy (Leica Sp5 Laser Scanning Confocal Microscope, GE).
AAV production
AAVDJ-A5b/A1 production was performed by the triple transfection method in HEK293T cells [50].
Spheroid formation and infection
5X104 MCF-7 cells were plated in a 24-well plate and immediately after plating, the cells were infected with AAVDJ-A5b or AAVDJ-A1 at a multiplicity of infection (MOI) of 107 viral genomes per cell. At 48 hours post infection, the cells were trypsinized and grown as spheroids using a procedure adapted from [51] . 200µl of a cell suspension containing 7500 cells were transferred into one well of Agarose-coated 96-well plate (1.5% agarose in DMEM wt/vol), after 4 hours GCV was added and one week later, each spheroid was re-infected with 5x108 viral particles.
Author contributions
MC and AF performed the experiments, AF RS and ID analysed data, AF and ID wrote the paper, AF conceived and designed the experiments.
Acknowledgments
We thank: T. Hiedmann for the NeoTet plasmid and J.L Garcia-Perez for the polyclonal ORF1p serum and the plasmids expressing wild-type and mutant ORF1/2, the IGBMC mono and polyclonal antibody facilities for generation of the ORF1 and HSV-TK antibodies. The strategy described here is covered by European Patent Application number EP 14 182 697 Synthetic Alu-Retrotransposon Vectors for Gene Therapy’ filed on August 28, 2014.
Conflicts of interest
The authors declare no conflict of interest.
Funding
This work was supported by grants from Connectus Alsace and the ANR programme Investissements d’Avenir ANR-10-LABX-0030-INRT. ID is ‘équipe labellisée’ of the Ligue Nationale contre le Cancer.
The RS laboratory is supported by the Helmholtz Gesellschaft and INSERM Plan Cancer (épigénetique et cancer).
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Naldini L. Gene therapy returns to centre stage. Nature. 2015; 526:351-360.
2. Fillat C, Carrio M, Cascante A, Sangro B. Suicide gene therapy mediated by the Herpes Simplex virus thymidine kinase gene/Ganciclovir system: fifteen years of application. Curr Gene Ther. 2003; 3:13-26.
3. Altaner C. Prodrug cancer gene therapy. Cancer Lett. 2008; 270:191-201.
4. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, et al. Initial sequencing and analysis of the human genome. Nature. 2001; 409:860-921.
5. Grimaldi G, Skowronski J, Singer MF. Defining the beginning and end of KpnI family segments. EMBO J. 1984; 3:1753-1759.
6. Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH Jr. Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci U S A. 2003; 100:5280-5285.
7. Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, Badge RM, Moran JV. LINE-1 retrotransposition activity in human genomes. Cell. 2010; 141:1159-1170.
8. Hancks DC, Kazazian HH Jr. Roles for retrotransposon insertions in human disease. Mob DNA. 2016; 7:9.
9. Richardson SR, Doucet AJ, Kopera HC, Moldovan JB, Garcia-Perez JL, Moran JV. The Influence of LINE-1 and SINE Retrotransposons on Mammalian Genomes. Microbiol Spectr. 2015; 3:MDNA3-0061-2014.
10. Beck CR, Garcia-Perez JL, Badge RM, Moran JV. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet. 2011; 12:187-215.
11. Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nat Genet. 2003; 35:41-48.
12. Hancks DC, Goodier JL, Mandal PK, Cheung LE, Kazazian HH Jr. Retrotransposition of marked SVA elements by human L1s in cultured cells. Hum Mol Genet. 2011; 20:3386-3400.
13. Raiz J, Damert A, Chira S, Held U, Klawitter S, Hamdorf M, Lower J, Stratling WH, Lower R, Schumann GG. The non-autonomous retrotransposon SVA is trans-mobilized by the human LINE-1 protein machinery. Nucleic Acids Res. 2012; 40:1666-1683.
14. Garcia-Perez JL, Morell M, Scheys JO, Kulpa DA, Morell S, Carter CC, Hammer GD, Collins KL, O’Shea KS, Menendez P, Moran JV. Epigenetic silencing of engineered L1 retrotransposition events in human embryonic carcinoma cells. Nature. 2010; 466:769-773.
15. Yang N, Kazazian HH Jr. L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat Struct Mol Biol. 2006; 13:763-771.
16. Kemp JR, Longworth MS. Crossing the LINE Toward Genomic Instability: LINE-1 Retrotransposition in Cancer. Front Chem. 2015; 3:68.
17. Elbarbary RA, Lucas BA, Maquat LE. Retrotransposons as regulators of gene expression. Science. 2016; 351:aac7247.
18. Belancio VP, Roy-Engel AM, Pochampally RR, Deininger P. Somatic expression of LINE-1 elements in human tissues. Nucleic Acids Res. 2010; 38:3909-3922.
19. Harris CR, Normart R, Yang Q, Stevenson E, Haffty BG, Ganesan S, Cordon-Cardo C, Levine AJ, Tang LH. Association of nuclear localization of a long interspersed nuclear element-1 protein in breast tumors with poor prognostic outcomes. GenesCancer. 2010; 1:115-124. doi: 10.1177/1947601909360812.
20. Nangia-Makker P, Sarvis R, Visscher DW, Bailey-Penrod J, Raz A, Sarkar FH. Galectin-3 and L1 retrotransposons in human breast carcinomas. Breast Cancer Res Treat. 1998; 49:171-183.
21. Ewing AD, Gacita A, Wood LD, Ma F, Xing D, Kim MS, Manda SS, Abril G, Pereira G, Makohon-Moore A, Looijenga LH, Gillis AJ, Hruban RH, et al. Widespread somatic L1 retrotransposition occurs early during gastrointestinal cancer evolution. Genome Res. 2015; 25:1536-1545.
22. Tubio JM, Li Y, Ju YS, Martincorena I, Cooke SL, Tojo M, Gundem G, Pipinikas CP, Zamora J, Raine K, Menzies A, Roman-Garcia P, Fullam A, et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 2014; 345:1251343.
23. Helman E, Lawrence MS, Stewart C, Sougnez C, Getz G, Meyerson M. Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res. 2014; 24:1053-1063.
24. Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ 3rd, Lohr JG, Harris CC, Ding L, Wilson RK, Wheeler DA, Gibbs RA, Kucherlapati R, et al. Landscape of somatic retrotransposition in human cancers. Science. 2012; 337:967-971.
25. Miki Y, Nishisho I, Horii A, Miyoshi Y, Utsunomiya J, Kinzler KW, Vogelstein B, Nakamura Y. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992; 52:643-645.
26. Rodic N, Steranka JP, Makohon-Moore A, Moyer A, Shen P, Sharma R, Kohutek ZA, Huang CR, Ahn D, Mita P, Taylor MS, Barker NJ, Hruban RH, et al. Retrotransposon insertions in the clonal evolution of pancreatic ductal adenocarcinoma. Nat Med. 2015; 21:1060-1064.
27. Shukla R, Upton KR, Munoz-Lopez M, Gerhardt DJ, Fisher ME, Nguyen T, Brennan PM, Baillie JK, Collino A, Ghisletti S, Sinha S, Iannelli F, Radaelli E, et al. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell. 2013; 153:101-111.
28. Solyom S, Ewing AD, Rahrmann EP, Doucet T, Nelson HH, Burns MB, Harris RS, Sigmon DF, Casella A, Erlanger B, Wheelan S, Upton KR, Shukla R, et al. Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res. 2012; 22:2328-2338.
29. Doucet-O’Hare TT, Rodic N, Sharma R, Darbari I, Abril G, Choi JA, Young Ahn J, Cheng Y, Anders RA, Burns KH, Meltzer SJ, Kazazian HH Jr. LINE-1 expression and retrotransposition in Barrett’s esophagus and esophageal carcinoma. Proc Natl Acad Sci U S A. 2015; 112:E4894-4900.
30. Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Genet Dev. 1999; 9:657-663.
31. Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002; 3:370-379.
32. Boeke JD. LINEs and Alus—the polyA connection. Nat Genet. 1997; 16:6-7.
33. Batzer MA, Kilroy GE, Richard PE, Shaikh TH, Desselle TD, Hoppens CL, Deininger PL. Structure and variability of recently inserted Alu family members. Nucleic Acids Res. 1990; 18:6793-6798.
34. Batzer MA, Rubin CM, Hellmann-Blumberg U, Alegria-Hartman M, Leeflang EP, Stern JD, Bazan HA, Shaikh TH, Deininger PL, Schmid CW. Dispersion and insertion polymorphism in two small subfamilies of recently amplified human Alu repeats. J Mol Biol. 1995; 247:418-427.
35. Deininger PL, Batzer MA. Alu repeats and human disease. Mol Genet Metab. 1999; 67:183-193.
36. Roy AM, Carroll ML, Nguyen SV, Salem AH, Oldridge M, Wilkie AO, Batzer MA, Deininger PL. Potential gene conversion and source genes for recently integrated Alu elements. Genome Res. 2000; 10:1485-1495.
37. Comeaux MS, Roy-Engel AM, Hedges DJ, Deininger PL. Diverse cis factors controlling Alu retrotransposition: what causes Alu elements to die? Genome Res. 2009; 19:545-555.
38. Esnault C, Casella JF, Heidmann T. A Tetrahymena thermophila ribozyme-based indicator gene to detect transposition of marked retroelements in mammalian cells. Nucleic Acids Res. 2002; 30:e49.
39. Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, Davidson MW, Wang J. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods. 2013; 10:407-409.
40. Dewannieux M, Heidmann T. Role of poly(A) tail length in Alu retrotransposition. Genomics. 2005; 86:378-381.
41. Ahl V, Keller H, Schmidt S, Weichenrieder O. Retrotransposition and Crystal Structure of an Alu RNP in the Ribosome-Stalling Conformation. Mol Cell. 2015; 60:715-727.
42. Kopera HC, Larson PA, Moldovan JB, Richardson SR, Liu Y, Moran JV. LINE-1 Cultured Cell Retrotransposition Assay. Methods Mol Biol. 2016; 1400:139-156.
43. Roy-Engel AM, Salem AH, Oyeniran OO, Deininger L, Hedges DJ, Kilroy GE, Batzer MA, Deininger PL. Active Alu element “A-tails”: size does matter. Genome Res. 2002; 12:1333-1344.
44. Black ME, Kokoris MS, Sabo P. Herpes simplex virus-1 thymidine kinase mutants created by semi-random sequence mutagenesis improve prodrug-mediated tumor cell killing. Cancer Res. 2001; 61:3022-3026.
45. Wei W, Gilbert N, Ooi SL, Lawler JF, Ostertag EM, Kazazian HH, Boeke JD, Moran JV. Human L1 retrotransposition: cis preference versus trans complementation. Mol Cell Biol. 2001; 21:1429-1439.
46. Zanoni M, Piccinini F, Arienti C, Zamagni A, Santi S, Polico R, Bevilacqua A, Tesei A. 3D tumor spheroid models for in vitro therapeutic screening: a systematic approach to enhance the biological relevance of data obtained. Sci Rep. 2016; 6:19103.
47. Weiswald LB, Bellet D, Dangles-Marie V. Spherical cancer models in tumor biology. Neoplasia. 2015; 17:1-15.
48. Samulski RJ, Muzyczka N. AAV-Mediated Gene Therapy for Research and Therapeutic Purposes. Annu Rev Virol. 2014; 1:427-451.
49. Brou C, Chaudhary S, Davidson I, Lutz Y, Wu J, Egly JM, Tora L, Chambon P. Distinct TFIID complexes mediate the effect of different transcriptional activators. EMBO J. 1993; 12:489-499.
50. Grieger JC, Choi VW, Samulski RJ. Production and characterization of adeno-associated viral vectors. Nat Protoc. 2006; 1:1412-1428.
51. Friedrich J, Seidel C, Ebner R, Kunz-Schughart LA. Spheroid-based drug screen: considerations and practical approach. Nat Protoc. 2009; 4:309-324.