
Haematological malignancies
Haematological malignancies (HMs) are tumours characterized by uncontrolled proliferation of cells from the immune and the haematopoietic system. In Europe, recent analyses show an age-standardized incidence rate of 32 tumours per 100.000 persons/year [1]. These heterogeneous pathologies are nowadays classified based on the affected cell lineage as myeloid or lymphoid. Neoplasias of lymphoid origin are the most frequently observed (75% of the total HMs) with multiple myeloma (MM), small B-cell lymphocytic lymphoma (SBLL) / chronic lymphatic leukaemia (CLL), diffuse large B-cell lymphoma (DLBCL) and Hodgkin lymphoma (HL) being the most common. Acute myeloid leukaemia (AML), myeloproliferative neoplasms (MPN) and myelodysplastic syndromes (MDS) account for the highest rate among the myeloid malignancies [2, 3]. Despite the differences in biology, clinical manifestations and outcome between disease subtypes, an overall significant progress in terms of diagnosis and cure rates has been achieved in the past decades. To date, the majority of paediatric acute lymphatic leukaemia (ALL) and chronic myeloid leukaemia (CML) cases are indeed cured or well controlled. Overall survival at 5 years is over 60% and around 80% in patients with non-Hodgkin and Hodgkin lymphoma, respectively [4, 5]. In contrast, CLL and AML have a high risk of relapse, and MM remains mostly incurable [6, 7, 8]. There is compelling evidence that a small population of stem-like cancer cells with the capacity for self-renewal and differentiation accounts for resistance and recurrence in many types of cancer [8, 9]. In the context of HMs, these cells are known as leukaemia-, lymphoma- or myeloma-initiating cells (from now on referred to as haematological cancer stem cells, HCSCs). Although there is some controversy on the phenotype of such a population(s), HCSCs are certainly involved in the initiation and maintenance of HMs [10-14].
Cancer research has been mostly focused on cancer cells themselves. However, it is now well accepted that tumours are complex tissues sustained by the dynamic interactions between cancer cells and their environment. This environment consists of a number of cell types (fibroblasts, endothelial cells, adipocytes, macrophages, antigen-presenting cells, etc) and many different molecules (growth factors, cytokines, chemokines, extracellular matrix, adhesion molecules, etc), which are commonly referred to as the tumour stroma [15]. Forty years ago, studies performed in the laboratory of Dr John Trentin demonstrated that stromal cells had an active role supporting haematopoiesis [16]. Furthermore, experimental evidence has demonstrated the interaction between stroma and haematologic cancer cells (HCCs) [17] (Figure 1). This is especially relevant in the case of HCSCs that house in niches contributing to a pro-tumourigenic environment [18, 19]. Therefore, the key challenges of HMs are to decipher in detail who participates and how in these complex networks that synergize to promote cancer progression.
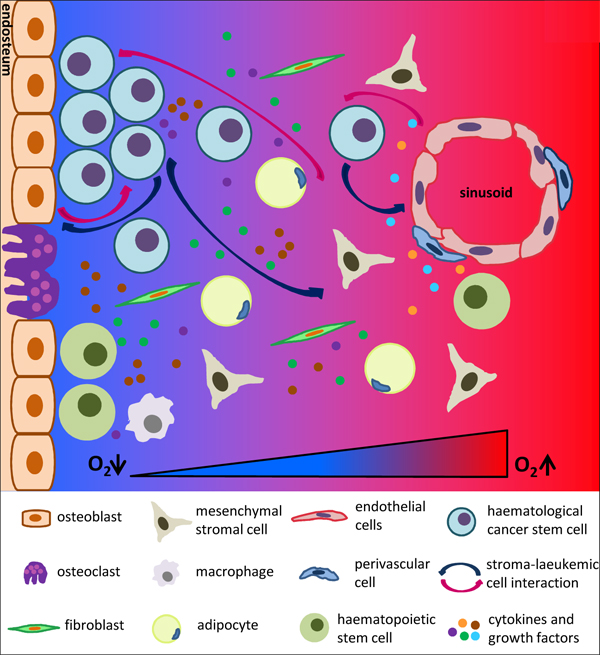
Figure 1: Picture of normal haematopoietic stem cells (HSCs) and haematological cancer stem /initiating cells (HCSCs) niches in the bone marrow. Both HSCs and HCSCs niches are composed of a collection of different cell types, growth factors and cytokines, which are localized close to the endosteum and sinusoids. Oxygen levels decrease from the sinusoids to the endosteum. HCSCs expand within the hypoxic endosteal niche while take advantage of the vascular niche to colonize distant organs.
Hypoxia in HMs and the stromal compartment
Hypoxia is, by definition, a state of reduced oxygenation that influences biological functions [20]. Since the initial indication made by Thomlinson and Gray, it is well established that hypoxia is a characteristic feature of solid tumours [21]. Unlike the normal tissue vasculature, the primitive and chaotic tumour neovasculature is unable to meet all the oxygen and nutrients demands. Nevertheless, this poor and hostile milieu drives cancer cell survival, cancer stem cell maintenance, metabolic reprogramming, angiogenesis and modulation of immune response, so that tumour hypoxia is associated with aggressiveness, metastatic spread and relapse [22].
Compared to most healthy tissues, the bone marrow (BM) environment is characterized by low oxygen availability. A relatively low level of oxygen is indeed a hallmark of the BM stem cell niche, and hypoxia induces the secretion of several growth factors and cytokines such as SDF-1/CXCL12 (stromal cell-derived factor), VEGF (vascular endothelial growth factor) and interleukin-6 involved in haematopoietic stem cells (HSCs) maintenance [23-27]. In vitro studies have shown that myelomatous BM environment is more hypoxic than the normal BM [28]. Jensen et al. remarked an increase in BM hypoxia during disease progression using a rat AML model [29]. Moreover, using pimonidazole staining for measuring hypoxia levels, Konopleva et al. elegantly demonstrated the high prevalence of hypoxia in human leukaemic BM [30]. Therefore, hypoxia certainly affects the different components of BM and modulates the highly complex and strictly regulated interactions between cancer and stromal cells. Hypoxia sustained AML- and CML-initiating cell maintenance, although contradictory effects on HCCs proliferation have been reported [29, 31-39]. In addition, neoangiogenesis and increased release of hypoxia-induced angiogenic cytokines such as VEGF, bFGF (basic-fibroblast growth factor) and angiopoietin 1 & 2 have been reported in AML and childhood ALL patients [40-42]. In vitro and in vivo studies have shown that angiogenic factors favour MM tumour development and lymphoma progression [43-53]. Hypoxia induces metabolic changes, enhances survival, reduces differentiation and promotes self-renewal of mesenchymal/stromal cells [54-56]. Furthermore, co-culture with these cells in hypoxia promotes maintenance and expansion of normal HSCs and human AML cells [39, 56, 57]. Finally, the poorly oxygenated niche and the hypoxia-induced glycololytic metabolism have been linked to chemoresistance in B-ALL, T-ALL, AML, lymphoma and MM cases [58-76].
HIF, the master hypoxia-signalling mediator: implications in HMs and the stromal compartment
The hypoxia-inducible transcription factors (HIFs) are central regulators of the cellular response to hypoxia [77]. HIF is a heterodimer composed of one of three oxygen-regulated HIF-α subunits (HIF-1α, HIF-2α and HIF-3α) and the constitutively expressed HIF-β subunit [78, 79]. HIF-1α and HIF-2α, also known as endothelial PAS protein (EPAS1), are the major activators of hypoxia-induced gene transcription, but, to date, little is known about expression and function of HIF-3α [80]. HIF-α proteins share similar structural domains such as an N-terminal basic helix-loop-helix (bHLH) domain involved in DNA binding, two Per-ARNT-Sim (PAS) domains allowing dimerisation, an oxygen-dependent degradation domain (ODDD), and the transactivation domain (TAD). While HIF-3α contains only one TAD, HIF-1α and HIF-2α contain an N-terminal (NTAD) and a C-terminal (CTAD) transactivation domain for recruitment of transcriptional coactivators [81]. In well-oxygenated cells, HIF-α subunits are hydroxylated by the family of prolyl hydroxylase domain-containing proteins (PHDs) on two conserved proline residues (Pro402 and Pro564 in the Human HIF-1α sequence) within the ODDD [82]. The hydroxylated motif allows the binding of the von Hippel-Lindau (VHL) protein, which mediates HIF-α ubiquitination and the further targeting to the proteasome for degradation [83]. While the PHD family consists of four PHDs, PHD1, 2 and 3 have been characterized much more extensively than PHD4, which is bound to the membrane of the reticulum endoplasmic [84-86]. Moreover, PHD2 has been described to be the main PHD controlling HIF-1α stability and levels in normoxia [87]. PHDs act as intracellular molecular sensors that use O2 as a substrate, and thus, their activity is compromised upon hypoxia [88, 89]. Thereby, HIF-α evades PHD/pVHL-mediated degradation, dimerises with HIF-1β and the HIF complex binds to specific HIF-response elements (HRE) of target genes. The oxygen-dependent hydroxylation of an asparagine residue in the CTAD of HIF-α (Asn803 in the Human HIF-1α sequence) by Factor Inhibiting HIF (FIH) negatively regulates HIF-target gene expression by impairing the recruitment of the co-activators CBP/p300 [90].
In addition to hypoxia, a number of studies have reported HIF-α sustained protein expression independently of oxygen availability. Growth factors and cytokines such as EGF (epidermal growth factor), FGF-2, heregulin, insulin, IGF1&2 (insulin-like growth factor 1 and 2), IL-1β, TNF-α (tumour necrosis factor α) and factors specifically involved in haematopoiesis such as SCF (stem cell factor) and thrombopoietin positively regulate HIF activity [91-98]. In addition to loss of function mutations in von Hippel-Lindau (VHL) and PTEN, or gain of function mutations in Myc, Ras and Raf [99-102], prevalent mutations found in HMs also promote HIF expression and activity. Hence, activating mutations of FLT3 (Fms-like tyrosine kinase 3), recognized as the most common molecular abnormality in AML [103], increase HIF-α accumulation via the PI3K/AKT/mTOR pathway [104, 105]. Bcr/Abl, an oncoprotein present in most CML cases but also found in ALL and AML patients, induces HIF-1α similarly to FLT3 [106]. Src, another proto-oncogen with a relevant role in HMs, activates HIF through the NADPH oxidase/Rac pathway [107]. NPM (nucleophosmin or nucleolar phosphoprotein B23), which is mutated and chromosomally translocated in many HMs, stimulates HIF activity by inactivating p14ARF [108, 109]. Moreover, contradictory results have been observed regarding IDH mutations and HIF-α accumulation [110-112].
HIF drives the transcription of genes involved in many pathways promoting angiogenesis and vascular remodelling, proliferation, survival and invasion of cancer cells and stem cell maintenance [113] (Figure 2). Overall, increased HIF-α expression is correlated with tumour growth and therapy resistance and, therefore, with disease relapse [114, 115]. Accordingly, sustained expression of HIF-α is a marker of poor prognosis not only in several types of solid tumours but also in HMs [116-129]. Therefore, inhibition of HIF-α (either by RNAi or small molecules) resulted in a failure of primary cells to form in vitro colonies and significantly increased disease-free survival in vivo [36, 118, 130-136]. However, the specific role of HIFs in HMs seems to be controversial and contradictory data have also been published. Thus HIF-1α has been reported to induce cell differentiation in AML, and loss of HIF-1α resulted in faster development of the disease and reduced survival [137-142]. Similarly, HIF-1α overexpression associated with increased survival in patients with diffuse large B-cell lymphoma [143]. Such an apparent controversy could be explained by the different system used (mouse versus human), the unspecificity of shRNA/drugs compared to knock-out models or the hypoxic exposure. Therefore, further studies will be certainly needed to clarify the oncogenic and/or tumour suppressor activity of HIF signalling in HMs, and particularly within HCSCs.
Homing and subsequent adhesion of cancer stem cells to the vascular and the endosteal niche triggers the in vivo tumour-stroma interactions. The chemokine receptor CXCR4 (C-X-C chemokine receptor type 4) and its ligand SDF-1/CXCL12 mediate this process and it is well known that HIF-1α regulates the expression of both [144, 145]. Moreover, the involvement of this axis in chemoresistance has been deeply demonstrated in HMs [146-151]. In this regard, it has been recently published that HIF-1α increases the interaction of CLL cells with the stroma and that stromal cells protect mantle cell lymphoma cells from the cytotoxic effect of chemotherapeutic agents [152, 153]. Furthermore, aberrant expression of HIF-1α in bone marrow endothelial cells has been linked to drug resistance and recurrence in patients with MM [126].
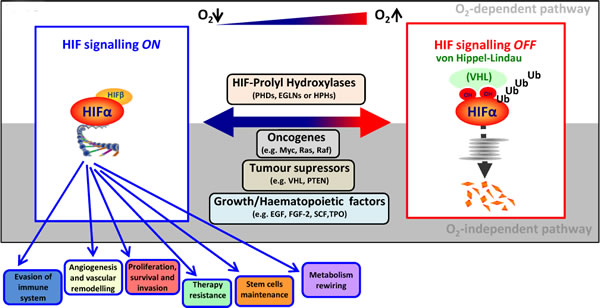
Figure 2: O2-dependent and -independent HIF signalling pathway. In well-oxygenated cells, the hydroxylation of HIF-α catalyzed by PHDs triggers VHL-mediated ubiquitination and subsequent degradation into the proteasome. In contrast, low oxygen availability stabilizes HIF-α by compromising PHD activity. Moreover, activation of oncogenes, loss of tumour suppressors, growth factors, specific prevalent mutations found in HMs and factors involved in haematopoiesis are also able to upregulate HIF-α independently of oxygen availability. Once stabilized, HIF-α translocates to the nucleus, binds to HIF-β and regulates the expression of genes promoting tumour progression.
Hypoxia and/or HIFs as targets to treat HMs
All the previously reported data emphasize the relevance of fighting against hypoxia and HIF signalling in HMs. Not only HCCs but also the stromal compartment should be targeted to fully frustrate the pro-tumourigenic environment promoted by their interaction. The therapeutic strategy aiming to directly target hypoxic cells within tumours remains a challenging approach, while success in clinical trials has so far proved elusive [154]. This approach relies mostly on the use of hypoxia-activated prodrugs (HAPs) or bioreductive drugs. The prodrugs are inactive medications that require metabolization before exhibiting pharmacological effects. In particular, HAPs require activation by oxygen-inhibited enzymes (typically by 1 or 2 electron oxidoreductases) to generate cytotoxic compounds [154]. One of the most extensively examined HAPs is evofosfamide (TH-302), the reductive activation of which generates bromo-isophosphoramide mustard (Br-IMP), a potent alkylating agent. TH-302 exhibited specific hypoxia-dependent cytotoxicity when tested in primary ALL and AML samples in vitro and reduced the AML stem cell pool in vivo [155, 156]. Similarly, TH-302 induced cell cycle arrest and triggered apoptosis in severely hypoxic conditions in MM, while had no effect at similar doses in normoxic conditions [38]. A phase I/II clinical trial in relapsed or refractory ALL or AML has been carried out using PR-104, which also results in the generation of a DNA-damaging metabolite. In this study, PR-104 demonstrated measurable clinical activity but also significant toxicity at the doses administered in the trial [30]. It is worth nothing that novel HAPs designed to release targeted therapeutics (pioneered by TH-4000 that releases EGFR tyrosine kinase inhibitor) have been recently developed, though to our knowledge no data related to HMs are available [154].
Regarding the use of drugs directly targeting HIF, several chemical inhibitors have been tested in different models. Echinomycin (NSC-13502) is an antibiotic derivative from the quinoxaline family, which inhibits HIF-1α/DNA binding activity. This inhibitor has been previously evaluated in clinical trials in solid tumours, though with disappointing results [157]. Interestingly, echinomycin abrogated in vitro and in vivo lymphoma and AML growth through preferential targeting of HCSCs [132, 133, 158]. HIF-1α inhibition by 2-methoxyestradiol (2ME2), and endogenous metabolite of oestrogen that disrupt microtubule architecture, and YC-1(3-(5’hydroxymethyl-2’-furyl)-1-benzy-lindazole) induced cell death in different HMs [159]. L-ascorbic acid was also able to specifically inhibit the proliferation of human CML cells via downregulation of HIF-1α transcription [135]. EZN-2968, a small 3rd generation antisense oligonucleotide against HIF1A mRNA, delayed acute promyelocytic leukaemia (APL) and MM progression [160, 161]. EZN-2968 has also been reported to block the interaction between MM cells and BM stromal cells through HIF-1α inhibition [161]. Furthermore, the combination of EZN-2088, a polyethylene glycol conjugate of irinotecan (PEG-SN38), with all-trans retinoic acid (ATRA) synergized to eradicate preclinical models of PML-RARα (promyelocytic leukaemia protein-retinoic receptor antagonist alpha) and PLZF (promyelocytic leukaemia zinc finger)-RARα-driven leukaemia [160]. More recently, it has been shown that chetomin, a small molecule able to disrupt HIF-1α binding to the p300 coactivator, exhibited antitumor activity in primary MM cells from patients [162]. Similarly, acriflavine, another FDA-approved HIF inhibitor, has demonstrated specificity towards CML stem cells [163]. Moreover, it should be pointed out the relevance of bortezomib (PS-341), a proteasome inhibitor, in the treatment of MM and mantle cell lymphoma patients. Relapse/refractory but also newly diagnosed cases benefit of bortezomib either as single agent or combined with other therapies [164, 165]. Indeed, bortezomib has been reported to repress HIF-1 (and not HIF-2)-dependent transcriptional activity by reinforcing FIH-mediated inhibition of p300 recruitment [166]. Supporting combinatorial therapeutic options, TH-302 together with bortezomib induced MM cell cycle arrest and triggered apoptosis in severe hypoxic conditions, while having no effect at similar doses in normoxic conditions [167]. More recently, PT2385 has been developed as a selective agent that blocks HIF-2α with potent anti-cancer activity in preclinical models of advanced clear cell renal cell carcinoma (ccRCC) [168-170]. However, this inhibitor has not been still tested for the treatment of HMs. Finally, it is worth mentioning that several agents in current clinical practice to treat HMs directly inhibit HIF-α, which might contribute to their therapeutic efficacy. This is indeed the case for imatinib or the topoisomerase I inhibitor, topotecan. Furthermore, the potential impact of rituximab on HIF1α expression levels might argue its favorable prognostic value in patients with DLBCL treated with this monoclonal antibody [123].
Conclusion
Data reported from several research laboratories claim that hypoxia and HIF-mediated signalling favour haematologic and lymphoid tumour progression and relapse. The results using hypoxia-activated prodrugs and HIF-α inhibitors in different preclinical and clinical models are really promising. Hence, these data give exciting perspectives to define new and better therapeutic approaches that may benefit patients suffering from HMs.
ACKNOWLEDGMENTS
The authors would like to thank all the laboratory members for discussions and comments. We apologize to the many research groups whose work was not cited due to space constraints.
CONFLICTs OF INTEREST
The authors declare that they have no conflict of interests.
FUNDING
Our research is supported by the Basque Department of Industry, Tourism and Trade (Etortek) and the MINECO grants. MI has been a recipient of a Lady Tata Memorial Trust postdoctoral fellowship.
REFERENCES
1. Sant M, Allemani C, Tereanu C, De Angelis R, Capocaccia R, Visser O, Marcos-Gragera R, Maynadié M, Simonetti A, Lutz JM, Berrino F, HAEMACARE Working Group. Incidence of hematologic malignancies in Europe by morphologic subtype: results of the HAEMACARE project. Blood. 2010; 116:3724–34.
2. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127:2391–405.
3. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, Jaffe ES. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016; 127:2375–90.
4. Barzenje DA, Holte H, Fosså A, Ghanima W, Liestøl K, Delabie J, Kolstad A. Long-term outcome for patients with early stage marginal zone lymphoma and mantle cell lymphoma. Leuk Lymphoma. 2017; 58:623–32.
5. Son CH, Chiu BC, Koshy M. Patterns of care and survival outcomes examining radiation therapy for advanced Hodgkin lymphoma. Leuk Lymphoma. 2017; 58:343–47.
6. Freireich EJ, Wiernik PH, Steensma DP. The leukemias: a half-century of discovery. J Clin Oncol. 2014; 32:3463–69.
7. Döhner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015; 373:1136–52.
8. Franqui-Machin R, Wendlandt EB, Janz S, Zhan F, Tricot G. Cancer stem cells are the cause of drug resistance in multiple myeloma: fact or fiction? Oncotarget. 2015; 6:40496–506. doi: 10.18632/oncotarget.5800.
9. Clappier E, Gerby B, Sigaux F, Delord M, Touzri F, Hernandez L, Ballerini P, Baruchel A, Pflumio F, Soulier J. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J Exp Med. 2011; 208:653–61.
10. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997; 3:730–37.
11. Martinez-Climent JA, Fontan L, Gascoyne RD, Siebert R, Prosper F. Lymphoma stem cells: enough evidence to support their existence? Haematologica. 2010; 95:293–302.
12. Kim SJ. Lymphoma stem cells: A step toward a new therapeutic target. Korean J Hematol. 2011; 46:211–13.
13. Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, Tanhehco Y, Smith BD, Civin CI, Jones RJ. Characterization of clonogenic multiple myeloma cells. Blood. 2004; 103:2332–36.
14. Kim D, Park CY, Medeiros BC, Weissman IL. CD19-CD45 low/- CD38 high/CD138+ plasma cells enrich for human tumorigenic myeloma cells. Leukemia. 2012; 26:2530–37.
15. Li H, Fan X, Houghton J. Tumor microenvironment: the role of the tumor stroma in cancer. J Cell Biochem. 2007; 101:805–15.
16. Trentin JJ. Determination of bone marrow stem cell differentiation by stromal hemopoietic inductive microenvironments (HIM). Am J Pathol. 1971; 65:621–28.
17. Schepers K, Campbell TB, Passegué E. Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell. 2015; 16:254–67.
18. Lane SW, Scadden DT, Gilliland DG. The leukemic stem cell niche: current concepts and therapeutic opportunities. Blood. 2009; 114:1150–57.
19. Basak GW, Srivastava AS, Malhotra R, Carrier E. Multiple myeloma bone marrow niche. Curr Pharm Biotechnol. 2009; 10:345–46.
20. Semenza GL. Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med. 2010; 2:336–61.
21. Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer. 1955; 9:539–49.
22. Wigerup C, Påhlman S, Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther. 2016; 164:152–69.
23. Ivanovic Z. Hypoxia or in situ normoxia: the stem cell paradigm. J Cell Physiol. 2009; 219:271–75.
24. Jing D, Wobus M, Poitz DM, Bornhäuser M, Ehninger G, Ordemann R. Oxygen tension plays a critical role in the hematopoietic microenvironment in vitro. Haematologica. 2012; 97:331–39.
25. Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, Sanchez-Martin M, Tsirigos A, Littman DR, Ferrando AA, Morrison SJ, Fooksman DR, Aifantis I, Schwab SR. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell. 2015; 27:755–68.
26. Narendran A, Ganjavi H, Morson N, Connor A, Barlow JW, Keystone E, Malkin D, Freedman MH. Mutant p53 in bone marrow stromal cells increases VEGF expression and supports leukemia cell growth. Exp Hematol. 2003; 31:693–701.
27. Chauhan D, Uchiyama H, Urashima M, Yamamoto K, Anderson KC. Regulation of interleukin 6 in multiple myeloma and bone marrow stromal cells. Stem Cells. 1995; 13 Suppl 2:35-9.
28. Asosingh K, De Raeve H, de Ridder M, Storme GA, Willems A, Van Riet I, Van Camp B, Vanderkerken K. Role of the hypoxic bone marrow microenvironment in 5T2MM murine myeloma tumor progression. Haematologica. 2005; 90:810–17.
29. Jensen PO, Mortensen BT, Hodgkiss RJ, Iversen PO, Christensen IJ, Helledie N, Larsen JK. Increased cellular hypoxia and reduced proliferation of both normal and leukaemic cells during progression of acute myeloid leukaemia in rats. Cell Prolif. 2000; 33:381–95.
30. Konopleva M, Thall PF, Yi CA, Borthakur G, Coveler A, Bueso-Ramos C, Benito J, Konoplev S, Gu Y, Ravandi F, Jabbour E, Faderl S, Thomas D, et al. Phase I/II study of the hypoxia-activated prodrug PR104 in refractory/relapsed acute myeloid leukemia and acute lymphoblastic leukemia. Haematologica. 2015; 100:927–34.
31. Desplat V, Faucher JL, Mahon FX, Dello Sbarba P, Praloran V, Ivanovic Z. Hypoxia modifies proliferation and differentiation of CD34(+) CML cells. Stem Cells. 2002; 20:347–54.
32. Giuntoli S, Rovida E, Barbetti V, Cipolleschi MG, Olivotto M, Dello Sbarba P. Hypoxia suppresses BCR/Abl and selects imatinib-insensitive progenitors within clonal CML populations. Leukemia. 2006; 20:1291–93.
33. Giuntoli S, Rovida E, Gozzini A, Barbetti V, Cipolleschi MG, Olivotto M, Dello Sbarba P. Severe hypoxia defines heterogeneity and selects highly immature progenitors within clonal erythroleukemia cells. Stem Cells. 2007; 25:1119–25.
34. Giuntoli S, Tanturli M, Di Gesualdo F, Barbetti V, Rovida E, Dello Sbarba P. Glucose availability in hypoxia regulates the selection of chronic myeloid leukemia progenitor subsets with different resistance to imatinib-mesylate. Haematologica. 2011; 96:204–12.
35. Drolle H, Wagner M, Vasold J, Kütt A, Deniffel C, Sotlar K, Sironi S, Herold T, Rieger C, Fiegl M. Hypoxia regulates proliferation of acute myeloid leukemia and sensitivity against chemotherapy. Leuk Res. 2015; 39:779–85.
36. Zou J, Li P, Lu F, Liu N, Dai J, Ye J, Qu X, Sun X, Ma D, Park J, Ji C. Notch1 is required for hypoxia-induced proliferation, invasion and chemoresistance of T-cell acute lymphoblastic leukemia cells. J Hematol Oncol. 2013; 6:3.
37. Muz B, de la Puente P, Azab F, Ghobrial IM, Azab AK. Hypoxia promotes dissemination and colonization in new bone marrow niches in Waldenström macroglobulinemia. Mol Cancer Res. 2015; 13:263–72.
38. Hu J, Handisides DR, Van Valckenborgh E, De Raeve H, Menu E, Vande Broek I, Liu Q, Sun JD, Van Camp B, Hart CP, Vanderkerken K. Targeting the multiple myeloma hypoxic niche with TH-302, a hypoxia-activated prodrug. Blood. 2010; 116:1524–27.
39. Griessinger E, Anjos-Afonso F, Pizzitola I, Rouault-Pierre K, Vargaftig J, Taussig D, Gribben J, Lassailly F, Bonnet D. A niche-like culture system allowing the maintenance of primary human acute myeloid leukemia-initiating cells: a new tool to decipher their chemoresistance and self-renewal mechanisms. Stem Cells Transl Med. 2014; 3:520–29.
40. Hussong JW, Rodgers GM, Shami PJ. Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood. 2000; 95:309–13.
41. Koomagi R, Zintl F, Sauerbrey A, Volm M. Vascular endothelial growth factor in newly diagnosed and recurrent childhood acute lymphoblastic leukemia as measured by real-time quantitative polymerase chain reaction. Clin Cancer Res. 2001; 7:3381–84.
42. Wellmann S, Guschmann M, Griethe W, Eckert C, von Stackelberg A, Lottaz C, Moderegger E, Einsiedel HG, Eckardt KU, Henze G, Seeger K. Activation of the HIF pathway in childhood ALL, prognostic implications of VEGF. Leukemia. 2004; 18:926–33.
43. Yata K, Otsuki T, Kurebayashi J, Uno M, Fujii T, Yawata Y, Takata A, Hyodoh F, Sugihara T. Expression of angiogenic factors including VEGFs and the effects of hypoxia and thalidomide on human myeloma cells. Int J Oncol. 2003; 22:165–73.
44. Vacca A, Scavelli C, Montefusco V, Di Pietro G, Neri A, Mattioli M, Bicciato S, Nico B, Ribatti D, Dammacco F, Corradini P. Thalidomide downregulates angiogenic genes in bone marrow endothelial cells of patients with active multiple myeloma. J Clin Oncol. 2005; 23:5334–46.
45. Dmoszyńska A, Bojarska-Junak A, Domański D, Roliński J, Hus M, Soroka-Wojtaszko M. Production of proangiogenic cytokines during thalidomide treatment of multiple myeloma. Leuk Lymphoma. 2002; 43:401–06.
46. Neben K, Moehler T, Egerer G, Kraemer A, Hillengass J, Benner A, Ho AD, Goldschmidt H. High plasma basic fibroblast growth factor concentration is associated with response to thalidomide in progressive multiple myeloma. Clin Cancer Res. 2001; 7:2675–81.
47. Terpos E, Anargyrou K, Katodritou E, Kastritis E, Papatheodorou A, Christoulas D, Pouli A, Michalis E, Delimpasi S, Gkotzamanidou M, Nikitas N, Koumoustiotis V, Margaritis D, et al, and Greek Myeloma Study Group, Greece. Circulating angiopoietin-1 to angiopoietin-2 ratio is an independent prognostic factor for survival in newly diagnosed patients with multiple myeloma who received therapy with novel antimyeloma agents. Int J Cancer. 2012; 130:735–42.
48. Zhang J, Sattler M, Tonon G, Grabher C, Lababidi S, Zimmerhackl A, Raab MS, Vallet S, Zhou Y, Cartron MA, Hideshima T, Tai YT, Chauhan D, et al. Targeting angiogenesis via a c-Myc/hypoxia-inducible factor-1alpha-dependent pathway in multiple myeloma. Cancer Res. 2009; 69:5082–90.
49. Minoia C, Quero C, Asselti M, Galise I, Marzano AL, Iacobazzi A, Rana A, Merchionne F, Serratì S, De Tullio G, Quintana G, Casiello M, Maiorano E, et al. Changes in angiogenesis and hypoxia-inducible factor-1α protein expression in relapsed/refractory indolent non-Hodgkin lymphomas. Br J Haematol. 2013; 163:640–45.
50. Dejean E, Renalier MH, Foisseau M, Agirre X, Joseph N, de Paiva GR, Al Saati T, Soulier J, Desjobert C, Lamant L, Prósper F, Felsher DW, Cavaillé J, et al. Hypoxia-microRNA-16 downregulation induces VEGF expression in anaplastic lymphoma kinase (ALK)-positive anaplastic large-cell lymphomas. Leukemia. 2011; 25:1882–90.
51. Passam FH, Alexandrakis MG, Kafousi M, Fotinou M, Darivianaki K, Tsirakis G, Roussou PA, Stathopoulos EN, Siafakas NM. Histological expression of angiogenic factors: VEGF, PDGFRalpha, and HIF-1alpha in Hodgkin lymphoma. Pathol Res Pract. 2009; 205:11–20.
52. Doussis-Anagnostopoulou IA, Talks KL, Turley H, Debnam P, Tan DC, Mariatos G, Gorgoulis V, Kittas C, Gatter KC. Vascular endothelial growth factor (VEGF) is expressed by neoplastic Hodgkin-Reed-Sternberg cells in Hodgkin’s disease. J Pathol. 2002; 197:677–83.
53. Frost P, Berlanger E, Mysore V, Hoang B, Shi Y, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors induce tumor cell apoptosis in vivo primarily by inhibiting VEGF expression and angiogenesis. J Oncol. 2013; 2013:897025.
54. Beegle J, Lakatos K, Kalomoiris S, Stewart H, Isseroff RR, Nolta JA, Fierro FA. Hypoxic preconditioning of mesenchymal stromal cells induces metabolic changes, enhances survival, and promotes cell retention in vivo. Stem Cells. 2015; 33:1818–28.
55. Holzwarth C, Vaegler M, Gieseke F, Pfister SM, Handgretinger R, Kerst G, Müller I. Low physiologic oxygen tensions reduce proliferation and differentiation of human multipotent mesenchymal stromal cells. BMC Cell Biol. 2010; 11:11.
56. Tamama K, Kawasaki H, Kerpedjieva SS, Guan J, Ganju RK, Sen CK. Differential roles of hypoxia inducible factor subunits in multipotential stromal cells under hypoxic condition. J Cell Biochem. 2011; 112:804–17.
57. Hammoud M, Vlaski M, Duchez P, Chevaleyre J, Lafarge X, Boiron JM, Praloran V, Brunet De La Grange P, Ivanovic Z. Combination of low O(2) concentration and mesenchymal stromal cells during culture of cord blood CD34(+) cells improves the maintenance and proliferative capacity of hematopoietic stem cells. J Cell Physiol. 2012; 227:2750–58.
58. Hulleman E, Kazemier KM, Holleman A, VanderWeele DJ, Rudin CM, Broekhuis MJ, Evans WE, Pieters R, Den Boer ML. Inhibition of glycolysis modulates prednisolone resistance in acute lymphoblastic leukemia cells. Blood. 2009; 113:2014–21.
59. Liu T, Kishton RJ, Macintyre AN, Gerriets VA, Xiang H, Liu X, Abel ED, Rizzieri D, Locasale JW, Rathmell JC. Glucose transporter 1-mediated glucose uptake is limiting for B-cell acute lymphoblastic leukemia anabolic metabolism and resistance to apoptosis. Cell Death Dis. 2014; 5:e1470.
60. Herranz D, Ambesi-Impiombato A, Sudderth J, Sánchez-Martín M, Belver L, Tosello V, Xu L, Wendorff AA, Castillo M, Haydu JE, Márquez J, Matés JM, Kung AL, et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med. 2015; 21:1182–89.
61. Song K, Li M, Xu X, Xuan LI, Huang G, Liu Q. Resistance to chemotherapy is associated with altered glucose metabolism in acute myeloid leukemia. Oncol Lett. 2016; 12:334–42.
62. Huang A, Ju HQ, Liu K, Zhan G, Liu D, Wen S, Garcia-Manero G, Huang P, Hu Y. Metabolic alterations and drug sensitivity of tyrosine kinase inhibitor resistant leukemia cells with a FLT3/ITD mutation. Cancer Lett. 2016; 377:149–57.
63. Ariës IM, Hansen BR, Koch T, van den Dungen R, Evans WE, Pieters R, den Boer ML. The synergism of MCL1 and glycolysis on pediatric acute lymphoblastic leukemia cell survival and prednisolone resistance. Haematologica. 2013; 98:1905–11.
64. Pang YY, Wang T, Chen FY, Wu YL, Shao X, Xiao F, Huang HH, Zhong H, Zhong JH. Glycolytic inhibitor 2-deoxy-d-glucose suppresses cell proliferation and enhances methylprednisolone sensitivity in non-Hodgkin lymphoma cells through down-regulation of HIF-1α and c-MYC. Leuk Lymphoma. 2015; 56:1821–30.
65. Gu L, Xie L, Zuo C, Ma Z, Zhang Y, Zhu Y, Gao J. Targeting mTOR/p70S6K/glycolysis signaling pathway restores glucocorticoid sensitivity to 4E-BP1 null Burkitt Lymphoma. BMC Cancer. 2015; 15:529.
66. Manerba M, Di Ianni L, Fiume L, Roberti M, Recanatini M, Di Stefano G. Lactate dehydrogenase inhibitors sensitize lymphoma cells to cisplatin without enhancing the drug effects on immortalized normal lymphocytes. Eur J Pharm Sci. 2015; 74:95–102.
67. Allegretti M, Ricciardi MR, Licchetta R, Mirabilii S, Orecchioni S, Reggiani F, Talarico G, Foà R, Bertolini F, Amadori S, Torrisi MR, Tafuri A. The pan-class I phosphatidyl-inositol-3 kinase inhibitor NVP-BKM120 demonstrates anti-leukemic activity in acute myeloid leukemia. Sci Rep. 2015; 5:18137.
68. Lindblad O, Cordero E, Puissant A, Macaulay L, Ramos A, Kabir NN, Sun J, Vallon-Christersson J, Haraldsson K, Hemann MT, Borg Å, Levander F, Stegmaier K, et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene. 2016; 35:5119–31.
69. Samuels AL, Heng JY, Beesley AH, Kees UR. Bioenergetic modulation overcomes glucocorticoid resistance in T-lineage acute lymphoblastic leukaemia. Br J Haematol. 2014; 165:57–66.
70. Meynet O, Bénéteau M, Jacquin MA, Pradelli LA, Cornille A, Carles M, Ricci JE. Glycolysis inhibition targets Mcl-1 to restore sensitivity of lymphoma cells to ABT-737-induced apoptosis. Leukemia. 2012; 26:1145–47.
71. Maxwell SA, Cherry EM, Bayless KJ. Akt, 14-3-3ζ, and vimentin mediate a drug-resistant invasive phenotype in diffuse large B-cell lymphoma. Leuk Lymphoma. 2011; 52:849–64.
72. Fujiwara S, Kawano Y, Yuki H, Okuno Y, Nosaka K, Mitsuya H, Hata H. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br J Cancer. 2013; 108:170–78.
73. Sanchez WY, McGee SL, Connor T, Mottram B, Wilkinson A, Whitehead JP, Vuckovic S, Catley L. Dichloroacetate inhibits aerobic glycolysis in multiple myeloma cells and increases sensitivity to bortezomib. Br J Cancer. 2013; 108:1624–33.
74. Yin L, Kufe T, Avigan D, Kufe D. Targeting MUC1-C is synergistic with bortezomib in downregulating TIGAR and inducing ROS-mediated myeloma cell death. Blood. 2014; 123:2997–3006.
75. Zub KA, Sousa MM, Sarno A, Sharma A, Demirovic A, Rao S, Young C, Aas PA, Ericsson I, Sundan A, Jensen ON, Slupphaug G. Modulation of cell metabolic pathways and oxidative stress signaling contribute to acquired melphalan resistance in multiple myeloma cells. PLoS One. 2015; 10:e0119857.
76. Maiso P, Huynh D, Moschetta M, Sacco A, Aljawai Y, Mishima Y, Asara JM, Roccaro AM, Kimmelman AC, Ghobrial IM. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015; 75:2071–82.
77. Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda). 2009; 24:97–106.
78. Benizri E, Ginouvès A, Berra E. The magic of the hypoxia-signaling cascade. Cell Mol Life Sci. 2008; 65:1133–49.
79. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012; 148:399–408.
80. Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011; 12:9–22.
81. Freedman SJ, Sun ZY, Poy F, Kung AL, Livingston DM, Wagner G, Eck MJ. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc Natl Acad Sci USA. 2002; 99:5367–72.
82. Berra E, Ginouvès A, Pouysségur J. The hypoxia-inducible-factor hydroxylases bring fresh air into hypoxia signalling. EMBO Rep. 2006; 7:41–45.
83. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999; 399:271–75.
84. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001; 107:43–54.
85. Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001; 294:1337–40.
86. Koivunen P, Tiainen P, Hyvärinen J, Williams KE, Sormunen R, Klaus SJ, Kivirikko KI, Myllyharju J. An endoplasmic reticulum transmembrane prolyl 4-hydroxylase is induced by hypoxia and acts on hypoxia-inducible factor alpha. J Biol Chem. 2007; 282:30544–52.
87. Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003; 22:4082–90.
88. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001; 292:464–68.
89. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001; 292:468–72.
90. Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002; 16:1466-71.
91. Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000; 60:1541–45.
92. Kihira Y, Yamano N, Izawa-Ishizawa Y, Ishizawa K, Ikeda Y, Tsuchiya K, Tamaki T, Tomita S. Basic fibroblast growth factor regulates glucose metabolism through glucose transporter 1 induced by hypoxia-inducible factor-1α in adipocytes. Int J Biochem Cell Biol. 2011; 43:1602–11.
93. Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001; 21:3995–4004.
94. Zelzer E, Levy Y, Kahana C, Shilo BZ, Rubinstein M, Cohen B. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1α/ARNT. EMBO J. 1998; 17:5085–94.
95. Feldser D, Agani F, Iyer NV, Pak B, Ferreira G, Semenza GL. Reciprocal positive regulation of hypoxia-inducible factor 1α and insulin-like growth factor 2. Cancer Res. 1999; 59:3915–18.
96. Hellwig-Bürgel T, Rutkowski K, Metzen E, Fandrey J, Jelkmann W. Interleukin-1β and tumor necrosis factor-α stimulate DNA binding of hypoxia-inducible factor-1. Blood. 1999; 94:1561–67.
97. Gibbs BF, Yasinska IM, Oniku AE, Sumbayev VV. Effects of stem cell factor on hypoxia-inducible factor 1 alpha accumulation in human acute myeloid leukaemia and LAD2 mast cells. PLoS One. 2011; 6:e22502.
98. Kirito K, Fox N, Komatsu N, Kaushansky K. Thrombopoietin enhances expression of vascular endothelial growth factor (VEGF) in primitive hematopoietic cells through induction of HIF-1alpha. Blood. 2005; 105:4258–63.
99. Blancher C, Moore JW, Robertson N, Harris AL. Effects of ras and von Hippel-Lindau (VHL) gene mutations on hypoxia-inducible factor (HIF)-1alpha, HIF-2alpha, and vascular endothelial growth factor expression and their regulation by the phosphatidylinositol 3′-kinase/Akt signaling pathway. Cancer Res. 2001; 61:7349–55.
100. Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, Gottschalk AR, Ryan HE, Johnson RS, Jefferson AB, Stokoe D, Giaccia AJ. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000; 14:391–96.
101. Doe MR, Ascano JM, Kaur M, Cole MD. Myc posttranscriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res. 2012; 72:949–57.
102. Kikuchi H, Pino MS, Zeng M, Shirasawa S, Chung DC. Oncogenic KRAS and BRAF differentially regulate hypoxia-inducible factor-1alpha and -2alpha in colon cancer. Cancer Res. 2009; 69:8499–506.
103. Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002; 100:1532–42.
104. Sathaliyawala T, O’Gorman WE, Greter M, Bogunovic M, Konjufca V, Hou ZE, Nolan GP, Miller MJ, Merad M, Reizis B. Mammalian target of rapamycin controls dendritic cell development downstream of Flt3 ligand signaling. Immunity. 2010; 33:597–606.
105. Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002; 22:7004–14.
106. Mayerhofer M, Valent P, Sperr WR, Griffin JD, Sillaber C. BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor-1α, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamycin. Blood. 2002; 100:3767–75.
107. Lee HY, Lee T, Lee N, Yang EG, Lee C, Lee J, Moon EY, Ha J, Park H. Src activates HIF-1α not through direct phosphorylation of HIF-1α specific prolyl-4 hydroxylase 2 but through activation of the NADPH oxidase/Rac pathway. Carcinogenesis. 2011; 32:703–12.
108. Korgaonkar C, Hagen J, Tompkins V, Frazier AA, Allamargot C, Quelle FW, Quelle DE. Nucleophosmin (B23) targets ARF to nucleoli and inhibits its function. Mol Cell Biol. 2005; 25:1258–71.
109. Fatyol K, Szalay AA. The p14ARF tumor suppressor protein facilitates nucleolar sequestration of hypoxia-inducible factor-1alpha (HIF-1alpha ) and inhibits HIF-1-mediated transcription. J Biol Chem. 2001; 276:28421–29.
110. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011; 19:17–30.
111. Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, Xiong Y. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009; 324:261–65.
112. Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Travins J, Weiss S, Looper R, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012; 483:484–88.
113. Semenza GL. Hypoxia-inducible factor 1 and cancer pathogenesis. IUBMB Life. 2008; 60:591–97.
114. Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999; 59:5830–35.
115. Trastour C, Benizri E, Ettore F, Ramaioli A, Chamorey E, Pouysségur J, Berra E. HIF-1alpha and CA IX staining in invasive breast carcinomas: prognosis and treatment outcome. Int J Cancer. 2007; 120:1451–58.
116. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003; 3:721–32.
117. Deeb G, Vaughan MM, McInnis I, Ford LA, Sait SN, Starostik P, Wetzler M, Mashtare T, Wang ES. Hypoxia-inducible factor-1α protein expression is associated with poor survival in normal karyotype adult acute myeloid leukemia. Leuk Res. 2011; 35:579–84.
118. Gao XN, Yan F, Lin J, Gao L, Lu XL, Wei SC, Shen N, Pang JX, Ning QY, Komeno Y, Deng AL, Xu YH, Shi JL, et al. AML1/ETO cooperates with HIF1α to promote leukemogenesis through DNMT3a transactivation. Leukemia. 2015; 29:1730–40.
119. Zhe N, Wang J, Chen S, Lin X, Chai Q, Zhang Y, Zhao J, Fang Q. Heme oxygenase-1 plays a crucial role in chemoresistance in acute myeloid leukemia. Hematology. 2015; 20:384–91.
120. Song K, Li M, Xu XJ, Xuan L, Huang GN, Song XL, Liu QF. HIF-1α and GLUT1 gene expression is associated with chemoresistance of acute myeloid leukemia. Asian Pac J Cancer Prev. 2014; 15:1823–29.
121. Radwan SM, Hamdy NM, Hegab HM, El-Mesallamy HO. Beclin-1 and hypoxia-inducible factor-1α genes expression: potential biomarkers in acute leukemia patients. Cancer Biomark. 2016; 16:619–26.
122. Silveira VS, Freire BM, Borges KS, Andrade AF, Cruzeiro GA, Sabino JP, Glass ML, Yunes JA, Brandalise SR, Tone LG, Scrideli CA. Hypoxia-related gene expression profile in childhood acute lymphoblastic leukemia: prognostic implications. Leuk Lymphoma. 2014; 55:1751–57.
123. Powell JR, Dojcinov S, King L, Wosniak S, Gerry S, Casbard A, Bailey H, Gallop-Evans E, Maughan T. Prognostic significance of hypoxia inducible factor-1α and vascular endothelial growth factor expression in patients with diffuse large B-cell lymphoma treated with rituximab. Leuk Lymphoma. 2013; 54:959–66.
124. Hernandez-Luna MA, Rocha-Zavaleta L, Vega MI, Huerta-Yepez S. Hypoxia inducible factor-1α induces chemoresistance phenotype in non-Hodgkin lymphoma cell line via up-regulation of Bcl-xL. Leuk Lymphoma. 2013; 54:1048–55.
125. Argyriou P, Papageorgiou SG, Panteleon V, Psyrri A, Bakou V, Pappa V, Spathis A, Economopoulou P, Papageorgiou E, Economopoulos T, Rontogianni D. Hypoxia-inducible factors in mantle cell lymphoma: implication for an activated mTORC1→HIF-1α pathway. Ann Hematol. 2011; 90:315–22.
126. Ria R, Catacchio I, Berardi S, De Luisi A, Caivano A, Piccoli C, Ruggieri V, Frassanito MA, Ribatti D, Nico B, Annese T, Ruggieri S, Guarini A, et al. HIF-1α of bone marrow endothelial cells implies relapse and drug resistance in patients with multiple myeloma and may act as a therapeutic target. Clin Cancer Res. 2014; 20:847–58.
127. Evens AM, Schumacker PT, Helenowski IB, Singh AT, Dokic D, Keswani A, Kordeluk E, Raji A, Winter JN, Jovanovic BD, Holmgren A, Nelson BP, Gordon LI. Hypoxia inducible factor-alpha activation in lymphoma and relationship to the thioredoxin family. Br J Haematol. 2008; 141:676–80.
128. Forristal CE, Brown AL, Helwani FM, Winkler IG, Nowlan B, Barbier V, Powell RJ, Engler GA, Diakiw SM, Zannettino AC, Martin S, Pattabiraman D, D’Andrea RJ, et al. Hypoxia inducible factor (HIF)-2α accelerates disease progression in mouse models of leukemia and lymphoma but is not a poor prognosis factor in human AML. Leukemia. 2015; 29:2075–85.
129. Giatromanolaki A, Bai M, Margaritis D, Bourantas KL, Koukourakis MI, Sivridis E, Gatter KC. Hypoxia and activated VEGF/receptor pathway in multiple myeloma. Anticancer Res. 2010; 30:2831–36.
130. Zhang H, Li H, Xi HS, Li S. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood. 2012; 119:2595–607.
131. Coltella N, Percio S, Valsecchi R, Cuttano R, Guarnerio J, Ponzoni M, Pandolfi PP, Melillo G, Pattini L, Bernardi R. HIF factors cooperate with PML-RARα to promote acute promyelocytic leukemia progression and relapse. EMBO Mol Med. 2014; 6:640–50.
132. Wang Y, Liu Y, Malek SN, Zheng P, Liu Y. Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell. 2011; 8:399–411.
133. Wang Y, Liu Y, Tang F, Bernot KM, Schore R, Marcucci G, Caligiuri MA, Zheng P, Liu Y. Echinomycin protects mice against relapsed acute myeloid leukemia without adverse effect on hematopoietic stem cells. Blood. 2014; 124:1127–35.
134. Giambra V, Jenkins CE, Lam SH, Hoofd C, Belmonte M, Wang X, Gusscott S, Gracias D, Weng AP. Leukemia stem cells in T-ALL require active Hif1α and Wnt signaling. Blood. 2015; 125:3917–27.
135. Kawada H, Kaneko M, Sawanobori M, Uno T, Matsuzawa H, Nakamura Y, Matsushita H, Ando K. High concentrations of L-ascorbic acid specifically inhibit the growth of human leukemic cells via downregulation of HIF-1α transcription. PLoS One. 2013; 8:e62717.
136. Rouault-Pierre K, Lopez-Onieva L, Foster K, Anjos-Afonso F, Lamrissi-Garcia I, Serrano-Sanchez M, Mitter R, Ivanovic Z, de Verneuil H, Gribben J, Taussig D, Rezvani HR, Mazurier F, Bonnet D. HIF-2α protects human hematopoietic stem/progenitors and acute myeloid leukemic cells from apoptosis induced by endoplasmic reticulum stress. Cell Stem Cell. 2013; 13:549–63.
137. Huang Y, Du KM, Xue ZH, Yan H, Li D, Liu W, Chen Z, Zhao Q, Tong JH, Zhu YS, Chen GQ. Cobalt chloride and low oxygen tension trigger differentiation of acute myeloid leukemic cells: possible mediation of hypoxia-inducible factor-1alpha. Leukemia. 2003; 17:2065–73.
138. Jiang Y, Xue ZH, Shen WZ, Du KM, Yan H, Yu Y, Peng ZG, Song MG, Tong JH, Chen Z, Huang Y, Lübbert M, Chen GQ. Desferrioxamine induces leukemic cell differentiation potentially by hypoxia-inducible factor-1 alpha that augments transcriptional activity of CCAAT/enhancer-binding protein-alpha. Leukemia. 2005; 19:1239–47.
139. Liu W, Guo M, Xu YB, Li D, Zhou ZN, Wu YL, Chen Z, Kogan SC, Chen GQ. Induction of tumor arrest and differentiation with prolonged survival by intermittent hypoxia in a mouse model of acute myeloid leukemia. Blood. 2006; 107:698–707.
140. Velasco-Hernandez T, Hyrenius-Wittsten A, Rehn M, Bryder D, Cammenga J. HIF-1α can act as a tumor suppressor gene in murine acute myeloid leukemia. Blood. 2014; 124:3597–607.
141. Velasco-Hernandez T, Tornero D, Cammenga J. Loss of HIF-1α accelerates murine FLT-3(ITD)-induced myeloproliferative neoplasia. Leukemia. 2015; 29:2366–74.
142. Vukovic M, Guitart AV, Sepulveda C, Villacreces A, O’Duibhir E, Panagopoulou TI, Ivens A, Menendez-Gonzalez J, Iglesias JM, Allen L, Glykofrydis F, Subramani C, Armesilla-Diaz A, et al. Hif-1α and Hif-2α synergize to suppress AML development but are dispensable for disease maintenance. J Exp Med. 2015; 212:2223–34.
143. Evens AM, Sehn LH, Farinha P, Nelson BP, Raji A, Lu Y, Brakman A, Parimi V, Winter JN, Schumacker PT, Gascoyne RD, Gordon LI. Hypoxia-inducible factor-1 {alpha} expression predicts superior survival in patients with diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol. 2010; 28:1017–24.
144. Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004; 10:858–64.
145. Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003; 425:307–11.
146. Wang Y, Miao H, Li W, Yao J, Sun Y, Li Z, Zhao L, Guo Q. CXCL12/CXCR4 axis confers adriamycin resistance to human chronic myelogenous leukemia and oroxylin A improves the sensitivity of K562/ADM cells. Biochem Pharmacol. 2014; 90:212–25.
147. Liou A, Delgado-Martin C, Teachey DT, Hermiston ML. The CXCR4/CXCL12 Axis Mediates Chemotaxis, Survival, and Chemoresistance in T-Cell Acute Lymphoblastic Leukemia. Blood. 2014; 124:3629.
148. Lee CG, Das B, Lin TL, Grimes C, Zhang X, Lavezzi T, Huang L, Cole J, Yau L, Li L. A rare fraction of drug-resistant follicular lymphoma cancer stem cells interacts with follicular dendritic cells to maintain tumourigenic potential. Br J Haematol. 2012; 158:79–90.
149. Chen Z, Teo AE, McCarty N. ROS-Induced CXCR4 Signaling Regulates Mantle Cell Lymphoma (MCL) Cell Survival and Drug Resistance in the Bone Marrow Microenvironment via Autophagy. Clin Cancer Res. 2016; 22:187–99.
150. Mirandola L, Apicella L, Colombo M, Yu Y, Berta DG, Platonova N, Lazzari E, Lancellotti M, Bulfamante G, Cobos E, Chiriva-Internati M, Chiaramonte R. Anti-Notch treatment prevents multiple myeloma cells localization to the bone marrow via the chemokine system CXCR4/SDF-1. Leukemia. 2013; 27:1558–66.
151. Azab AK, Runnels JM, Pitsillides C, Moreau AS, Azab F, Leleu X, Jia X, Wright R, Ospina B, Carlson AL, Alt C, Burwick N, Roccaro AM, et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009; 113:4341–51.
152. Valsecchi R, Coltella N, Belloni D, Ponente M, Ten Hacken E, Scielzo C, Scarfò L, Bertilaccio MT, Brambilla P, Lenti E, Martinelli Boneschi F, Brendolan A, Ferrero E, et al. HIF-1α regulates the interaction of chronic lymphocytic leukemia cells with the tumor microenvironment. Blood. 2016; 127:1987–97.
153. Kurtova AV, Tamayo AT, Ford RJ, Burger JA. Mantle cell lymphoma cells express high levels of CXCR4, CXCR5, and VLA-4 (CD49d): importance for interactions with the stromal microenvironment and specific targeting. Blood. 2009; 113:4604–13.
154. Phillips RM. Targeting the hypoxic fraction of tumours using hypoxia-activated prodrugs. Cancer Chemother Pharmacol. 2016; 77:441–57.
155. Portwood S, Lal D, Hsu YC, Vargas R, Johnson MK, Wetzler M, Hart CP, Wang ES. Activity of the hypoxia-activated prodrug, TH-302, in preclinical human acute myeloid leukemia models. Clin Cancer Res. 2013; 19:6506–19.
156. Benito J, Ramirez MS, Millward NZ, Velez J, Harutyunyan KG, Lu H, Shi YX, Matre P, Jacamo R, Ma H, Konoplev S, McQueen T, Volgin A, et al. Hypoxia-Activated Prodrug TH-302 Targets Hypoxic Bone Marrow Niches in Preclinical Leukemia Models. Clin Cancer Res. 2016; 22:1687–98.
157. Cobb WR, Bogden AE, Reich SD, Griffin TW, Kelton DE, LePage DJ. Activity of two phase I drugs N-methylformamide (NSC-3051) and Echinomycin (NSC-526417) against fresh surgical explants of human tumors in the 6-day subrenal capsule (SRC) assay. Invest New Drugs. 1983; 1:5–9.
158. Yonekura S, Itoh M, Okuhashi Y, Takahashi Y, Ono A, Nara N, Tohda S. Effects of the HIF1 inhibitor, echinomycin, on growth and NOTCH signalling in leukaemia cells. Anticancer Res. 2013; 33:3099–103.
159. Zhe N, Chen S, Zhou Z, Liu P, Lin X, Yu M, Cheng B, Zhang Y, Wang J. HIF-1α inhibition by 2-methoxyestradiol induces cell death via activation of the mitochondrial apoptotic pathway in acute myeloid leukemia. Cancer Biol Ther. 2016; 17:625–34.
160. Coltella N, Valsecchi R, Ponente M, Ponzoni M, Bernardi R. Synergistic Leukemia Eradication by Combined Treatment with Retinoic Acid and HIF Inhibition by EZN-2208 (PEG-SN38) in Preclinical Models of PML-RARα and PLZF-RARα-Driven Leukemia. Clin Cancer Res. 2015; 21:3685–94.
161. Borsi E, Perrone G, Terragna C, Martello M, Zamagni E, Tacchetti P, Pantani L, Brioli A, Dico AF, Zannetti BA, Rocchi S, Cavo M. HIF-1α inhibition blocks the cross talk between multiple myeloma plasma cells and tumor microenvironment. Exp Cell Res. 2014; 328:444–55.
162. Viziteu E, Grandmougin C, Goldschmidt H, Seckinger A, Hose D, Klein B, Moreaux J. Chetomin, targeting HIF-1α/p300 complex, exhibits antitumour activity in multiple myeloma. Br J Cancer. 2016; 114:519–23.
163. Cheloni G, Tanturli M, Ho-Ngoc D, Shan Y, Tusa I, Rovida E, Li S, Dello Sbarba P. Targeting Chronic Myeloid Leukemia Stem Cells through Pharmacological Inhibition of HIF-1α. ASH Annual Meeting Abstracts. 2016; 128: 4235.
164. Scott K, Hayden PJ, Will A, Wheatley K, Coyne I. Bortezomib for the treatment of multiple myeloma. Cochrane Database Syst Rev. 2016; 4:CD010816.
165. Hambley B, Caimi PF, William BM. Bortezomib for the treatment of mantle cell lymphoma: an update. Ther Adv Hematol. 2016; 7:196–208.
166. Shin DH, Chun YS, Lee DS, Huang LE, Park JW. Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood. 2008; 111:3131–36.
167. Hu J, Van Valckenborgh E, Xu D, Menu E, De Raeve H, De Bruyne E, Xu S, Van Camp B, Handisides D, Hart CP, Vanderkerken K. Synergistic induction of apoptosis in multiple myeloma cells by bortezomib and hypoxia-activated prodrug TH-302, in vivo and in vitro. Mol Cancer Ther. 2013; 12:1763–73.
168. Wallace EM, Rizzi JP, Han G, Wehn PM, Cao Z, Du X, Cheng T, Czerwinski RM, Dixon DD, Goggin BS, Grina JA, Halfmann MM, Maddie MA, et al. A Small-Molecule Antagonist of HIF2α Is Efficacious in Preclinical Models of Renal Cell Carcinoma. Cancer Res. 2016; 76:5491–500.
169. Cho H, Du X, Rizzi JP, Liberzon E, Chakraborty AA, Gao W, Carvo I, Signoretti S, Bruick RK, Josey JA, Wallace EM, Kaelin WG. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature. 2016; 539:107–11.
170. Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, Homayoun F, Ma Y, Patel N, Yell P, Hao G, Yousuf Q, Joyce A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature. 2016; 539:112–17.