
HOX genes in cancer
The HOX genes
The HOX genes are a family of homeodomain-containing transcription factors that were originally identified due to their pivotal roles in early development [1]. These are reflected in a range of striking developmental changes in HOX-mutant animals, most notably Drosophila. One of the best known examples of this is the ectopic expression of the Antennapedia (Antp) gene in cells that would normally give rise to antennae; a near perfect pair of legs replaces the antennae of adult flies that develop from these embryos [2]. These observations revealed that HOX genes can determine the identity of cells and tissues, and hence also help regulate the proliferation, differentiation, and survival of these cells. The identification of additional mutants revealed a family of 8 HOX genes in the fly, which are expressed in an anterior to posterior pattern along the main embryonic axis and specify the identity of different embryonic structures [1].
An unusual feature of HOX gene organization is their existence in clusters within a single chromosome, and 2 further rounds of duplication in the course of vertebrate evolution have given rise to 4 chromosomal clusters in mammals, referred to as A, B, C, and D. The cluster names are also used to denote specific genes in conjunction with the position of the gene in the cluster, thus for example HOXD1 is the most 3’ member of the D cluster [3]. The genes within clusters share enhancer regions and this plays a significant role in the regulation of HOX gene expression, as do a number of microRNAs encoded within each cluster [4]. A further unusual feature of HOX regulation, which is in part enabled by enhancer sharing, is temporal and spatial collinearity during development, whereby each HOX gene in a cluster is expressed earlier in development and with a more anterior border of expression than its 5’ neighbour [1].
In total mammals have 39 HOX genes that play key roles in patterning both the main embryonic anterior to posterior axis at a very early stage of development and embryonic structures that develop later, for example the limbs and many of the organs [3]. The high level of sequence identity between HOX genes is reflected in a high level of functional redundancy during development [5], although there are also many examples of HOX genes playing specific roles in the embryo, for example during limb development where members of the HOXD cluster define specific structures [6].
Although HOX genes were originally characterized as developmental genes they also play a number of important roles in the adult, and indeed their original embryonic expression patterns are sometimes maintained, at least to a limited extent [7]. The most notable examples of HOX gene function in the adult include the maintenance of hematopoietic stem cells (HSCs) [8], the specification of different blood cell lineages [9], and regulation of tissue identity during implantation and the menstrual cycle [10]. In addition, and as discussed in detail below, the HOX genes become highly dysregulated (and often over expressed) in a wide range of both solid and haematological cancers.
HOX cofactors
Although HOX proteins can bind to DNA through their homeodomain, this binding is relatively non-specific as it generally involves only a 4 base pair recognition sequence. Greater specificity is conferred by the binding of cofactors such Pre-B-cell Leukemia Homeobox (PBX) and Myeloid Ecotropic Viral Integration Site 1 Homolog (MEIS) proteins [11]. The latter bind to HOX proteins 9-13 [12], whilst PBX family members bind to HOX proteins 1-11 [13-15]. In addition to increasing the complexity of the DNA binding sequence, these cofactors also influence key transcriptional events such as the recruitment of RNA polymerase II or III, or transcriptional inhibitors such as HDAC. Hence, for example, HOXC6 can recruit RNA polymerase II through a TAAT site in the promoter of the S100B gene to promote its transcription in neuroblastoma cell lines [16], whilst HOXD3 needs to bind as a heterodimer with PBX1B to recruit RNA polymerase II and activate transcription of the ITGB3 gene (encoding Integrin β3) during angiogenesis [17]. Conversely, HOXB6 and HOXD4 can bind to and inhibit the histone acetyl transferase CBP on the TWIST1 enhancer, thereby repressing TWIST1 transcription [18].
In addition to determining target gene specificity and the mode of transcriptional regulation, HOX cofactors also play a role in post-translational regulation through facilitating the entry of HOX proteins into the nucleus (considered in more detail below).
The role of HOX genes in cancer
A potential role for HOX genes in cancer first became apparent from their frequent inclusion in chimeric, oncogenic gene fusions that drive the formation of haematological malignancies [9]. It has subsequently become apparent that HOX genes are profoundly dysregulated in a wide range of both solid and haematological malignancies, most frequently showing very high levels of over expression. There is now a vast amount of data available on HOX gene expression in different malignancies, and it is not within the scope of this review to detail this. The reader is instead referred to a number of recent specialist reviews that examine the expression and possible function of HOX genes in specific cancer types such as melanoma [19], and head and neck [4], prostate [20], breast [21], ovarian [22], and pancreatic cancer [23]. Despite the very high degree of HOX dysregulation in cancer, relatively few HOX genes have been shown to act as oncogenes in the strictest sense, i.e. that their forced expression in phenotypically normal cells is sufficient to cause a switch to a malignant phenotype, although an important example is HOXA9, which can immortalize normal bone marrow cells in mice when overexpressed [24]. Current evidence broadly indicates that other HOX genes have a general supportive role in malignancy, both at the cellular level (for example in promoting proliferation and blocking apoptosis) [25], and at the tumour level, where they have been shown to variously induce angiogenesis [26], drive metastasis [27], and facilitate drug [21, 28, 29] and radiation resistance [30]. These pro-oncogenic roles are reflected in numerous reports of elevated HOX expression being associated with a poor clinical outcome and prognosis (considered in more detail below).
In addition to a pro-oncogenic role, a number of HOX genes also act as tumour suppressors. Examples include HOXA4, the expression of which can block the spread of ovarian cancer cells [31], HOXA5 that has been shown to stabilize the P53 protein in breast cancer cells [32] and promote an epithelial phenotype [33], and HOXC8 which is inversely related to progression in ovarian cancer [31]. Intriguingly it seems that the regulation of HOX tumour suppressor targets may involve HOX binding to the promoter or enhancer region without PBX, as the identified binding sites are often for HOX only [33, 34], or the HOX protein might have an additional function independent of that as a transcription factor [35].
HOX genes as biomarkers
The differential expression of HOX genes in cancer make the products of these genes potential biomarkers for both diagnosis and prognosis, and for precision medicine applications such as predicting treatment response (reviewed by Morgan and El-Tanani, 2016 [36]). One of the best characterized examples is Engrailed-2 (EN2), a gene that is very closely related to the HOX genes but which is not located within the 4 main chromosomal clusters described above [37]. EN2 is overexpressed in a number of different malignancies, most notably prostate and bladder cancer, and urinary EN2 protein has been shown to be a potential diagnostic marker of both of these diseases [38, 39]. In addition, HOXC6 RNA has been shown to be a potential diagnostic marker for prostate cancer as part of a multi gene panel [40]. HOX gene expression at the RNA level has also been shown to have prognostic significance in acute myeloid leukaemia (AML) [41], and in several solid malignancies including mesothelioma (HOXB4) [42], breast cancer (HOXB7) [43], ovarian cancer [44], oral squamous cell carcinoma (HOXD13) [45], thyroid cancer (HOXC10) [46], clear cell renal cell carcinoma (HOXC11) [47], gastric cancer (HOXC6) [48], and bladder cancer (HOXB13) [49]. Discreet patterns of HOX gene expression apparently exist within different types of cancer, indicating that they could be used to distinguish between cancer types when the primary tumour type is unknown, for example in circulating tumour cells (CTCs) [36].
PBX genes in cancer
The PBX genes
PBX genes are homologues of the Drosophila extradenticle gene (Exd) and 4 are encoded in the human genome (PBX1-4). Like the HOX genes they encode homeodomain-containing transcription factors, but do not exist in chromosomal clusters. In addition to the homeodomain, PBX proteins contain other highly conserved regions, one of which is required for binding to a number of closely related transcription factors, MEIS and PREP [11]. PBX proteins also include 2 nuclear localization signals (NLSs) in the homeodomain region and a distinct nuclear export sequence (NES) [50]. The extent of nuclear localization of PBX proteins is determined by the balance between import and export pathways mediated by the NLSs and NES, respectively [51]. Unusually, the NES of PBX does not bind to exportin to directly promote nuclear exportation, but instead blocks access to the NLSs through binding to the homeodomain [52].
PBX proteins form strong complexes with HOX1-11 proteins in the presence of a HOX/PBX DNA binding consensus [53-55]. X-ray crystallography data for HOXB1-PBX1 and HOXB9-PBX1 complexes on DNA have revealed that each of the homeodomains binds to one half of an octameric consensus sequence, and that this interaction is stabilized by a HOX/PBX interaction mediated by a conserved hexapeptide sequenced in HOX proteins [15, 56-58]. Full length PBX1 alone cannot activate transcription, while a portion of it (amino acids 39-232) can specifically block transcriptional activation by the SP1 transcription factor [59, 60]. However, when complexed with HOXB1, PBX1 switches from a transcriptional repressor to a transcriptional activator [61].
The role of PBX genes in cancer
Although less studied than the HOX genes in the context of cancer, the PBX gene family is known to have a number of oncogenic functions. Best characterised of these is as a chimeric fusion partner in various leukaemias and lymphomas, for example PBX1 and E2A in pre-B cell acute lymphoblastic leukaemia [62]. It has also been shown that the upregulation of PBX3 together with MEIS1 is necessary to drive leukemogenesis efficiently in mouse models; PBX3 protein is required both to stabilize MEIS1 and induce transcription of the MEIS1 gene [63]. Correspondingly, a 4-gene signature consisting of PBX3, HOXA7, HOXA9, and HOXA11 has been shown to be an independent predictor of poor survival in patients with cytogenetically abnormal AML (CA-AML), and that PBX3 (but not PBX1 or PBX2) is frequently co-expressed with HOXA9 in various subtypes of CA-AML, particularly MLL-rearranged AML, and may thus be a potential pathologic cofactor of HOXA9. This is further supported by the finding that knock-down of PBX3 prevents it from exerting a synergistic effect with HOXA9 in promoting leukemogenesis [64].
The PBX genes are also over expressed in a number of solid tumours. These include colorectal cancer (CRC), in which PBX3 expression was shown to be correlated with invasive potential in vitro, and significantly associated with lymph node invasion, distant metastasis, advanced TNM stage and poor overall survival of patients. Furthermore, the forced expression of PBX3 in cells with a low metastatic potential was shown to promote migration and invasion, at least in part through the upregulation of phosphorylated extracellular signal-regulated kinase (ERK)1/2 [65]. PBX3 has also been shown to be upregulated in gastric cancer cells, and that this was associated with greater invasion depth, and advanced clinical stage and tumour grade. The overexpression of PBX3 in gastric cancer cell lines with low endogenous PBX3 expression accelerated cell proliferation and increased colony formation and cell-invading ability [66]. Similarly, increased levels of PBX3 expression have been reported in prostate tumours and this was found to have prognostic significance in this cancer. PBX3 expression is promoted by androgen signalling through a pathway that is negatively regulated in part by the Lethal-7 family of microRNAs (let-7), which are in turn downstream targets of the Myc protoncogene [67]. Although the majority of reports implicate PBX3 as the most oncogenic PBX gene, a recent study demonstrated that increased PBX1 expression in ovarian cancer was associated with shorter post-chemotherapy survival and increased resistance to platinum-based drugs through the maintenance of a stem cell-like phenotype [68].
Targeting HOX/PBX dimers
Strategies for interfering with HOX/PBX dimer formation: HXR9 and other peptides
The key roles that HOX and PBX proteins play in cancer indicate that they are potential therapeutic targets. However, the high level of functional redundancy amongst HOX proteins and the general difficulty in producing effective small molecule inhibitors against transcription factors have proved significant barriers to this approach. As an alternative, it was proposed that the interaction between HOX and PBX proteins could be targeted, as this is mediated by a highly conserved hexapeptide sequence in HOX proteins and a hydrophobic binding pocket within PBX. Although a small molecule inhibitor of this interaction has previously been described, its Kd was in the micromolar range and it does not seem to have been adopted for experimental or clinical use [69]. To date, a more useful set of inhibitors have proved to be peptides that employ the hexapeptide sequence to act as a competitive antagonist of HOX/PBX binding. Several peptides have been described, but the one most frequently used is HXR9, an 18 amino acid peptide containing the hexapeptide sequence together with 9 arginine residues that promote cellular uptake by endocytosis. HXR9 was originally shown to be cytotoxic to melanoma cell lines and primary melanoma cells and was reported to reduce the growth of B16F10 murine melanoma tumours in an orthotropic model [25]. Subsequently, HXR9 was shown to inhibit the growth of a range of tumour types in mouse xenograft models, including non-small cell lung [70], breast [34], ovarian [71], and prostate cancer [72], and mesothelioma [42], melanoma [73], and meningioma [74] (Table 1).
Table 1: Previous in vivo studies of HOX/PBX inhibition using HXR9
Cancer (cell line) |
Delivery (study duration) |
Max % inhibition of tumour growth |
Biomarkers investigated |
Reference |
Prostate (LNCaP) |
100 mg/kg IT single dose when tumour volume > 100 mm3 (52 days) |
530 |
cFos - tumour response |
[72] |
Breast (SKBR3) |
20 mg/kg IT on weeks 1 and 2 (50 days) |
340 |
cFos - tumour response Mean HOXB1-9 expression - sensitivity |
[34] |
NSCLC (A549) |
100 mg/kg IT single dose when tumour volume > 100 mm3 (18 days) or 10 mg/kg IP twice weekly (18 days) |
880 (IT) 160 (IP) |
cFos, EGR1, EGR4 - tumour response |
[70] |
Mesothelioma (MSTO-211H) |
25 mg/kg IP every 4 days, max 5 doses (37 days) |
230 |
cFos - tumour response Ratio of mean “tumour suppressor” and “oncogenic” HOX expression - sensitivity |
[42] |
Melanoma (B16F10) |
10 mg/kg IV twice weekly (28 days) |
480 |
cFos, DUSP1, ATF3 - tumour response |
[25] |
Melanoma (A375M) |
100 mg/kg IT single dose when tumour volume > 100 mm3 (21 days) |
440 |
cFos - tumour response |
[73] |
Ovarian (SKOV-3) |
1 × 100 mg/kg IT (week 1) and then 10 mg/kg IT twice weekly, with or without cisplatin IP 3 mg/kg weekly (29 days) |
140 (300 when combined with cisplatin) |
[44] |
|
Ovarian (SK-OV3) |
1 × 100 mg/kg IV (week 1) and then 10 mg/kg IV weekly (32 days) |
200 |
cFos - tumour response |
[71] |
Meningioma (IOMM-Lee) |
30 mg/kg IV on days 7, 9, 13, 16, 19 (21 days) |
170 |
[74] |
Abbreviations: IT, intratumoral; IP, intraperitoneal; IV, intravenous; NSCLC, non-small cell lung cancer; “tumour response”, actual response of tumour to treatment; “sensitivity”, prediction of tumour sensitivity to treatment
The mechanism by which HOX/PBX inhibition causes cell death remains to be fully elucidated (Figure 1). In most solid tumours cell death is mediated by apoptosis [25, 34, 42, 71, 72], although in a number of cancers, including some types of renal cancer, necrosis instead plays an important role [75]. However, both these events seem to be activated, at least in part, by a rapid increase in cFos expression, as preventing this using gene knock down strategies can achieve a partial rescue from HXR9-mediated cell killing [25]. The upregulation of cFos has been shown to cause apoptosis in a number of different cancers [76-83], in a mechanism that might involve activation of Fas ligand (FasL) transcription through the AP1 transcriptional activator consisting of the Fos/Jun heterodimer, which in turn promotes apoptosis through the FasL/Fas receptor pathway [76, 79-82]. High levels of cFos tumour expression are associated with longer survival in ovarian cancer patients [84], and, in addition to promoting apoptosis, it has also been shown to reduce the growth of ovarian xenograft tumours in mice through changes in cell adhesion [85]. cFos is an early response gene to many types of cellular stress, although there is now evidence to suggest that HOX/PBX dimers repress cFos expression both directly, through HOX/PBX binding sites in its promoter, and indirectly through HOX/PBX mediated regulation of a cFos-targeting microRNA [73].
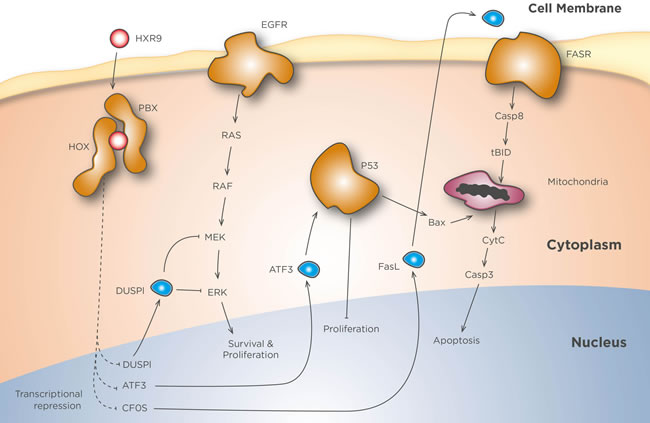
Figure 1: HXR9 mechanisms of action. In the absence of inhibition HOX and PBX dimerize, enter the nucleus, and bind to HOX/PBX consensus sequences present in numerous target genes. These include cFos, DUSP1, and ATF3, all of which are inhibited by HOX/PBX dimers. HXR9 mimics the hexapeptide loop of HOX proteins that binds to a pocket in PBX, thereby acting as a competitive inhibitor of this interaction. HOX/PBX inhibition greatly increases cFos transcription, and the resulting protein can dimerize with Jun (not shown) to activate FasL transcription. FasL protein then binds to the Fas receptor (FasR) and activates the extrinsic apoptotic pathway. The transcription of ATF3 and DUSP1 is also enhanced by HXR9. ATF3 stabilizes p53, which in turn promotes mitochondria-mediated apoptosis and blocks proliferation, while DUSP1 dephosphorylates MEK and ERK, silencing Ras mediated signalling.
Predicting sensitivity to HOX/PBX inhibition
As with other types of targeted therapy, companion diagnostics are becoming increasingly important for patient stratification in clinical trials as well as personalized therapies. The identification of markers that can predict which tumours will be sensitive to HOX/PBX inhibition is an important preclinical goal, although to date there is relatively little evidence for a single or even multiplex marker that can fill this role outside of limited, cancer-specific settings. In a study on breast cancer, the sensitivity of breast cancer cell lines to killing by HXR9 was shown to be strongly associated with the combined expression of HOX genes HOXB1 through to HOXB9 [34]. However, this relationship has not been reported in other cancer types, and indeed it was not found amongst cell lines derived from mesothelioma, where instead there was an apparent relationship between HXR9-mediated cell killing and the ratio of expression in HOX genes with reported pro-oncogenic functions to those with tumour-suppressor functions [42]. Neither of these markers has been validated in primary tumours, although in breast cancer [34] and mesothelioma [42] a subgroup of primary tumours was identified that showed a very high level of the combined HOXB1-9 expression and a high HOX oncogenic to tumour suppressor ratio, respectively. Furthermore, in bone marrow samples from primary AML patients, high levels of HOXA gene and PBX3 expression were found to be associated with greater sensitivity to HXR9 [64].
The lack of a robust and generalized marker of tumour sensitivity to HOX/PBX inhibition might be related to the difficulty in quantifying these dimers in cells and tissues, as opposed to measuring HOX gene expression at the RNA or protein level, which is at best an indirect measure of this. Work is continuing to develop a quantitative assay for HOX/PBX dimers, and it is hoped that this will in turn lead to a more accurate test for tumour sensitivity to HOX/PBX inhibition.
Biomarkers of response to HOX/PBX inhibition
Surrogate markers of tumour response to treatment, which allow an earlier assessment of the efficacy of treatment than conventional clinical endpoints (for example survival and disease free survival) are of growing importance both in clinical trials and in precision medicine. The regulatory function of HOX/PBX dimers means that there are a number of immediate transcriptional targets that change in response to HOX/PBX inhibition. These include cFos, as described above, together with a number of other targets that might act as a readout of the efficacy of inhibitors such as HXR9 [25]. The best characterised of these are dual specificity phosphatase 1 (DUSP1) and activating transcription factor 3 (ATF3). DUSP1 can dephosphorylate serine, threonine and tyrosine residues in a wide range of substrates, and block signalling through the mitogen activated protein kinase pathway, thereby preventing cellular proliferation [86], and decreased DUSP1 expression is associated with higher histological grades in multiple tumour types [87]. ATF3 also has a potential role in tumour suppression as it can stabilize p53 protein and prevent its ubiquination, induce apoptosis, and promote cell cycle arrest [88-90].
Future directions
There is now considerable in vitro and in vivo data supporting the therapeutic potential of inhibiting HOX/PBX dimer formation in cancer. To date, however, the only effective inhibitors of HOX/PBX binding are the HXR9 peptide and its derivatives. Peptide-based therapeutics are already used in cancer therapy though [91], including Carfilzomib for multiple myeloma [92], and indeed a variant of HXR9 for intratumoral injection is expected to enter clinical trials during 2018. Developing a small molecule inhibitor of HOX/PBX binding remains a significant challenge, and the only previously reported small molecule inhibitors of this interaction are unlikely to have any therapeutic application as the minimum reported Kd for PBX binding was 65 µM [69]. Development of a small molecule inhibitor therefor remains an important clinical goal.
Conclusions
Since the publication of the initial study in 2007 it has become increasingly apparent that the HOX/PBX dimer is a potential therapeutic target in both solid and haematological malignancies. Clinical exploitation of this target is aided by the co-development of markers that predict the sensitivity of tumours to HOX/PBX inhibitors, and which allow the initial response to treatment to be monitored. The outcome of clinical trials for peptide and small molecule inhibitors of HOX/PBX are eagerly awaited.
Abbreviations
AML, acute myeloid leukemia; ATF3, activating transcription factor 3; CA-AML, cytogenetically abnormal AML; CTC, circulating tumour cell; CRC, colorectal cancer; DUSP1, dual specificity phosphatase 1; EN2, Engrailed-2; FasL, Fas ligand; HSC, hematopoietic stem cell; MEIS, Myeloid Ecotropic Viral Integration Site 1 Homolog; NES, nuclear export sequence; NLS, nuclear localization signal; PBX, Pre-B-cell Leukemia Homeobox.
Author contributions
RM wrote the first draft of the manuscript; HS helped plan the review and advised on potential clinical applications; MELT advised on the potential use of HOX genes as biomarkers; KDH helped with the discussion of HOX and PBX molecular biology; KJH contributed to the discussion of cell death pathways.
Acknowledgments
The authors wish to thank Inprint+Design (University of Bradford) for helping to prepare Figure 1.
Conflicts of interest
The authors have no conflicts of interest to declare.
References
1. Mallo M, Wellik DM, Deschamps J. Hox genes and regional patterning of the vertebrate body plan. Dev Biol. 2010; 344: 7-15. doi: 10.1016/j.ydbio.2010.04.024.
2. Gehring WJ. Homeo boxes in the study of development. Science. 1987; 236: 1245-52.
3. Holland PW, Booth HA, Bruford EA. Classification and nomenclature of all human homeobox genes. BMC Biol. 2007; 5: 47. doi: 10.1186/1741-7007-5-47.
4. Platais C, Hakami F, Darda L, Lambert DW, Morgan R, Hunter KD. The role of HOX genes in head and neck squamous cell carcinoma. J Oral Pathol Med. 2016; 45: 239-47. doi: 10.1111/jop.12388.
5. Di-Poi N, Koch U, Radtke F, Duboule D. Additive and global functions of HoxA cluster genes in mesoderm derivatives. Dev Biol. 2010; 341: 488-98. doi: 10.1016/j.ydbio.2010.03.006.
6. Pineault KM, Wellik DM. Hox genes and limb musculoskeletal development. Curr Osteoporos Rep. 2014; 12: 420-7. doi: 10.1007/s11914-014-0241-0.
7. Morgan R. Hox genes: a continuation of embryonic patterning? Trends Genet. 2006; 22: 67-9. doi: 10.1016/j.tig.2005.11.004.
8. Lebert-Ghali CE, Fournier M, Dickson GJ, Thompson A, Sauvageau G, Bijl JJ. HoxA cluster is haploinsufficient for activity of hematopoietic stem and progenitor cells. Exp Hematol. 2010; 38: 1074-86 e1-5. doi: 10.1016/j.exphem.2010.07.006.
9. Alharbi RA, Pettengell R, Pandha HS, Morgan R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia. 2013; 27: 1000-8. doi: 10.1038/leu.2012.356.
10. Xu B, Geerts D, Bu Z, Ai J, Jin L, Li Y, Zhang H, Zhu G. Regulation of endometrial receptivity by the highly expressed HOXA9, HOXA11 and HOXD10 HOX-class homeobox genes. Hum Reprod. 2014; 29: 781-90. doi: 10.1093/humrep/deu004.
11. Longobardi E, Penkov D, Mateos D, De Florian G, Torres M, Blasi F. Biochemistry of the tale transcription factors PREP, MEIS, and PBX in vertebrates. Dev Dyn. 2014; 243: 59-75. doi: 10.1002/dvdy.24016.
12. Williams TM, Williams ME, Innis JW. Range of HOX/TALE superclass associations and protein domain requirements for HOXA13:MEIS interaction. Dev Biol. 2005; 277: 457-71. doi: 10.1016/j.ydbio.2004.10.004.
13. Allen TD, Zhu YX, Hawley TS, Hawley RG. TALE homeoproteins as HOX11-interacting partners in T-cell leukemia. Leuk Lymphoma. 2000; 39: 241-56. doi: 10.3109/10428190009065824.
14. Brendolan A, Ferretti E, Salsi V, Moses K, Quaggin S, Blasi F, Cleary ML, Selleri L. A Pbx1-dependent genetic and transcriptional network regulates spleen ontogeny. Development. 2005; 132: 3113-26. doi: 10.1242/dev.01884.
15. Piper DE, Batchelor AH, Chang CP, Cleary ML, Wolberger C. Structure of a HoxB1-Pbx1 heterodimer bound to DNA: role of the hexapeptide and a fourth homeodomain helix in complex formation. Cell. 1999; 96: 587-97.
16. Zhang X, Hamada J, Nishimoto A, Takahashi Y, Murai T, Tada M, Moriuchi T. HOXC6 and HOXC11 increase transcription of S100 beta gene in BrdU-induced in vitro differentiation of GOTO neuroblastoma cells into Schwannian cells. J Cell Mol Med. 2007; 11: 299-306. doi: 10.1111/j.1582-4934.2007.00020.x.
17. Charboneau A, East L, Mulholland N, Rohde M, Boudreau N. Pbx1 is required for Hox D3-mediated angiogenesis. Angiogenesis. 2005; 8: 289-96. doi: 10.1007/s10456-005-9016-7.
18. Shen WF, Krishnan K, Lawrence HJ, Largman C. The HOX homeodomain proteins block CBP histone acetyltransferase activity. Mol Cell Biol. 2001; 21: 7509-22. doi: 10.1128/MCB.21.21.7509-7522.2001.
19. Pierard GE, Pierard-Franchimont C. HOX Gene Aberrant Expression in Skin Melanoma: A Review. J Skin Cancer. 2012; 2012: 707260. doi: 10.1155/2012/707260.
20. Javed S, Langley SE. Importance of HOX genes in normal prostate gland formation, prostate cancer development and its early detection. BJU Int. 2014; 113: 535-40. doi: 10.1111/bju.12269.
21. Jin K, Sukumar S. HOX genes: Major actors in resistance to selective endocrine response modifiers. Biochim Biophys Acta. 2016; 1865: 105-10. doi: 10.1016/j.bbcan.2016.01.003.
22. Kelly ZL, Michael A, Butler-Manuel S, Pandha HS, Morgan RG. HOX genes in ovarian cancer. J Ovarian Res. 2011; 4: 16. doi: 10.1186/1757-2215-4-16.
23. Gray S, Pandha HS, Michael A, Middleton G, Morgan R. HOX genes in pancreatic development and cancer. JOP. 2011; 12: 216-9.
24. Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J. 1998; 17: 3714-25. doi: 10.1093/emboj/17.13.3714.
25. Morgan R, Pirard PM, Shears L, Sohal J, Pettengell R, Pandha HS. Antagonism of HOX/PBX dimer formation blocks the in vivo proliferation of melanoma. Cancer Res. 2007; 67: 5806-13. doi: 10.1158/0008-5472.CAN-06-4231.
26. Kachgal S, Mace KA, Boudreau NJ. The dual roles of homeobox genes in vascularization and wound healing. Cell Adh Migr. 2012; 6: 457-70. doi: 10.4161/cam.22164.
27. Hong CS, Jeong O, Piao Z, Guo C, Jung MR, Choi C, Park YK. HOXB5 induces invasion and migration through direct transcriptional up-regulation of beta-catenin in human gastric carcinoma. Biochem J. 2015; 472: 393-403. doi: 10.1042/BJ20150213.
28. Li N, Jia X, Wang J, Li Y, Xie S. Knockdown of homeobox A5 by small hairpin RNA inhibits proliferation and enhances cytarabine chemosensitivity of acute myeloid leukemia cells. Mol Med Rep. 2015; 12: 6861-6. doi: 10.3892/mmr.2015.4331.
29. Xiao F, Bai Y, Chen Z, Li Y, Luo L, Huang J, Yang J, Liao H, Guo L. Downregulation of HOXA1 gene affects small cell lung cancer cell survival and chemoresistance under the regulation of miR-100. Eur J Cancer. 2014; 50: 1541-54. doi: 10.1016/j.ejca.2014.01.024.
30. Rubin E, Wu X, Zhu T, Cheung JC, Chen H, Lorincz A, Pandita RK, Sharma GG, Ha HC, Gasson J, Hanakahi LA, Pandita TK, Sukumar S. A role for the HOXB7 homeodomain protein in DNA repair. Cancer Res. 2007; 67: 1527-35. doi: 10.1158/0008-5472.CAN-06-4283.
31. Klausen C, Leung PC, Auersperg N. Cell motility and spreading are suppressed by HOXA4 in ovarian cancer cells: possible involvement of beta1 integrin. Mol Cancer Res. 2009; 7: 1425-37. doi: 10.1158/1541-7786.MCR-08-0466.
32. Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E, Marks J, Sukumar S. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature. 2000; 405: 974-8. doi: 10.1038/35016125.
33. Teo WW, Merino VF, Cho S, Korangath P, Liang X, Wu RC, Neumann NM, Ewald AJ, Sukumar S. HOXA5 determines cell fate transition and impedes tumor initiation and progression in breast cancer through regulation of E-cadherin and CD24. Oncogene. 2016; 35: 5539-51. doi: 10.1038/onc.2016.95.
34. Morgan R, Boxall A, Harrington KJ, Simpson GR, Gillett C, Michael A, Pandha HS. Targeting the HOX/PBX dimer in breast cancer. Breast Cancer Res Treat. 2012; 136: 389-98. doi: 10.1007/s10549-012-2259-2.
35. Lambert B, Vandeputte J, Remacle S, Bergiers I, Simonis N, Twizere JC, Vidal M, Rezsohazy R. Protein interactions of the transcription factor Hoxa1. BMC Dev Biol. 2012; 12: 29. doi: 10.1186/1471-213X-12-29.
36. Morgan R, El-Tanani M. HOX Genes as Potential Markers of Circulating Tumour Cells. Curr Mol Med. 2016; 16: 322-7.
37. Morgan R. Engrailed: complexity and economy of a multi-functional transcription factor. FEBS Lett. 2006; 580: 2531-3. doi: 10.1016/j.febslet.2006.04.053.
38. Morgan R, Boxall A, Bhatt A, Bailey M, Hindley R, Langley S, Whitaker HC, Neal DE, Ismail M, Whitaker H, Annels N, Michael A, Pandha H. Engrailed-2 (EN2): a tumor specific urinary biomarker for the early diagnosis of prostate cancer. Clin Cancer Res. 2011; 17: 1090-8. doi: 10.1158/1078-0432.CCR-10-2410.
39. Morgan R, Bryan RT, Javed S, Launchbury F, Zeegers MP, Cheng KK, James ND, Wallace DM, Hurst CD, Ward DG, Knowles MA, Pandha H. Expression of Engrailed-2 (EN2) protein in bladder cancer and its potential utility as a urinary diagnostic biomarker. Eur J Cancer. 2013; 49: 2214-22. doi: 10.1016/j.ejca.2013.01.019.
40. Leyten GH, Hessels D, Smit FP, Jannink SA, de Jong H, Melchers WJ, Cornel EB, de Reijke TM, Vergunst H, Kil P, Knipscheer BC, Hulsbergen-van de Kaa CA, Mulders PF, et al. Identification of a Candidate Gene Panel for the Early Diagnosis of Prostate Cancer. Clin Cancer Res. 2015; 21: 3061-70. doi: 10.1158/1078-0432.CCR-14-3334.
41. Gao L, Sun J, Liu F, Zhang H, Ma Y. Higher expression levels of the HOXA9 gene, closely associated with MLL-PTD and EZH2 mutations, predict inferior outcome in acute myeloid leukemia. Onco Targets Ther. 2016; 9: 711-22. doi: 10.2147/OTT.S95279.
42. Morgan R, Simpson G, Gray S, Gillett C, Tabi Z, Spicer J, Harrington KJ, Pandha HS. HOX transcription factors are potential targets and markers in malignant mesothelioma. BMC Cancer. 2015; 16: 85. doi: 10.1186/s12885-016-2106-7.
43. Jin K, Kong X, Shah T, Penet MF, Wildes F, Sgroi DC, Ma XJ, Huang Y, Kallioniemi A, Landberg G, Bieche I, Wu X, Lobie PE, et al. The HOXB7 protein renders breast cancer cells resistant to tamoxifen through activation of the EGFR pathway. Proc Natl Acad Sci U S A. 2012; 109: 2736-41. doi: 10.1073/pnas.1018859108.
44. Kelly Z, Moller-Levet C, McGrath S, Butler-Manuel S, Kavitha Madhuri T, Kierzek AM, Pandha H, Morgan R, Michael A. The prognostic significance of specific HOX gene expression patterns in ovarian cancer. Int J Cancer. 2016; 139: 1608-17. doi: 10.1002/ijc.30204.
45. Aquino G, Franco R, Sabatino R, Mantia EL, Scognamiglio G, Collina F, Longo F, Ionna F, Losito NS, Liguori G, Botti G, Cantile M. Deregulation of paralogous 13 HOX genes in oral squamous cell carcinoma. Am J Cancer Res. 2015; 5: 3042-55.
46. Feng X, Li T, Liu Z, Shi Y, Peng Y. HOXC10 up-regulation contributes to human thyroid cancer and indicates poor survival outcome. Mol Biosyst. 2015; 11: 2946-54. doi: 10.1039/c5mb00253b.
47. Liu YJ, Zhu Y, Yuan HX, Zhang JP, Guo JM, Lin ZM. Overexpression of HOXC11 homeobox gene in clear cell renal cell carcinoma induces cellular proliferation and is associated with poor prognosis. Tumour Biol. 2015; 36: 2821-9. doi: 10.1007/s13277-014-2909-6.
48. Zhang Q, Jin XS, Yang ZY, Wei M, Liu BY, Gu QL. Upregulated Hoxc6 expression is associated with poor survival in gastric cancer patients. Neoplasma. 2013; 60: 439-45. doi: 10.4149/neo_2013_057.
49. Marra L, Cantile M, Scognamiglio G, Perdona S, La Mantia E, Cerrone M, Gigantino V, Cillo C, Caraglia M, Pignata S, Facchini G, Botti G, Chieffi S, et al. Deregulation of HOX B13 expression in urinary bladder cancer progression. Curr Med Chem. 2013; 20: 833-9.
50. Abu-Shaar M, Ryoo HD, Mann RS. Control of the nuclear localization of Extradenticle by competing nuclear import and export signals. Genes Dev. 1999; 13: 935-45.
51. Stevens KE, Mann RS. A balance between two nuclear localization sequences and a nuclear export sequence governs extradenticle subcellular localization. Genetics. 2007; 175: 1625-36. doi: 10.1534/genetics.106.066449.
52. Saleh M, Huang H, Green NC, Featherstone MS. A conformational change in PBX1A is necessary for its nuclear localization. Exp Cell Res. 2000; 260: 105-15. doi: 10.1006/excr.2000.5010.
53. Chan SK, Jaffe L, Capovilla M, Botas J, Mann RS. The DNA binding specificity of Ultrabithorax is modulated by cooperative interactions with extradenticle, another homeoprotein. Cell. 1994; 78: 603-15.
54. Johnson FB, Parker E, Krasnow MA. Extradenticle protein is a selective cofactor for the Drosophila homeotics: role of the homeodomain and YPWM amino acid motif in the interaction. Proc Natl Acad Sci U S A. 1995; 92: 739-43. doi:
55. Merabet S, Hudry B, Saadaoui M, Graba Y. Classification of sequence signatures: a guide to Hox protein function. Bioessays. 2009; 31: 500-11. doi: 10.1002/bies.200800229.
56. Gehring WJ, Qian YQ, Billeter M, Furukubo-Tokunaga K, Schier AF, Resendez-Perez D, Affolter M, Otting G, Wuthrich K. Homeodomain-DNA recognition. Cell. 1994; 78: 211-23.
57. LaRonde-LeBlanc NA, Wolberger C. Structure of HoxA9 and Pbx1 bound to DNA: Hox hexapeptide and DNA recognition anterior to posterior. Genes Dev. 2003; 17: 2060-72. doi: 10.1101/gad.1103303.
58. Passner JM, Ryoo HD, Shen L, Mann RS, Aggarwal AK. Structure of a DNA-bound Ultrabithorax-Extradenticle homeodomain complex. Nature. 1999; 397: 714-9. doi: 10.1038/17833.
59. Knoepfler PS, Lu Q, Kamps MP. Pbx-1 Hox heterodimers bind DNA on inseparable half-sites that permit intrinsic DNA binding specificity of the Hox partner at nucleotides 3’ to a TAAT motif. Nucleic Acids Res. 1996; 24: 2288-94.
60. Lu Q, Kamps MP. Heterodimerization of Hox proteins with Pbx1 and oncoprotein E2a-Pbx1 generates unique DNA-binding specifities at nucleotides predicted to contact the N-terminal arm of the Hox homeodomain--demonstration of Hox-dependent targeting of E2a-Pbx1 in vivo. Oncogene. 1997; 14: 75-83. doi: 10.1038/sj.onc.1200799.
61. Saleh M, Rambaldi I, Yang XJ, Featherstone MS. Cell signaling switches HOX-PBX complexes from repressors to activators of transcription mediated by histone deacetylases and histone acetyltransferases. Mol Cell Biol. 2000; 20: 8623-33.
62. Aspland SE, Bendall HH, Murre C. The role of E2A-PBX1 in leukemogenesis. Oncogene. 2001; 20: 5708-17. doi: 10.1038/sj.onc.1204592.
63. Garcia-Cuellar MP, Steger J, Fuller E, Hetzner K, Slany RK. Pbx3 and Meis1 cooperate through multiple mechanisms to support Hox-induced murine leukemia. Haematologica. 2015; 100: 905-13. doi: 10.3324/haematol.2015.124032.
64. Li Z, Zhang Z, Li Y, Arnovitz S, Chen P, Huang H, Jiang X, Hong GM, Kunjamma RB, Ren H, He C, Wang CZ, Elkahloun AG, et al. PBX3 is an important cofactor of HOXA9 in leukemogenesis. Blood. 2013; 121: 1422-31. doi: 10.1182/blood-2012-07-442004.
65. Han HB, Gu J, Ji DB, Li ZW, Zhang Y, Zhao W, Wang LM, Zhang ZQ. PBX3 promotes migration and invasion of colorectal cancer cells via activation of MAPK/ERK signaling pathway. World J Gastroenterol. 2014; 20: 18260-70. doi: 10.3748/wjg.v20.i48.18260.
66. Li Y, Sun Z, Zhu Z, Zhang J, Sun X, Xu H. PBX3 is overexpressed in gastric cancer and regulates cell proliferation. Tumour Biol. 2014; 35: 4363-8. doi: 10.1007/s13277-013-1573-6.
67. Ramberg H, Grytli HH, Nygard S, Wang W, Ogren O, Zhao S, Lovf M, Katz B, Skotheim RI, Bjartell A, Eri LM, Berge V, Svindland A, et al. PBX3 is a putative biomarker of aggressive prostate cancer. Int J Cancer. 2016; 139: 1810-20. doi: 10.1002/ijc.30220.
68. Jung JG, Shih IM, Park JT, Gerry E, Kim TH, Ayhan A, Handschuh K, Davidson B, Nickles Fader A, Selleri L, Wang TL. Ovarian cancer chemoresistance relies on the the stem cell reprogramming factor PBX1. Cancer Res. 2016. doi: 10.1158/0008-5472.CAN-16-0980.
69. Ji T, Lee M, Pruitt SC, Hangauer DG. Privileged scaffolds for blocking protein-protein interactions: 1,4-disubstituted naphthalene antagonists of transcription factor complex HOX-PBX/DNA. Bioorg Med Chem Lett. 2004; 14: 3875-9. doi: 10.1016/j.bmcl.2004.05.068.
70. Plowright L, Harrington KJ, Pandha HS, Morgan R. HOX transcription factors are potential therapeutic targets in non-small-cell lung cancer (targeting HOX genes in lung cancer). Br J Cancer. 2009; 100: 470-5. doi: 10.1038/sj.bjc.6604857.
71. Morgan R, Plowright L, Harrington KJ, Michael A, Pandha HS. Targeting HOX and PBX transcription factors in ovarian cancer. BMC Cancer. 2010; 10: 89. doi: 10.1186/1471-2407-10-89.
72. Morgan R, Boxall A, Harrington KJ, Simpson GR, Michael A, Pandha HS. Targeting HOX transcription factors in prostate cancer. BMC Urol. 2014; 14: 17. doi: 10.1186/1471-2490-14-17.
73. Errico MC, Felicetti F, Bottero L, Mattia G, Boe A, Felli N, Petrini M, Bellenghi M, Pandha HS, Calvaruso M, Tripodo C, Colombo MP, Morgan R, et al. The abrogation of the HOXB7/PBX2 complex induces apoptosis in melanoma through the miR-221&222-c-FOS pathway. Int J Cancer. 2013; 133: 879-92. doi: 10.1002/ijc.28097.
74. Ando H, Natsume A, Senga T, Watanabe R, Ito I, Ohno M, Iwami K, Ohka F, Motomura K, Kinjo S, Ito M, Saito K, Morgan R, et al. Peptide-based inhibition of the HOXA9/PBX interaction retards the growth of human meningioma. Cancer Chemother Pharmacol. 2014; 73: 53-60. doi: 10.1007/s00280-013-2316-5.
75. Shears L, Plowright L, Harrington K, Pandha HS, Morgan R. Disrupting the interaction between HOX and PBX causes necrotic and apoptotic cell death in the renal cancer lines CaKi-2 and 769-P. J Urol. 2008; 180: 2196-201. doi: 10.1016/j.juro.2008.07.018.
76. Eichhorst ST, Muller M, Li-Weber M, Schulze-Bergkamen H, Angel P, Krammer PH. A novel AP-1 element in the CD95 ligand promoter is required for induction of apoptosis in hepatocellular carcinoma cells upon treatment with anticancer drugs. Mol Cell Biol. 2000; 20: 7826-37.
77. Grimm C, Wenzel A, Behrens A, Hafezi F, Wagner EF, Reme CE. AP-1 mediated retinal photoreceptor apoptosis is independent of N-terminal phosphorylation of c-Jun. Cell Death Differ. 2001; 8: 859-67. doi: 10.1038/sj.cdd.4400871.
78. Hafezi F, Grimm C, Wenzel A, Abegg M, Yaniv M, Reme CE. Retinal photoreceptors are apoptosis-competent in the absence of JunD/AP-1. Cell Death Differ. 1999; 6: 934-6. doi: 10.1038/sj.cdd.4400574.
79. Kasibhatla S, Brunner T, Genestier L, Echeverri F, Mahboubi A, Green DR. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol Cell. 1998; 1: 543-51.
80. Kolbus A, Herr I, Schreiber M, Debatin KM, Wagner EF, Angel P. c-Jun-dependent CD95-L expression is a rate-limiting step in the induction of apoptosis by alkylating agents. Mol Cell Biol. 2000; 20: 575-82.
81. Le-Niculescu H, Bonfoco E, Kasuya Y, Claret FX, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol. 1999; 19: 751-63.
82. Matsui K, Xiao S, Fine A, Ju ST. Role of activator protein-1 in TCR-mediated regulation of the murine fasl promoter. J Immunol. 2000; 164: 3002-8.
83. Rich KA, Zhan Y, Blanks JC. Aberrant expression of c-Fos accompanies photoreceptor cell death in the rd mouse. J Neurobiol. 1997; 32: 593-612.
84. Mahner S, Baasch C, Schwarz J, Hein S, Wolber L, Janicke F, Milde-Langosch K. C-Fos expression is a molecular predictor of progression and survival in epithelial ovarian carcinoma. Br J Cancer. 2008; 99: 1269-75. doi: 10.1038/sj.bjc.6604650.
85. Oliveira-Ferrer L, Rossler K, Haustein V, Schroder C, Wicklein D, Maltseva D, Khaustova N, Samatov T, Tonevitsky A, Mahner S, Janicke F, Schumacher U, Milde-Langosch K. c-FOS suppresses ovarian cancer progression by changing adhesion. Br J Cancer. 2014; 110: 753-63. doi: 10.1038/bjc.2013.774.
86. Ducruet AP, Vogt A, Wipf P, Lazo JS. Dual specificity protein phosphatases: therapeutic targets for cancer and Alzheimer’s disease. Annu Rev Pharmacol Toxicol. 2005; 45: 725-50. doi: 10.1146/annurev.pharmtox.45.120403.100040.
87. Shen J, Zhang Y, Yu H, Shen B, Liang Y, Jin R, Liu X, Shi L, Cai X. Role of DUSP1/MKP1 in tumorigenesis, tumor progression and therapy. Cancer Med. 2016; 5: 2061-8. doi: 10.1002/cam4.772.
88. Lu D, Wolfgang CD, Hai T. Activating transcription factor 3, a stress-inducible gene, suppresses Ras-stimulated tumorigenesis. J Biol Chem. 2006; 281: 10473-81. doi: 10.1074/jbc.M509278200.
89. Yan C, Boyd DD. ATF3 regulates the stability of p53: a link to cancer. Cell Cycle. 2006; 5: 926-9. doi: 10.4161/cc.5.9.2714.
90. Yan C, Lu D, Hai T, Boyd DD. Activating transcription factor 3, a stress sensor, activates p53 by blocking its ubiquitination. EMBO J. 2005; 24: 2425-35. doi: 10.1038/sj.emboj.7600712.
91. Kaspar AA, Reichert JM. Future directions for peptide therapeutics development. Drug Discov Today. 2013; 18: 807-17. doi: 10.1016/j.drudis.2013.05.011.
92. Hajek R, Masszi T, Petrucci MT, Palumbo A, Rosinol L, Nagler A, Yong KL, Oriol A, Minarik J, Pour L, Dimopoulos MA, Maisnar V, Rossi D, et al. A randomized phase III study of carfilzomib vs low-dose corticosteroids with optional cyclophosphamide in relapsed and refractory multiple myeloma (FOCUS). Leukemia. 2016. doi: 10.1038/leu.2016.176.