
INTRODUCTION
Gastric cancer (GC) is the second most common malignant cancer and the fourth leading cause of cancer-related deaths worldwide [1]. In China, approximately three hundred thousand cases of gastric cancer are diagnosed annually, and GC has become a major public health threat [2, 3]. GC is a multistep and slow progressing disease whose stages include chronic gastritis, intestinal metaplasia, hyperplasia, early gastric cancer and advanced cancer. As a precancerous lesion of the stomach, gastric intestinal metaplasia (GIM) has attracted much attention in recent years [4, 5].
In GIM, normal gastric epithelium cells are replaced with intestinal cell types [6, 7]. One study suggested that the incidence of GIM advancing to gastric cancer was 1.8% in a separate cohort of patients followed up for 1 to 10 years [8]. Moreover, this study showed that the rate was higher in type III GIM than that in type I non-cancerous GIM [8]. A recent study also showed that the origin of intestinal metaplasia of gastric mucosa cells is derived from basal stem cells [9]. Under normal conditions, basal stem cells could differentiate into gastric epithelial cells to maintain their active capabilities. The caudal-related homeobox gene CDX2 (caudal type homeobox 2), plays a critical role in the progression of basal stem cell differentiation [10–13]. In the normal adult gastric epithelium, the expression of CDX2 is low, but when the gastric epithelium is stimulated by adverse external factors, the expression of CDX2 could be up-regulated, leading to the formation of GIM [14–17]. However, the molecular mechanism of CDX2 up-regulation remains unclear.
Human telomerase, which consists of human telomerase associated protein (hTP1), human telomerase RNA (hTR) and human telomerase reverse transcriptase (hTERT), hTERT is a central factor based on a rate-limiting step in telomerase activity [18]. hTERT promotes the proliferation of different types of cancer [19–22]. Previously, we observed that hTERT promotes the proliferation and invasion of gastric cancer via interactions with different genes [23–25]. In addition, the suppression of hTERT could also inhibit the malignant activity of gastric cancer [26, 27]. Previous studies have shown that the expression of hTERT in human precancerous gastric lesions was higher compared with the normal gastric epithelium, while hTR and hTP1 did not show significant changes [28]. Consistent with a previous study, there is a close association between hTERT and GIM, but the detailed mechanism needs further study.
In this paper, we investigated the role of hTERT in IM. Furthermore, we found that hTERT promotes gastric intestinal metaplasia by up-regulating CDX2 expression and its potential mechanism.
RESULTS
hTERT was closely related with the expression of CDX2 in GIM and gastric cancer cells
In our study, we first detected the expression of hTERT and CDX2 in GIM tissue and corresponding normal gastric mucosa using immunohistochemistry. The expression of hTERT was increased in GIM tissues compared with that in normal gastric mucosa (Figure 1A). In addition, we measured the levels of CDX2 and its downstream gene MUC2, which was increased in GIM as a positive control (Figure 1A). We found that CDX2 and hTERT are highly expressed in GIM, which indicated that there was a strong relationship between hTERT and CDX2 in GIM tissue.
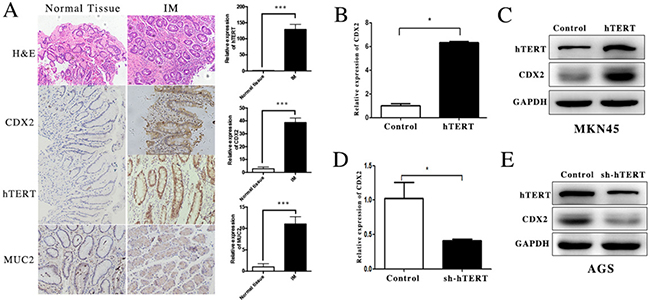
Figure 1: hTERT was closely related with the expression of CDX2 in GIM and gastric cancer cells. (A) The expression of hTERT, CDX2 and MUC2 was detected in GIM tissues and normal gastric mucosa using IHC (paired t-test was used to analyze data, ***p < 0.001). (B and C) The expression of CDX2 in MKN45 cell was detected using qRT-PCR and WB (unpaired t-test was used to analyze data, p < 0.05). (D and E) The expression of CDX2 in AGS cell was measured using qRT-PCR and WB (unpaired t-test was used to analyze data,*p < 0.05).
To further investigate the effect of hTERT on CDX2 expression, the hTERT over-expression plasmid was transfected into MKN45 cell line and hTERT sh-RNA plasmid was also transfected into the AGS cell line. A significant increase in CDX2 was observed at both the mRNA and protein levels after hTERT was overexpressed (Figure 1B and 1C). After suppressing the expression of hTERT in the AGS cell line, we found that the CDX2 mRNA and protein levels were decreased, as expected (Figure 1D and 1E). According to these results, hTERT could increase the expression of CDX2 at both the clinical and cellular levels.
hTERT increase the activity of CDX2 promoter partly through NF-κB signaling pathway
In a previous study, we showed that the expression of CDX2 was closely related with the activity of its promoter [10]. To investigate whether the increased expression of CDX2 was induced by the activation of hTERT on the CDX2 promoter, we transfected a CDX2 reporter plasmid into MKN45 and AGS cells. The results showed that hTERT has a provoking effect on CDX2 promoter activity in transcriptional activation in the MKN45 cell line, and its depletion inhibited the activity of the CDX2 promoter (Figure 2A and 2B). Based on the present results, we concluded that hTERT could increase the expression of CDX2 by increasing CDX2 promoter. However, a recent study showed that hTERT was not a transcriptional factor, which indicated that hTERT could not directly regulate the promoter of gene. Thus, we speculated that hTERT could bind with other transcription factors to stimulate CDX2 expression.
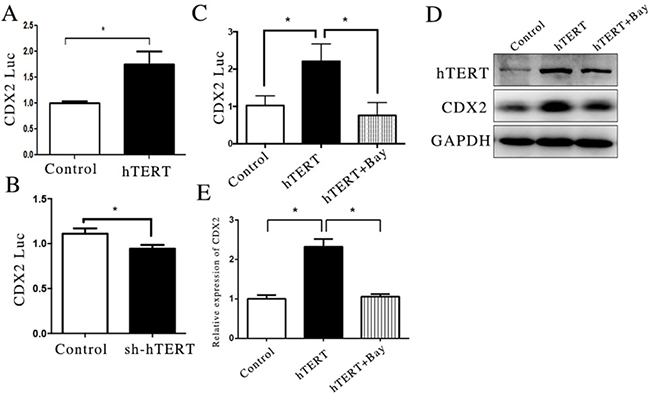
Figure 2: hTERT increase the activity of CDX2 promoter partly through NF-κB signaling pathway. (A and B) The luciferase activity of CDX2 in MKN45 cells and AGS cells, after hTERT was over-expressed or suppressed, respectively (unpaired t-test was used to analyze data, *p < 0.05). (C) The luciferase activity of CDX2 in MKN45 cells, after the NF-κB signaling pathway was blocked (unpaired t-test was used to analyze data, *p < 0.05). (D and E) The mRNA and protein levels of CDX2 were measured using qRT-PCR and WB, after the NF-KB signaling pathway was blocked (unpaired t-test was used to analyze data, *p < 0.05).
A recent study showed the involvement of the NF-κB signaling pathway in the formation of Barrett’s esophageal development [29, 30]. Moreover, recent studies have shown that hTERT could interact with the NF-κB signaling pathway [31]. According to these results, we explored the role of the NF-κB signaling pathway in GIM. Interestingly, the inhibition of NF-κB suppressed the increased CDX2 promoter activity induced by hTERT in the MKN45 cell line (Figure 2C). In addition, this phenomenon was also observed at both the CDX2 RNA and protein levels (Figure 2D and 2E). These results and those of a previous study suggest that NF-κB signaling pathway plays an important role in the up-regulation of CDX2, which is induced by hTERT. However, the detail mechanism remains unknown.
hTERT binds to the CDX2 promoter in the gastric intestinal metaplasia cell line MKN45
Both p65 and p50 were important components in the NF-κB signaling pathway, and these two transcription factors have shown multiple functions in various bioactivities. According to these results, we assessed whether p65 and p50 also participated in the regulation of CDX2 promoter activity via interactions with hTERT. In certain cancer cell lines, the p65 and p50 subunits of NF-κB generate opposing effects on CDX2 expression [32, 33]. Therefore, to determine the relationship between p65/p50 and CDX2 promoter activity, we constructed p65 and p50 eukaryotic plasmids and transfected these vectors into MKN45 cells. We found that both p65 and p50 increased CDX2 promoter activity (Figure 3A). The suppression of p65 and p50 could decrease CDX2 expression at the protein level (Figure 3B). Thus, we determined whether hTERT could directly bind p65 and/or p50 to the CDX2 promoter in GIM. To confirm this hypothesis, we used a Co-IP assay to assess whether hTERT could bind with p50 and p65. Interestingly, both p65 and p50 could bind to hTERT in the MKN45 cell line (Figure 3C). Furthermore, a ChIP assay was used to detect whether p65 and p50 could bind to the CDX2 promoter. The results showed that p50 could bind to the CDX2 promoter, whereas p65 did not (Figure 3D). This result indicated that hTERT binds to p50 to confirm the complex, which could directly activate the promoter of CDX2. However, the role for p65 in the regulation of the CDX2 promoter remains unclear.
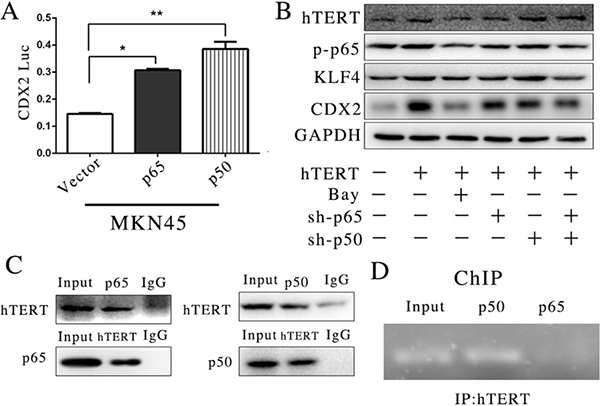
Figure 3: hTERT could bind with CDX2 promoter in intestinal metaplasia of gastric in the MKN45 cell line. (A) The luciferase activity of CDX2 in MKN45 cells, after p65 and p50 was over-expressed (unpaired t-test was used to analyze data, *p < 0.05, **p < 0.01). (B) The expression of CDX2, hTERT and KLF4 was detected using WB, after p65 and p50 were suppressed or the signaling pathway was completely suppressed by Bay11-7085. (C) The association of hTERT with p65 or p50 was measured by Co-IP. (D) The ChIP assay was applied to detect the binding relationship with the CDX2 promoter.
hTERT interacts with p65 to indirectly stimulate CDX2 expression
In the previous experiment, we found that the suppression of NF-KB signaling pathway could reduce the expression of CDX2. It has previously been reported that hTERT has a relationship with NF-κB in the nucleus and promotes the expression of NF-κB downstream gene transcription [28]. However, p65 did not bind and influence the promoter activity of CDX2 (Figure 3D). To explore the effect of hTERT on p65 protein phosphorylation in the nucleus, we used western blotting as a detection method, which showed that the over-expression of hTERT could increase phosphorylated p65 (Figure 4A), indicating that hTERT could activate p65.
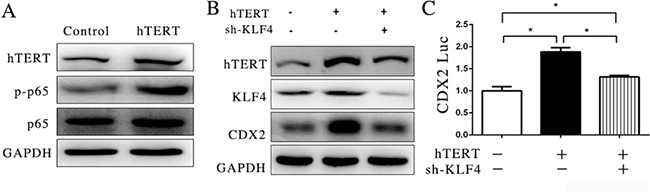
Figure 4: hTERT could interact with p65 to indirectly stimulate CDX2 expression. (A) The expression of p65 and p-p65 was detected using WB after hTERT was overexpressed in MKN45 cells. (B) The expression of CDX2 was measured after KLF4 was suppressed. (C) Luciferase activity of CDX2 promoter was measured after KLF4 was suppressed (unpaired t-test was used to analyze data, *p < 0.05).
KLF4 (Kruppel-like factor 4) is a transcription factor that is important in tumorigenesis. It has been reported that the KLF4 and CDX2 play an important role in Barrett’s epithelium development [34]. A previous study also showed that KLF4 could be activated through p65 [27]. Thus, we wondered whether KLF4 is sufficient to promote the expression of CDX2 induced by hTERT in the GIM. To confirm this hypothesis, we transfected shRNA to reduce the expression of KLF4 in MKN45 cells. The results showed that the upregulation of CDX2 induced by hTERT could be reduced when KLF4 expression was suppressed (Figure 4B). Moreover, we found that KLF4 suppression could decrease the activity of the CDX2 promoter (Figure 4C). These results indicated that hTERT could activate phosphorylated p65 and KLF4 expression.
hTERT activated KLF4 transcription via NF-κB pathway
We found that hTERT stimulated CDX2 promoter activity through KLF4 in a previous experiment. However, the role of KLF4 in GIM remains unclear. To understand the key role of KLF4 in GIM, we first used immunohistochemical staining to detect KLF4 levels in GIM tissue. KLF-positive cells with nuclear staining were clearly observed in GIM tissue (Figure 5A). Thus, we also assessed whether the overexpression of hTERT could activate the expression of KLF4. Western blotting and qRT-PCR assays were applied, showing that KLF4 mRNA and protein increased after hTERT was overexpressed in the MKN45 cell line (Figure 5B and 5C). Next, we focused on the regulatory mechanism of KLF4 induced by hTERT, we transfected a KLF4 reporter plasmid into MKN45 cells, and a Luciferase Reporter Assay was used to measure the activity of this promoter. We found that hTERT had a positive effect on KLF4 promoter activity because the activity of KLF4 promoter was highly increased (Figure 5D). A clear link between KLF4 and NF-κB pathway has been reported [27]. To examine the potential regulatory mechanism of KLF4 up-regulated by hTERT, we transfected a p65-overexpression plasmid in the MKN45 cell line and found that the overexpression of p65 increased the KLF4 expression at the mRNA, protein and promoter activity levels (Figure 5E, 5F and 5G). Moreover, we found that BAY 11-7085, an inhibitor of the NF-κB pathway, could significantly suppress the expression of KLF4 protein induced by hTERT (Figure 5H). These results suggest that hTERT activated KLF4 expression via the NF-κB pathway.
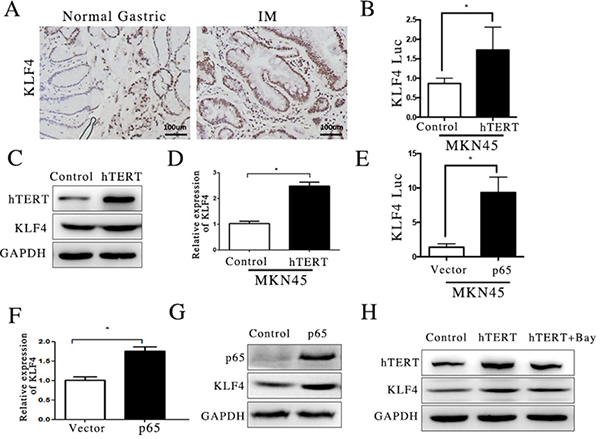
Figure 5: hTERT activated KLF4 transcription via NF-κB pathway. (A) The expression of KLF4 in normal gastric mucosa and GIM was measured using IHC. (B and C) The expression of KLF4 was measured using qRT-PCR and WB after hTERT was overexpressed (unpaired t-test was used to analyze data, *p < 0.05). (D) The luciferase activity of KLF4 promoter was measured after hTERT was overexpressed (unpaired t-test was used to analyze data, *p < 0.05). (E) The luciferase activity of KLF4 was detected after p65 was overexpressed (unpaired t-test was used to analyze data, *p < 0.05). (F and G) The mRNA and protein levels of KLF4 were measured after p65 was overexpressed (unpaired t-test was used to analyze data, *p < 0.05). (H) The level of KLF4 was detected using WB, after NF-κB signaling pathway was suppressed by Bay11-7085.
DISCUSSION
The process of GIM is stimulated by destructive factors, such as HP infection [35–37]. In the present study, the relationship between gastric cancer and GIM is clear, particularly type III of IM, which is associated with the incidence of the aggressive form of stomach cancer [38–41]. As a premalignant lesion of gastric carcinoma, GIM has received much attention [42, 43]. However, little information is known about the process and mechanism of GIM. Thus revealing its mechanism is important in preventing the tumorigenesis of gastric cancer.
CDX2 plays a pivotal role in the differentiation of stem cells [44–47]. Under normal conditions, the expression of CDX2 was detected in IM but was absent in normal gastric cells [48]. In previous studies, researchers showed that the up-regulation of CDX2 expression during IM is important in the determination of the incomplete type of IM (type IIb, III) and early-stage GC, which had also been demonstrated in animals [49–52]. However, the regulation of CDX2 in GIM remains largely unknown.
Previous studies have suggested that hTERT serves as a critical character in the proliferation of tissue progenitor cells and human gastric cancer cells [20–23]. Interestingly, hTERT expression was higher in GIM compared with normal gastric epithelial cells. However, few studies have reported the molecular mechanism of hTERT in GIM. In this study, we detected the expression of hTERT and CDX2 at both the mRNA and protein levels in normal gastric mucosa and GIM, and hTERT expression was obviously increased in GIM (Figure 1A). In addition, the same trend was observed in the expression of CDX2 (Figure 1A).
To investigate whether hTERT could directly activate the expression of CDX2, we used qRT-PCR and western blot assays to detect the expression of CDX2, after overexpression and suppression of hTERT were procced in different cells. The results showed that hTERT could activate CDX2 expression at both the mRNA and protein levels, whereas CDX2 expression was decreased in AGS cells under knockout of hTERT (Figure 2). In addition, hTERT increased the promoter activity of CDX2 (Figure 2). These results indicated that hTERT could stimulate the level of CDX2 expression via controlling transcription activity, which promoted the process of GIM.
In addition to maintaining the elongation of telomere, Ghosh et al reported that telomerase could regulate NF-κB-dependent transcription, promoting the phosphorylation of p65 [31]. Another study reported that TERT interacts with NF-κB/p65 and activates MMP expression as a transcriptional regulator [53]. Consistent with previous studies, we showed that hTERT interacts with p65 in the nucleus and promotes the level of phosphorylated p65, which is likely associated with the suppression of degradation processes (Figure 4). These results revealed that hTERT enhanced NF-κB dependent transcription.
Study has reported that KLF4-induced activation of the CDX2-promoter is controlled by a site located between-94 and +52, by dual luciferase assay and ChIP assay in Barrett’s epithelium development [29]. We also found that inhibition of KLF4 could lead to decrease of CDX2, at both protein and promoter level in our study (Figure 4), suggesting that KLF4 could influce the activity of CDX2 promoter. Subsequently, we also examined the effect of hTERT on the expression of KLF4 in our study. The results showed that both the mRNA and protein levels of KLF4 and reporter were augmented in the MKN45 cell line after transfecting hTERT (Figure 5). This result suggests that hTERT could stimulate the expression of KLF4. The promoter of KLF4 was positively regulated by p50 and p65 following exposure to bile acids in rat primary cultured keratinocytes [29]. In the present study, the NF-κB inhibitor can significantly suppress the expression of KLF4, although hTERT was over-expressed, which suggests that hTERT increased KLF4 expression via the NF-κB signaling pathway. To examine whether KLF4 increases the CDX2 expression induced by hTERT, we suppressed the expression of KLF4 and found that KLF4 could reduce the expression of CDX2, at least in part.
P65 and p50 are key factors in the NF-κB signaling pathway [54], but their function in GIM largely remained unclear [55, 56]. In the present study, we investigated the effect of p65/p50 on CDX2 expression in IM. The results showed that both p65 and p50 positively affect CDX2 promoter activity, whereas the suppression of p65 and p50 decreased the CDX2 protein levels (Figure 3). In the MKN45 cell line, only p50 could bind to the CDX2 promoter (Figure 3). These findings suggested that hTERT increases CDX2 expression through transcriptional activation via nuclear translocation and the binding of the p50 subunit of NF-κB in GIM.
In conclusion, we have shown that hTERT was increased in GIM, which may promote GIM through the NF-κB signaling pathway in a direct/indirect manner. As shown in the present study, hTERT bound to p65 to improve the expression of KLF4, which could activate the CDX2 promoter; in addition, hTERT could promote the expression of CDX2 through the direct binding of p50 to the CDX2 promoter (Figure 6).
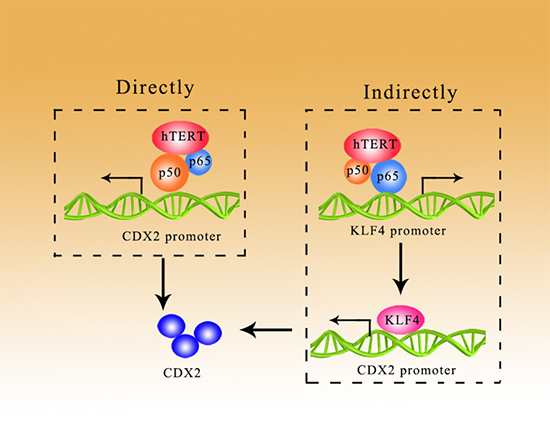
Figure 6: hTERT could promote the expression of CDX2 in a direct/indirect manner. Direct manner: hTERT binds with p50 to increase CDX2 expression at the promoter level. Indirect manner: hTERT binds with p65 to improve the expression of KLF4, which activates the CDX2 promoter.
MATERIALS AND METHODS
Patient tissue samples
Nineteen gastric intestinal metaplasia and paired normal gastric mucosa tissue samples were obtained from the Department of Gastroenterology, the Second Affiliated Hospital of Third Military Medical University during October 2013 and July 2014. For this study, inclusion criteria were age 18–70 years, gastric intestinal metaplasia was diagnosed by one experienced endoscopic doctor, written informed consent was obtained from all patient samples. The exclusion criteria were patients with gastric cancer, coagulopathy and gastric variceal. Detailed information (such as age, sex, histological and clinical stage, and pathological data) was obtained from the hospital records. This study was approved by the Institutional Review Board of the Third Military Medical University. Two independent pathologists determined the pathological types of intestinal metaplasia according to the criteria established by classification.
Immunohistochemistry
Gastric intestinal metaplasia and paired normal gastric epithelium tissue samples were fixed with 10% neutral formalin and embedded in paraffin, and 4-μm-thick sections were prepared. Immunostaining was performed using the avidin–biotin–peroxidase complex method (Ultrasensitive™, MaiXin, Fuzhou, China). The sections were deparaffinized in xylene, rehydrated with graded alcohol, and subsequently boiled in 0.01-M citrate buffer (pH 6.0) for 2 min in an autoclave. Hydrogen peroxide (0.3 %) was applied to block endogenous peroxide activity, and the sections were incubated with normal goat serum to reduce nonspecific binding. The tissue sections were incubated with anti-hTERT rabbit polyclonal antibody (1:100 dilution, Abcam, USA), anti-CDX2 rabbit polyclonal antibody (1:100 dilution, CST, USA), and anti-MUC2 rabbit polyclonal antibody (1:100 dilution; Abcam, USA). Rabbit immunoglobulin was used as a negative control. Staining for both antibodies was performed at room temperature for 2 h. Biotinylated goat anti-mouse serum IgG was used as a secondary antibody. After washing, the sections were incubated with streptavidin/biotin conjugated with horseradish peroxidase, and the peroxidase reaction was developed with 3, 3′-diaminobenzidine tetrahydrochloride. Counterstaining with hematoxylin was performed, and the sections were dehydrated in ethanol before mounting. The immunostaining criteria were scored on a semiquantitative scale by evaluating representative tumor areas. According to previous reports, we counted 400 cells and calculated the percentage of positively stained cells. The expression was scored as positive when >10 % of the cells in a specimen were stained. Image Pro Plus 6.0 (Media Cybernetics, USA) was used to analyze the absorbance in each sight.
Vector construction
A total of 2881 bp of the CDX2 promoter (Gene ID: 1045) was amplified using PCR and cloned into the Kpn I and Xho I sites in a pGL3-basic luciferase vector (Promega, Madison, WI, USA) to generate p CDX2/2881-Luc. The +1 position refers to the major transcription start site identified in the CDX2 gene. Furthermore, 2012 bp of the KLF4 promoter (Gene ID: 9314) was amplified using PCR, cloned into the Sac I and Hind III sites of a pGL3-basic luciferase vector to generate pKLF4/2012-Luc. Position +1 refers to the major transcription start site identified in the KLF4 gene. As an internal control for the dual luciferase assay, pRL-TATA-Renilla-Luc was used.
cDNA encoding full-length human telomerase (Gene ID: 7015) was amplified by PCR and cloned into the pIERS2-EGFP Vector (Invitrogen, Carlsbad, CA, USA). The pIERS2-EGFP Vector without hTERT sequences was used as a negative control, as previously reported. According to the present method, p65 (Gene ID: 5970) and p50 (Gene ID: 4790) expression vectors were also constructed in the M95 vector (Funeng, Guangzhou, China).
Construction of small interfering RNA plasmid vector
Two shRNA constructs of KLF4 were selected based on two complementary oligonucleotides
(5′-caccgaccaggcactaccgtaaacattcaagagatgtttacggta gtgcctggtctttttt-3′ and 5′-gatccaaaaaagaccaggcactaccgtaaa catctcttgaatgtttacggtagtgcctggtc-3′). Two shRNA sequences targeting p65 were also selected: 5′-caccgcccatggaattccagtacctttcaagagaaggtactggaattccatg ggc-3′ and 5′-gatccaaaaaagcccatggaattccagtaccttctcttgaa aggtactggaattccatgggc-3′. Furthermore, two shRNA sequences for p50 were listed as follows: 5′-caccggacagtactacctacgatggttcaagagaccatcgtaggtagtactgt ccttttttg-3′ and 5′-gatccaaaaaaggacagtactacctacgatggtctctt gaaccatcgtaggtagtactgtcc-3′. sh-RNA specific targeting hTERT sequence: 5′-caccgaccaggcactaccgtaaacattcaagag atgtttacggtagtgcctggtctttttt-3′ and 5′-gatccaaaaaagaccaggca ctaccgtaaacatctcttgaatgtttacggtagtgcctggtc-3’.
Cell culture and cell transfection
Human gastric cancer cell lines MKN45 and AGS were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China), and cultured in Dulbecco’s Modified Eagle Medium (Sigma, USA) and supplemented with 10% heat-inactivated fetal bovine serum (FBS, Tianjin, China). MKN45 and AGS cells were separately cultured onto six-well plates and transfected with 2 μg of each plasmid in each well using 4 μl of Lipofectamine 200 (Invitrogen, USA), according to the manufacturer’s instructions. Bay11-7085 (Sigma, USA), a pharmacologic inhibitor, was used to block IκB phosphorylation. Cells were pretreated with Bay11-7085 at a concentration of 5 μM for 24 hours after transfection.
phosphorylation. Cells were pretreated with Bay11-7085 at a concentration of 5 μM for 24 hours after transfection.
RNA extraction and real-time PCR
Total RNA was extracted using the Trizol method (TaKaRa, Japan), according to a standard protocol. cDNA was synthesized using a reverse transcription kit (TaKaRa, Japan). A real-time fluorescence PCR assay based on SYBR Premix Ex Taq (TaKaRa, Japan) was then performed. Sangon Biotech synthesized the primer sequence (Sangon Biotech, China).
Protein extraction and western blot analysis
All the proteins were extracted using RIPA buffer containing PMSF (Beyotime, China). The protein samples were loaded onto the SDS-PAGE gels and electrotransferred to PVDF membranes (Milipore, America). The membranes were blocked with 5% nonfat milk for 2 hours, and incubated overnight at 4°C with specific primary antibodies, i.e., anti-hTERT (1:1000, Abcam, USA), anti-KLF4 (1:1000, Abcam, USA), anti-p65 (1:1000, Abcam, USA), anti-p50 (1:1000, Abcam, USA), anti- CDX2 (1:1000, Abcam, USA), anti-MUC2 (1:1000, Abcam, USA) and anti-GAPDH (1:500,Boaoshen, China) antibodies. The membranes were subsequently incubated with a secondary antibody. The bands were quantified using Quantity One software (Bio-Rad, USA), and the gray value was analyzed using GraphPad Prism software (GraphPad, USA).
Luciferase reporter assay
Various reporter plasmids (0.1 μg) and different types of overexpression plasmids (0.1 μg) were cotransfected into MKN45 cell line. Renilla luciferase plasmids (0.1 μg) were cotransfected as an internal control for transfection efficiency. At 24 h after transfection of the luciferase vectors, a dual luciferase reporter assay (Promega, USA) was performed according to the manufacturer’s instructions. Each transfection was performed in triplicate.
Chromatin immunoprecipitation
The chromatin immunoprecipitation (Ch-IP) analysis was performed using an EpiQuik Chromatin Immunoprecipitation Kit (Epigentek Group, USA). MKN45 cells were transiently transfected with a CDX2 promoter vector for 48 h, after which a ChIP analysis was performed. Total DNA prior to immunoprecipitation was used as the input value. Chromatin was immunoprecipitated with anti-p50 and anti-p65 antibodies and IgG antibody was used as a negative control. The primers were designed according to pervious study [57], and listed as follows: forward primer 5-GAGGGGTTGTGCGTAGAGTG-3 reverse primer 5-CCTTCCGTGATTAACGAGTGT-3
Statistical analysis
The data were expressed as the means ± standard deviation (SD) and analyzed using GraphPad Prism5 software (GraphPad, America). Values were compared using Student’s t-test or one-way ANOVA. p < 0.05 was considered statistically significant. Three biological replicates were applied in each experiment.
ACKNOWLEDGMENTS AND FUNDING
This work was nancially supported by grants from the National Nature Science Foundation of China (No. 81572460), the Nature Science Foundation of Chongqing (CSTSC2013jjb10017).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics. 2012. CA Cancer J Clin. 2015; 65:87–108.
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China 2015. CA Cancer J Clin. 2016; 66:115–132.
3. Chen W. Cancer statistics: updated cancer burden in China. Chin J Cancer Res. 2015; 27:1.
4. Liu KS, Wong IO, Leung WK. Helicobacter pylori associated gastric intestinal metaplasia: Treatment and surveillance. World J Gastroenterol. 2016; 22:1311–1320.
5. Park YH, Kim N. Review of atrophic gastritis and intestinal metaplasia as a premalignant lesion of gastric cancer. J Cancer Prev. 2015; 20:25–40.
6. Weledji EP, Enow Orock G, Ngowe MN. Intestinal metaplasia and anastomotic recurrence of gastric carcinoma. J Gastrointest Oncol. 2014; 5:474–480.
7. Kang JM, Lee BH, Kim N, Lee HS, Lee HE, Park JH, Kim JS, Jung HC, Song IS. CDX1 and CDX2 expression in intestinal metaplasia, dysplasia and gastric cancer. J Korean Med Sci. 2011; 26:647–653.
8. Silva F, Carvalho F, Peixoto A, Teixeira A, Almeida R, Reis C, Bravo LE, Realpe L, Correa P, David L. MUC1 polymorphism confers increased risk for intestinal metaplasia in a Colombian population with chronic gastritis. Eur J Hum Genet. 2003; 11:380–384.
9. Bjerknes M, Cheng H. Multipotential stem cells in adult mouse gastric epithelium. Am J Physiol Gastrointest Liver Physiol. 2002; 283:G767–777.
10. Barros R, da Costa LT, Pinto-de-Sousa J, Duluc I, Freund JN, David L, Almeida R. CDX2 autoregulation in human intestinal metaplasia of the stomach: impact on the stability of the phenotype. Gut. 2011; 60:290–298.
11. Mesquita P, Jonckheere N, Almeida R, Ducourouble MP, Serpa J, Silva E, Pigny P, Silva FS, Reis C, Silberg D, Van Seuningen I, David L. Human MUC2 mucin gene is transcriptionally regulated by Cdx homeodomain proteins in gastrointestinal carcinoma cell lines. J Biol Chem. 2003; 278:51549–51556.
12. Friis-Hansen L, Rieneck K, Nilsson HO, Wadstrom T, Rehfeld JF. Gastric inflammation, metaplasia, and tumor development in gastrin-deficient mice. Gastroenterology. 2006; 131:246–258.
13. Barros R, Freund JN, David L, Almeida R. Gastric intestinal metaplasia revisited: function and regulation of CDX2. Trends Mol Med. 2012; 18:555–563.
14. Asano N, Imatani A, Watanabe T, Fushiya J, Kondo Y, Jin X, Ara N, Uno K, Iijima K, Koike T, Strober W, Shimosegawa T. Cdx2 Expression and Intestinal Metaplasia Induced by H. pylori Infection of Gastric Cells Is Regulated by NOD1-Mediated Innate Immune Responses. Cancer Res. 2016; 76:1135–1145.
15. Barros R, Pereira B, Duluc I, Azevedo M, Mendes N, Camilo V, Jacobs RJ, Paulo P, Santos-Silva F, van Seuningen I, van den Brink GR, David L, Freund JN, Almeida R. Key elements of the BMP/SMAD pathway co-localize with CDX2 in intestinal metaplasia and regulate CDX2 expression in human gastric cell lines. J Pathol. 2008; 215:411–420.
16. Shin CM, Kim N, Chang H, Kim JS, Lee DH, Jung HC. Follow-Up Study on CDX1 and CDX2 mRNA Expression in Noncancerous Gastric Mucosae After Helicobacter pylori Eradication. Dig Dis Sci. 2016; 61:1051–1059.
17. Masood MA, Loya A, Yusuf MA. CDX2 as a prognostic marker in gastric cancer. Acta Gastroenterol Belg. 2016; 79:197–200.
18. Qin Y, Guo H, Tang B, Yang SM. The non-reverse transcriptase activity of the human telomerase reverse transcriptase promotes tumor progression (review). Int J Oncol. 2014; 45: 525–531.
19. Catarino R, Araujo A, Coelho A, Gomes M, Nogueira A, Lopes C, Medeiros RM. Prognostic significance of telomerase polymorphism in non-small cell lung cancer. Clin Cancer Res. 2010; 16:3706–3712.
20. Listerman I, Sun J, Gazzaniga FS, Lukas JL, Blackburn EH. The major reverse transcriptase-incompetent splice variant of the human telomerase protein inhibits telomerase activity but protects from apoptosis. Cancer Res. 2013; 73:2817–2828.
21. Yasukawa M, Ochi T, Fujiwara H. Relapse of renal cell carcinoma with disappearance of HLA class I following hTERT peptide vaccination. Ann Oncol. 2010; 21:2122–2124.
22. Qin Y, Tang B, Hu CJ, Xiao YF, Xie R, Yong X, Wu YY, Dong H, Yang SM. An hTERT/ZEB1 complex directly regulates E-cadherin to promote epithelial-to-mesenchymal transition (EMT) in colorectal cancer. Oncotarget. 2016; 7:351–361. doi: 10.18632/oncotarget.5968.
23. He B, Xiao YF, Tang B, Wu YY, Hu CJ, Xie R, Yang X, Yu ST, Dong H, Zhao XY, Li JL, Yang SM. hTERT mediates gastric cancer metastasis partially through the indirect targeting of ITGB1 by microRNA-29a. Sci Rep. 2016; 6:21955.
24. Tang B, Xie R, Qin Y, Xiao YF, Yong X, Zheng L, Dong H, Yang SM. Human telomerase reverse transcriptase (hTERT) promotes gastric cancer invasion through cooperating with c-Myc to upregulate heparanase expression. Oncotarget. 2016; 7:11364–11379. doi: 10.18632/oncotarget.6575.
25. Hu C, Ni Z, Li BS, Yong X, Yang X, Zhang JW, Zhang D, Qin Y, Jie MM, Dong H, Li S, He F, Yang SM. hTERT promotes the invasion of gastric cancer cells by enhancing FOXO3a ubiquitination and subsequent ITGB1 upregulation. Gut. 2017; 66:31–42.
26. Zhang D, Xiao YF, Zhang JW, Xie R, Hu CJ, Tang B, Wang SM, Wu YY, Hao NB, Yang SM. miR-1182 attenuates gastric cancer proliferation and metastasis by targeting the open reading frame of hTERT. Cancer Lett. 2015; 360:151–159.
27. Chen L, Lu MH, Zhang D, Hao NB, Fan YH, Wu YY, Wang SM, Xie R, Fang DC, Zhang H, Hu CJ, Yang SM. miR-1207-5p and miR-1266 suppress gastric cancer growth and invasion by targeting telomerase reverse transcriptase. Cell Death Dis. 2014; 5:e1034.
28. Duarte MC, Babeto E, Leite KR, Miyazaki K, Borim AA, Rahal P, Silva AE. Expression of TERT in precancerous gastric lesions compared to gastric cancer. Braz J Med Biol Res. 2011; 44:100–104.
29. Kazumori H, Ishihara S, Takahashi Y, Amano Y, Kinoshita Y. Roles of Kruppel-like factor 4 in oesophageal epithelial cells in Barrett's epithelium development. Gut. 2011; 60:608–617.
30. Rau TT, Rogler A, Frischauf M, Jung A, Konturek PC, Dimmler A, Faller G, Sehnert B, El-Rifai W, Hartmann A, Voll RE, Schneider-Stock R. Methylation-dependent activation of CDX1 through NF-kappaB: a link from inflammation to intestinal metaplasia in the human stomach. Am J Pathol. 2012; 181:487–498.
31. Ghosh A, Saginc G, Leow SC, Khattar E, Shin EM, Yan TD, Wong M, Zhang Z, Li G, Sung WK, Zhou J, Chng WJ, Li S, et al. Telomerase directly regulates NF-kappaB-dependent transcription. Nat Cell Biol. 2012; 14:1270–1281.
32. Debruyne PR, Witek M, Gong L, Bribe R, Chervoneva I, Jin T, Domon-Cell C, Palazzo JP, Freund JN, Li P, Pitari GM, Schulz S, Waldman SA. Bile acids induce ectopic expression of intestinal guanylyl cyclase C Through nuclear factor-kappaB and Cdx2 in human esophageal cells. Gastroenterology. 2006; 130:1191–1206.
33. Obon-Santacana M, Kaaks R, Slimani N, Lujan-Barroso L, Freisling H, Ferrari P, Dossus L, Chabbert-Buffet N, Baglietto L, Fortner RT, Boeing H, Tjønneland A, Olsen A, et al. Dietary intake of acrylamide and endometrial cancer risk in the European Prospective Investigation into Cancer and Nutrition cohort. Br J Cancer. 2014; 111:987–997.
34. Kim S, Domon-Dell C, Wang Q, Chung DH, Di Cristofano A, Pandolfi PP, Freund JN, Evers BM. PTEN and TNF-alpha regulation of the intestinal-specific Cdx-2 homeobox gene through a PI3K, PKB/Akt, and NF-kappaB-dependent pathway. Gastroenterology. 2002; 123:1163–1178.
35. El-Zimaity HM, Ota H, Graham DY, Akamatsu T, Katsuyama T. Patterns of gastric atrophy in intestinal type gastric carcinoma. Cancer. 2002; 94:1428–1436.
36. Sue S, Shibata W, Maeda S. Helicobacter pylori-Induced Signaling Pathways Contribute to Intestinal Metaplasia and Gastric Carcinogenesis. Biomed Res Int. 2015; 2015:737621.
37. Nakajima N, Kozu K, Kobayashi S, Nishiyama R, Okubo R, Akai Y, Moriyama M, Kinukawa N. The expression of IGF-1R in Helicobacter pylori-infected intestinal metaplasia and gastric cancer. J Clin Biochem Nutr. 2016; 59:53–57.
38. Higa JT, Hwang JH. Gastric intestinal metaplasia: An irreversible risk factor for gastric cancer? Gastrointest Endosc. 2016; 84:625–627.
39. Jiang JX, Liu Q, Zhao B, Zhang HH, Sang HM, Djaleel SM, Zhang GX, Xu SF. Risk factors for intestinal metaplasia in a southeastern Chinese population: an analysis of 28,745 cases. J Cancer Res Clin Oncol. 2016.
40. O'Connor A, McNamara D, O'Morain CA. Surveillance of gastric intestinal metaplasia for the prevention of gastric cancer. Cochrane Database Syst Rev. 2013:CD009322.
41. Reddy KM, Chang JI, Shi JM, Wu BU. Risk of Gastric Cancer Among Patients With Intestinal Metaplasia of the Stomach in a US Integrated Health Care System. Clin Gastroenterol Hepatol. 2016; 14:1420–1425.
42. Shichijo S, Hirata Y, Niikura R, Hayakawa Y, Yamada A, Ushiku T, Fukayama M, Koike K. Histologic intestinal metaplasia and endoscopic atrophy are predictors of gastric cancer development after Helicobacter pylori eradication. Gastrointest Endosc. 2016; 84:618–624.
43. De Re V, Orzes E, Canzonieri V, Maiero S, Fornasarig M, Alessandrini L, Cervo S, Steffan A, Zanette G, Mazzon C, De Paoli P, Cannizzaro R. Pepsinogens to Distinguish Patients With Gastric Intestinal Metaplasia and Helicobacter pylori Infection Among Populations at Risk for Gastric Cancer. Clin Transl Gastroenterol. 2016; 7:e183.
44. Phillips RW, Frierson HF, Jr., Moskaluk CA. Cdx2 as a marker of epithelial intestinal differentiation in the esophagus. Am J Surg Pathol. 2003; 27:1442–1447.
45. Stringer EJ, Duluc I, Saandi T, Davidson I, Bialeka M, Sato T, Barker N, Clevers H, Pritchard CA, Winton DJ, Wright NA, Freund JN, Deschamps J, Beck F. Cdx2 determines the fate of postnatal intestinal endoderm. Development. 2012; 139:465–474.
46. Camilo V, Garrido M, Valente P, Ricardo S, Amaral AL, Barros R, Chaves P, Carneiro F, David L, Almeida R. Differentiation reprogramming in gastric intestinal metaplasia and dysplasia: role of SOX2 and CDX2. Histopathology. 2015; 66:343–350.
47. Jang BG, Lee BL, Kim WH. Intestinal Stem Cell Markers in the Intestinal Metaplasia of Stomach and Barrett's Esophagus. PLoS One. 2015; 10:e0127300.
48. Uesaka T, Kageyama N. Cdx2 homeodomain protein regulates the expression of MOK, a member of the mitogen-activated protein kinase superfamily, in the intestinal epithelial cells. FEBS Lett. 2004; 573:147–154.
49. Xiao ZY, Ru Y, Sun JT, Gao SG, Wang YF, Wang LD, Feng XS. Expression of CDX2 and villin in gastric cardiac intestinal metaplasia and the relation with gastric cardiac carcinogenesis. Asian Pac J Cancer Prev. 2012; 13:247–250.
50. Khor TS, Alfaro EE, Ooi EM, Li Y, Srivastava A, Fujita H, Park Y, Kumarasinghe MP, Lauwers GY. Divergent expression of MUC5AC, MUC6, MUC2, CD10, and CDX-2 in dysplasia and intramucosal adenocarcinomas with intestinal and foveolar morphology: is this evidence of distinct gastric and intestinal pathways to carcinogenesis in Barrett Esophagus? Am J Surg Pathol. 2012; 36:331–342.
51. Tatsugami M, Ito M, Tanaka S, Yoshihara M, Matsui H, Haruma K, Chayama K. Bile acid promotes intestinal metaplasia and gastric carcinogenesis. Cancer Epidemiol Biomarkers Prev. 2012; 21:2101–2107.
52. Bornschein J, Toth K, Selgrad M, Kuester D, Wex T, Molnar B, Tulassay Z, Malfertheiner P. Dysregulation of CDX1, CDX2 and SOX2 in patients with gastric cancer also affects the non-malignant mucosa. J Clin Pathol. 2013; 66:819–822.
53. Ding D, Xi P, Zhou J, Wang M, Cong YS. Human telomerase reverse transcriptase regulates MMP expression independently of telomerase activity via NF-kappaB-dependent transcription. FASEB J. 2013; 27:4375–4383.
54. Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 2010; 1799:775–787.
55. Huang X, Lv B, Zhang S, Dai Q, Chen BB, Meng LN. Effects of radix curcumae-derived diterpenoid C on Helicobacter pylori-induced inflammation and nuclear factor kappa B signal pathways. World J Gastroenterol. 2013; 19:5085–5093.
56. Li S, Chen X, Zhou L, Wang BM. Farnesoid X receptor signal is involved in deoxycholic acid-induced intestinal metaplasia of normal human gastric epithelial cells. Oncol Rep. 2015; 34:2674–2682.
57. Hou XF, Zhang HY, Zhang X, Lynch JP, Strauch ED, Wang JY, Melton SD, Genta RM, Wang DH, Spechler SJ, Souza RF. Acid and Bile Salt Induced CDX2 Expression Differs in Squamous Cells from Patients with and without Barrett’s Esophagus. Gastroenterology. 2010; 139:194–203.