
INTRODUCTION
Many cancer treatment modalities either interfere with DNA metabolism or with the repair of DNA damage. Combination of different, carefully selected modalities can enhance clinical outcomes. Here, we investigate the effectiveness of different combinations of three modalities which interfere with DNA integrity or DNA repair: cisplatin (cDDP), mild hyperthermia (heating the tumour to 40–43°C for 1 h) and Poly-(ADP-ribose)-polymerase1 inhibitors (PARP1-i).
A high percentage of breast and ovarian cancers (~50–85% and ~30–66 %, respectively) are caused by mutations in BRCA1 or BRCA2 genes [1–4]. Tumours with mutations in either of these genes require homologous recombination (HR) for repair [5]. Inactive HR can be due to mutations in BRCA1 or BRCA2, which may result in potentially lethal accumulation of DNA double strand breaks (DSBs). HR-deficient (c.q. BRCA-deficient) cells are thus exquisitely sensitive to PARP1-i [6]. Importantly, this also implies that healthy, HR-proficient cells are not targeted by PARP1-i, which makes this therapy particularly desirable for patients with HR-deficient tumours [5, 7]. Clinical trials have indeed confirmed the effectiveness of PARP1-i as a single treatment against BRCA-deficient tumours [8, 9].
In HR-proficient tumours, synthetic lethality can also be induced by combining PARP1-i with a local treatment of mild hyperthermia [5, 6, 10–15], which causes degradation of BRCA2 for several hours [13] and thereby HR deficiency at the heated tumour site. Combination of hyperthermia (HT) with PARP1-i thus creates a possibility to induce synthetic lethality in every tumour type that can be heated locally [13, 16]. Cisplatin (cDDP) is a widely used chemotherapeutic agent that is combined with HT (hence called thermochemotherapy) as standard treatment for previously irradiated patients with recurrent cervical a . behind [17–19] cDDP induces DSBs that are usually repaired by HR, because cDDP disrupts the non-homologous end joining (NHEJ), the other major DSB repair pathway [20, 21]. In absence of HR and NHEJ, a PARP1-dependent back-up NHEJ (b-NHEJ) pathway can take over the repair of DSBs [22]. As a consequence, a combination of HT, cDDP and PARP1-i could potentially cause an overload of DSBs while simultaneously interfering with all major DSB repair pathways [23]. The accumulation of unrepaired DSBs can result in cell death.
In this study, HR-proficient cell lines (R1, SiHa, HeLa) and a HR-proficient rhabdomyosarcoma allograft model were used to investigate the effectiveness of treatments combining PARP1-i, with mild HT and cDDP. Cell survival as well as cell cycle analyses, quantification of γ-H2AX foci and apoptotic assays were performed. Finally, the effects of the different treatments on tumour outgrowth were measured in vivo.
RESULTS
Addition of PARP1-i to cDDP-based thermochemotherapy diminishes cell survival
The response of cells to HT was investigated for BRCA2 levels (Figure 1A). After HT, BRCA2 levels are downregulated. Effects of HT on homologous recombination were studied by scoring co-localization of Rad51 and γ-H2AX (Figure 1B). After RT alone, a clear co-localization of γ-H2AX, a protein detecting DNA double strand breaks and Rad51, a protein involved in homologous recombination is observed. After RT + HT, Rad51 foci are absent, indicating homologous recombination is not active anymore. To directly evaluate the effect of combinations of treatments with HT, cDDP and PARP1-i, clonogenic assays were conducted using the R1, SiHa and HeLa cells (Figure 1C). For all cell lines, therapy with the PARP1-i alone killed 30–40% of the cells. As such, treatment with PARP1-i was only slightly more effective than HT as a single treatment. cDDP was the most effective monotherapy. The combination treatment of PARP1-i with HT was equally effective as cDDP alone, and more effective than PARP1-i or HT alone. PARP1-i combined with cDDP was more effective than cDDP alone in the R1 cell line. In SiHa and HeLa cells, PARP1-i plus cDDP demonstrated a small decrease in cell survival, compared to cDDP alone. Combinational treatment of cDDP and HT was highly toxic and around 80–90% of the cells did not survive this treatment.
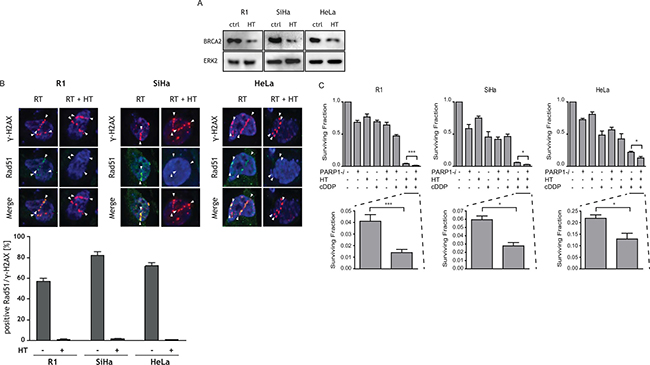
Figure 1: The effects of PARP1-i, HT, cDDP and combined treatments on BRCA2, Rad51 and cell survival are shown. (A) Western blots demonstrating R1, SiHa and HeLa cells are BRCA2 proficient. After HT, BRCA2 is downregulated. (B) γ-H2AX and Rad51 co-localization, to investigate activity of homologous recombination. (C) Clonogenic assays were performed to study the effect of the different treatment combinations, 10–12 days after treatments. The addition of PARP1-i to cDDP-based thermochemotherapy resulted in a significantly lower cell survival compared to cDDP-based thermochemotherapy alone. R1: p = 0.0008, SiHa: p = 0.034, HeLa: p = 0.021. The bar graph shows the mean of at least five independent experiments. From left to right: R1, SiHa, Hela cells. *p < 0.05, **p < 0.01, ***p < 0.001.
The addition of PARP1-i to cDDP-based thermochemotherapy caused a higher than 2-fold reduction in cell survival in R1 cells, an almost 2-fold reduction in SiHa cells and a ~1.5-fold reduction in HeLa cells.
Triple modality treatment leads to accumulation of DNA damage
Formation of γ-H2AX, which represents unrepaired DSBs, was analysed by flow cytometry, in order to identify a possible mechanism for differences in cell survival analyses after the triple modality treatment (Figure 2A). Cells grown on cover slips, treated with different combinations of cDDP, HT and PARP1-i were used for immunocytochemistry. For each condition one representative cell is depicted in Figure 2B. An up to 1.5-fold increase in γ-H2AX intensity was found after any of the single- and double-treatments. The load of DNA damage after addition of PARP1-i to cDDP-based thermochemotherapy was significantly higher than after cDDP-based thermochemotherapy alone.
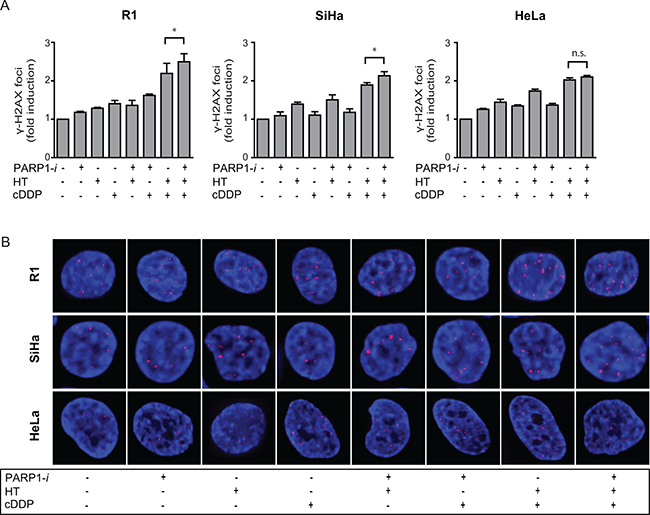
Figure 2: DSBs were analysed using the γ-H2AX assay. (A) The induction of DSBs in R1 and SiHa was significantly higher after addition of PARP1-i to cDDP-based thermochermotherapy. In HeLa cells this was not found to be significant, although a trend is seen. R1: p = 0.048, SiHa: p = 0.035, HeLa: p = 0.068 From left to right: R1, SiHa, Hela cells. (B) One representative cell is depicted for each condition. Bars represent the mean of three independent experiments with the standard error of the mean (SEM). *p < 0.05.
Triple modality treatment increases the fraction of cells in S-phase
Cell cycle distribution was studied by incorporation of BrdU. In the untreated samples, ~50% of R1, SiHa and HeLa cells were in G1-phase, ~40% in S-phase and ~10% in G2-phase of the cell cycle (Figure 3). Treatment with PARP1-i caused modest changes in cell cycle distribution, while after HT a slight decrease in G1 cells was observed, combined with a minor increase of cells in the G2-phase. Of all monotherapies, cDDP had the strongest effects on cell cycle distribution in R1 cells, causing increased fraction of S-phase cells, as compared to the control. Moreover, in all cell lines, an increased proportion of cells in S-phase was found after a single treatment with cDDP and hyperthermia and after the triple modality.
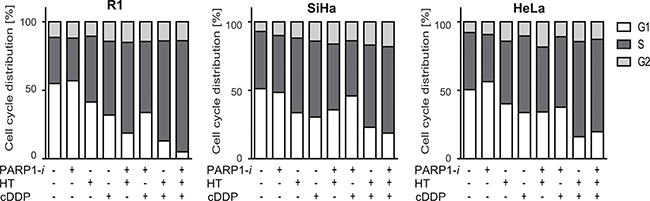
Figure 3: Cell cycle distribution using BrdU incorporation. While the monotherapies hardly ever changed the cell cycle analyses, any treatment containing cDDP resulted in a larger proportion of cells in S-phase. The differences found after adding PARP1-i to thermochemotherapy were not significant. From left to right: R1, SiHa, Hela cells.
Addition of PARP1-i to cDDP and hyperthermia increases apoptosis
To examine whether cell death detected in the clonogenic assays is due to induction of apoptosis, a Nicoletti assay was performed. The results of this assay demonstrate a 2 to 5-fold induction in apoptotic levels in R1 cells after any combination of treatment, except after the triple modality, for which there was an almost 9-fold induction of apoptosis (Figure 4). The degree of induction differed per cell line, but the trend was similar for all tested cell lines.
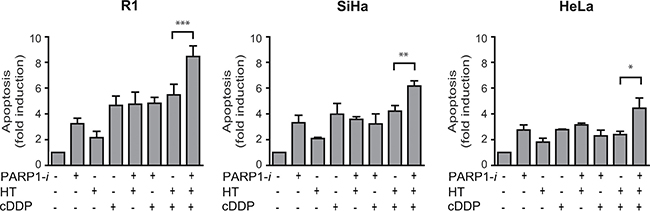
Figure 4: Apoptotic levels measured using the Nicoletti assay. An ~1.5-fold induction in apoptosis is found after treatment with PARP1-i, cDDP and HT compare to cDDP with HT in R1 and SiHa cells (R1: p = 0.0007, SiHa: p = 0.0054). A smaller, though significant effect was found in HeLa cells after addition of a PARP1-i (HeLa: p = 0.026). From left to right: R1, SiHa, Hela cells. *p < 0.05, **p < 0.01, ***p < 0.001.
The triple modality treatment delays tumour outgrowth in vivo
In vivo, tumours were treated with any combination of PARP1-i, HT and cDDP to test whether the therapy induces similar effects in vivo as in vitro. In untreated rats the tumour volume increased approximately exponentially, without a significant change in the slope of the growth curve. A treatment with cDDP alone, cDDP combined with either PARP1-i or HT, or cDDP combined with both induced a change in the steepness of the curve (Figure 5A), indicating that these treatments contributed to tumour control. Effects on tumour growth delay range from modest in the groups treated with PARP1-i, HT and HT+PARP1-i to substantial after cDDP alone, PARP1-i + cDDP, PARP1-i combined with HT and the triple modality. The differences in tumour growth were also calculated as the period of time needed to reach 10 times the tumour starting volume (T10 × SV) (Figure 5B). While the mono-therapies had only marginal influence on reaching T10 × SV, compared to the untreated animals, the double treatments did cause a delay of a few days. The highest level of tumour control was seen after the triple modality treatment, where the tumours took, on average, almost twice as long to reach the T10 × SV, as compared to the single therapies and approximately 1.5 times longer compared to cDDP-based thermochemotherapy. Importantly, no (acute) toxicities were detected in any of the rats.
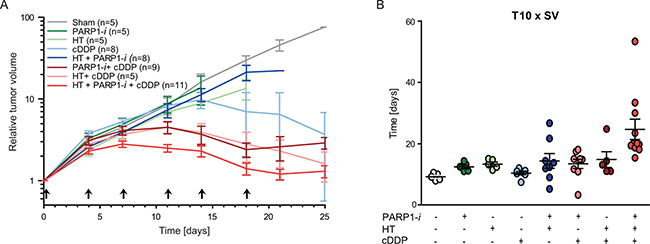
Figure 5: In vivo experiments performed on rats. (A) Relative tumour volume demonstrated that PARP1-i enhanced the effectiveness of cDDP-based thermochemotherapy and reduced the in vivo tumour outgrowth. Arrows present the time of treatment. (B) Time needed to achieve ten times the start volume (T10 × SV). Animals treated with the triple modality showed a delay in reaching T10 × SV compared to thermochemotherapy or any single treatment. All animal experiments included at least five rats per group.
DISCUSSION AND CONCLUSIONS
Our data demonstrated that addition of PARP1-i to cDDP-based thermochemotherapy results in an almost 2-fold reduction in cell survival, as compared to cDDP-based thermochemotherapy alone. This can be explained in part by an increased induction of apoptosis, which might be due to the higher DNA damage load, as indicated by γ-H2AX analysis. This, in turn, could result in increased activation of the S/G2 checkpoint, as we observed a higher fraction of cells in S-phase after cDDP-based thermochemotherapy and an even more profound increase after the triple modality treatment. Prolonged cell cycle block could eventually lead to apoptosis. It is important to stress that all these effects were caused by a single treatment session and we expect that a repeated treatment regimen, more akin to clinical settings, would lead to stronger responses.
After the last treatment the tumour growth recovered. Although the double treatment with HT+cDDP and PARP1-i+cDDP were almost as effective in controlling tumour growth (Figure 5A), in terms of reaching ten times the start volume, the triple modality considerably delays tumour growth.
Clinical trials that have been performed on patient with BRCA-deficient tumours received daily Olaparib, sometimes even twice a day [8]. However, in our study, we mimic the BRCA-deficiency by applying HT. This results in a temporary downregulation of BRCA and within this timeframe, the PARP1-i should be administered. Therefore, the animals in our study were treated twice a week with two times Olaparib and one dose of cisplatin on the day of HT treatment. Thus, although it is not an identical schedule to the previous clinical trials with Olaparib, it is the most optimal scheme for treating BRCA-proficient tumours with HT and PARP1-i. As a consequence, since we have not treated daily, and only for a period of three weeks, it may be a valid consideration to extend the treatments with a couple of weeks, as HT sessions in patients are normally given once or twice a week for a period of five weeks in our center. Therefore, the outcomes of this study are promising, because we have treated these tumours with just 60% of the normal HT scheme and already the tumour shrank to its starting volume. However, an additional agent to standard thermochemotherapy may increase toxicity levels. Whether this occurs should be investigated.
There is growing evidence that platinum-based agents can also contribute in interfering with HR, resulting in HR-deficiency [24]. Subsequently, it might be interesting to combine platinum-based agents with PARP1-i. However, some people state that the use of PARP1-i may be limited in the clinic due to an increase in observed toxicities in combination with chemotherapeutic agents [25, 26]. The triple modality that we propose, PARP1-i combined with cDDP-based thermochemotherapy might have more potential, because HT can make chemotherapeutic agents penetrate more deeply into the tumour and HT targets hypoxic areas, which are generally least sensitive to other anti-cancer therapies. Thus, lower chemotherapeutic dose may be as effective as normal concentrations without HT, but at a reduced toxicity [16].
In summary, there have been many studies that use the HR-deficiency in BRCA-mutated tumours to induce potentially lethal DNA damages by inhibition of PARP1. The advantage of our study is the possibility to treat BRCA-proficient cells with an already successful clinically applied therapy that can temporarily inhibit the HR. Therefore, we may induce synthetic lethality for any tumour type.
MATERIALS AND METHODS
Cell lines and cell culture
Rat rhabdomyosarcoma cells (R1, a cell line developed in our own institute for in vitro cultures as well as for in vivo growing tumors [27]) were grown in MEM. The cervical carcinoma cells (SiHa and HeLa) were obtained from the American Type Culture Collection (ATCC) and grown in EMEM. All cell lines are BRCAwt. All media contained 25 mM Hepes (Gibco-BRL life technologies, Breda, The Netherlands) supplemented with 10% heat-inactivated foetal bovine serum (FBS) and 2 mM glutamine. Cells were maintained in a 37°C incubator with humidified air supplemented with 2% and 5% CO2, respectively. The cell division time of R1 cells was approximately 16 h and of SiHa and HeLa cells 24 h.
In vitro treatments
Cells were treated for approximately 60 min with 5 μM cisplatin (cDDP; Platosin®, Pharmachemie B.V., Haarlem, The Netherlands) and/or continuously with 100 μM of PARP1-i (dissolved in DMSO; NU1025, Tocris Bioscience, Bristol, UK). cDDP was added 5 minutes before HT, while PARP1-i was added 30 min prior to the HT treatment. Immediately after HT, medium was refreshed and PARP1-i was re-added until the end of the experiment.
HT was performed by partially submerging the culture dishes in a thermostatically controlled water bath (Lauda aqualine AL12, Beun de Ronde, Abcoude, The Netherlands) for 1 h at 42°C. In order to check the temperature, thermocouples were placed in parallel culture dishes; the desired temperature (± 0.1°C) was reached in approximately 5 min. HT was performed in an atmosphere containing 2% (R1 cells) or 5% (SiHa and HeLa cells) CO2 with gas mixture inflow rate of 2 L/min.
Western blotting
To identify the BRCA2 status of these cell lines, western blots were conducted. Also, the response to HT was tested. Cells were harvested 30 min post treatment. Next, pellets were lysed on ice in ice-cold RIPA buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM Na2EDTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin) with protein inhibitors for 30 min [28]. Laemmli buffer containing fresh added 2-mercaptoethanol (355 mM) was mixtued to the supernatant (1:1), before heated in boiling water for 2–5 min. Finally, samples were sonificated (Sonics & Materials Inc). One μg of protein was loaded on 4–15% SDS-PAGE gradient precast gels (BioRad) and transferred to PVDF membranes. Loading of protein were checked by Ponceau S staining. Immunodetection was performed for anti-BRCA2 (antibodies-online) and anti-ERK2 (Bethyl Laboratories), and a secondary anti-rabbit (Invitrogen Life Technologies). All samples were enhanced using chemoluminescence (Amersham Pharmacia Biotech). Eventually, blots were analysed using LAS4000 (GE, Healthcare life sciences).
Co-localization of Rad51 and γ-H2AX foci
Activity of homologous recombination has been investigated by co-localization of Rad51 and γ-H2AX foci. Cells were plated on 1.8 μm mylar membrane dishes. One day later, cells were treated with or without HT prior to a min irradiation of α-particles. Afterwards, 30 min after treatment, cells were fixed with 2% paraformaldehyde. Cells were washed with PBS and permeabilized during a 30-minute incubation with TNBS (PBS containing 0.1% Triton X-100 and 1% FCS), before staining with γ-H2AX (Millipore, dilution 1:100 in TNBS) and Rad51 antibody (dilution 1:25 in TNBS) as described by Bergs et al [29]. Eventually, a drop of vectashield containing DAPI (Life technologies, USA) was pipetted in the slide and the coverslip is turned upside down on the slide. Samples were scored under the fluorescence microscope.
Cell survival assay
Cell survival was studied for R1, SiHa and HeLa cells using different combinations of HT, cDDP and PARP1-i. Clonogenic assays were conducted as described by Franken et al. [30]. Cells were plated at 4 h prior to treatment into 6-well culture plates (Costar, USA). Dishes were placed in a 37°C incubator with the desired percentages of CO2 at 37°C until sufficiently large clones were formed, which took approximately 10–12 days. Afterwards, the medium was removed and cells were washed with PBS before a 30-min incubation in a 2–3 ml of a mixture containing 6.0% glutaraldehyde and 0.5% crystal violet, at room temperature. Finally, plates were washed with tap water and dried at room temperature. Colonies were counted under a light microscope [31]. Surviving fractions were calculated by dividing the plating efficiency of treated cells by that of control cells [32].
Detection of DNA DSBs quantifying γ-H2AX foci and flow cytometric detection of γ-H2AX
To identify a possible mechanism for the differences in cell survival after the triple modality treatment, we quantified the formation of γ-H2AX, representing unrepaired DSBs, by flow cytometry. For immunocytochemistry, to present a representative image, cells were plated 24 h before any combination of cDDP, PARP1-i and HT treatment on sterile coverslips. On the next day, cells were fixed for 10 min in PBS containing 2% paraformaldehyde, three times washed with PBS. Then, cells were permeabilised during a 30-min incubation in TNBS (PBS containing 0.1% Triton X-100 and 1% FCS). Afterwards, cells were stained for 90 min at room temperature with 50 μl primary antibody of a mouse monoclonal anti-γ-H2AX (Millipore, dilution 1:100 in TNBS). Cells were washed once in PBS and two times in TNBS before staining for 30 min at room temperature with a secondary antibody anti-mouse Cy3 (Jackson, diluted 1:100 in TNBS). After three times washing with TNBS, vectashield-containing DAPI (Life technologies, USA) was dropped onto glass slides, before placing the coverslips upside-down on the slides. γ-H2AX foci were scored under a fluorescence microscope.
To quantify γ-H2AX using flow cytometry, the staining was performed at 24 h after treatment. Cells were centrifuged (1200 rpm, 10 min), prior to fixation in a mixture containing 2 ml PBS and 6 ml absolute ethanol. Next day, cells were washed and incubated on ice for 10 min with PBS + 0.1% triton and 4% BSA. After centrifugation, cells were stained with antibodies against γ-H2AX-FITC (4 μg/ml, Merck Millipore, USA). Samples were then analysed using FACS Canto (BD Biosciences, USA).
Cell cycle analysis
The thymidine analogue 5-Bromo-2′-deoxy-uridine (BrdU, Sigma Aldrich, USA) was used to analyse cell cycle distribution. At 16 h after treatments, BrdU (diluted 1:100) was added for 60 min at 37°C, before fixating cells in a mixture containing 2 ml PBS and 6 ml of absolute ethanol. Cells were stored at 4°C. The next day, after spinning down samples at 1200 rpm for 2 min, pellets were resuspended in pepsin-HCl (0.4 mg/ml, 0.1 N HCl) and cells were incubated for 30 min at room temperature. Cells were then washed with PBT (PBS containing 0.5% Tween-20 (Sigma Aldrich USA)). After centrifugation, pellets were resuspended in HCl (2 N, Merck) and incubated for 30 min at 37°C. Next, cells were stained with rat-anti-BrdU primary antibody (Abcam, UK) diluted 1:100 in PBTb (1% bovine serum albumin, Sigma, in PBT) for 60 min at 37°C. After washing cells two times with PBT and once with PBTg (PBT containing 1% normal goat serum (Dako, USA), cells were stained with a goat-anti-rat secondary antibody labelled with FITC (Abcam, UK) diluted 1:100 in PBTg for 60 min at 37°C. Cells were washed three times with PBS before resuspending the pellets in PBS. Finally, propidium iodide (Sigma-Aldrich, USA) was added and cell suspensions were vortexed. Analysis was performed using the FACS Canto flow cytometer (BD Biosciences, USA).
Apoptosis assay
To study apoptosis after any combination of treatments, the Nicoletti assay [30] was performed. Cells were collected 48 h after the different treatments and pellets were resuspended in Nicoletti buffer (0.1% w/v Sodium citrate, 0.1% v/v Triton X-100 in demi water, pH 7.4), containing propidium iodide (Sigma-Aldrich, USA). Analyses were performed using the FACS Canto flow cytometer (BD Biosciences, USA).
Animal experiments
Female WAG-Rij rats were implanted with 1 mm3 piece of rhabdomyosarcoma tumour tissue in the right hind leg. After approximately three weeks, when the tumours had grown to 50–100 mm3, the animals were treated twice a week for a period of 3 weeks. The treatment involved HT for 1.5 h at 42°C in a thermostatically controlled water bath. In order to monitor the temperature thermocouples were placed next to the tumours. Earlier experiments [13, 34] demonstrated that when the water bath was set to 42.7°C, the intra-tumour temperature was 42°C. Animals were cooled to prevent them from receiving whole body hyperthermia. The PARP1 inhibitor Olaparib was dissolved in a mixture of 10% 2-hydroxy-propyl-β-cyclodextrin and 10% DMSO in PBS and administrated at a dosage of 50 mg/kg p.o. 16 h and 2 h prior to HT. cDDP (Platosin®, Pharmachemie B.V., Haarlem, The Netherlands) was given i.p. at the dosage of 2.0 mg/kg 2 h prior to hyperthermia. Animal weight was monitored every two days and tumour sizes were measured using a Vernier caliper every two days, until the end of the experiment. Animals were sacrificed when the tumour volume exceeded 3500 mm³. The treated hind leg of rats was checked every two days for skin irritation. All animal experiments included at least five rats per group.
Statistical analyses
All in vitro data represent means (of at least three experiments) with standard error of the mean (SEM) of at least three independent experiments. SPSS (Chicago, IL, USA) statistical software using a non-parametric Mann-Whitney test was used to analyse cell survival, γ-H2AX foci and apoptosis. P-values lower than 0.05 were considered statistically significant.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the Dutch Cancer Society (UVA 2012-5540).
CONFLICTS OF INTEREST
The authors report no conflicts of interest.
REFERENCES
1. Easton DF, Ford D, Bishop DT. Breast and ovarian cancer incidence in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. American journal of human genetics. 1995; 56:265–271.
2. Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet. 1994; 343:692–695.
3. Easton DF, Steele L, Fields P, Ormiston W, Averill D, Daly PA, McManus R, Neuhausen SL, Ford D, Wooster R, Cannon-Albright LA, Stratton MR, Goldgar DE. Cancer risks in two large breast cancer families linked to BRCA2 on chromosome 13q12-13. American journal of human genetics. 1997; 61:120–128.
4. Antoniou AC, Gayther SA, Stratton JF, Ponder BA, Easton DF. Risk models for familial ovarian and breast cancer. Genetic epidemiology. 2000; 18:173–190.
5. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913–917.
6. Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Molecular oncology. 2011; 5:387–393.
7. Girolimetti G, Perrone AM, Santini D, Barbieri E, Guerra F, Ferrari S, Zamagni C, De Iaco P, Gasparre G, Turchetti D. BRCA-associated ovarian cancer: from molecular genetics to risk management. BioMed research international. 2014; 2014:787143.
8. Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, Lu K, Schmutzler RK, Matulonis U, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010; 376:245–251.
9. Huehls AM, Wagner JM, Huntoon CJ, Karnitz LM. Identification of DNA repair pathways that affect the survival of ovarian cancer cells treated with a poly(ADP-ribose) polymerase inhibitor in a novel drug combination. Molecular pharmacology. 2012; 82:767–776.
10. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434:917–921.
11. Dedes KJ, Wilkerson PM, Wetterskog D, Weigelt B, Ashworth A, Reis-Filho JS. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle. 2011; 10:1192–1199.
12. Eppink B, Krawczyk PM, Stap J, Kanaar R. Hyperthermia-induced DNA repair deficiency suggests novel therapeutic anti-cancer strategies. International journal of hyperthermia. 2012; 28:509–517.
13. Krawczyk PM, Eppink B, Essers J, Stap J, Rodermond H, Odijk H, Zelensky A, van Bree C, Stalpers LJ, Buist MR, Soullie T, Rens J, Verhagen HJ, et al. Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:9851–9856.
14. Schaaf L, Schwab M, Ulmer C, Heine S, Murdter TE, Schmid JO, Sauer G, Aulitzky WE and van der Kuip H. Hyperthermia Synergizes with Chemotherapy by Inhibiting PARP1-Dependent DNA Replication Arrest. Cancer research. 2016; 76:2868–2875.
15. van den Tempel N, Horsman MR, Kanaar R. Improving efficacy of hyperthermia in oncology by exploiting biological mechanisms. International journal of hyperthermia. 2016; 32:446–454.
16. Oei AL, Vriend LE, van Leeuwen CM, Rodermond HM, Ten Cate R, Westermann AM, Stalpers LJ, Crezee J, Kanaar R, Kok HP, Krawczyk PM, Franken NA. Sensitizing thermochemotherapy with a PARP1-inhibitor. Oncotarget. 2017; 8:16303–16312.
17. Rietbroek RC, Schilthuis MS, Bakker PJ, van Dijk JD, Postma AJ, Gonzalez Gonzalez D, Bakker AJ, van der Velden J, Helmerhorst TJ, Veenhof CH. Phase II trial of weekly locoregional hyperthermia and cisplatin in patients with a previously irradiated recurrent carcinoma of the uterine cervix. Cancer. 1997; 79:935–943.
18. de Wit R, van der Zee J, van der Burg ME, Kruit WH, Logmans A, van Rhoon GC, Verweij J. A phase I/II study of combined weekly systemic cisplatin and locoregional hyperthermia in patients with previously irradiated recurrent carcinoma of the uterine cervix. British journal of cancer. 1999; 80:1387–1391.
19. Franckena M, De Wit R, Ansink AC, Notenboom A, Canters RA, Fatehi D, Van Rhoon GC, Van Der Zee J. Weekly systemic cisplatin plus locoregional hyperthermia: an effective treatment for patients with recurrent cervical carcinoma in a previously irradiated area. International journal of hyperthermia. 2007; 23:443–450.
20. Poklar N, Pilch DS, Lippard SJ, Redding EA, Dunham SU, Breslauer KJ. Influence of cisplatin intrastrand crosslinking on the conformation, thermal stability, and energetics of a 20-mer DNA duplex. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93:7606–7611.
21. Rudd GN, Hartley JA, Souhami RL. Persistence of cisplatin-induced DNA interstrand crosslinking in peripheral blood mononuclear cells from elderly and young individuals. Cancer chemotherapy and pharmacology. 1995; 35:323–326.
22. Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. The Journal of biological chemistry. 2004; 279:55117–55126.
23. Metzger MJ, Stoddard BL, Monnat RJ, Jr. PARP-mediated repair, homologous recombination, and back-up non-homologous end joining-like repair of single-strand nicks. DNA repair. 2013; 12:529–534.
24. McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O'Connor MJ, Tutt AN, Zdzienicka MZ, Smith GC, Ashworth A. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer research. 2006; 66:8109–8115.
25. Kummar S, Chen A, Ji J, Zhang Y, Reid JM, Ames M, Jia L, Weil M, Speranza G, Murgo AJ, Kinders R, Wang L, Parchment RE, et al. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer research. 2011; 71:5626–5634.
26. Rajan A, Carter CA, Kelly RJ, Gutierrez M, Kummar S, Szabo E, Yancey MA, Ji J, Mannargudi B, Woo S, Spencer S, Figg WD, Giaccone G. A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clinical cancer research. 2012; 18:2344–2351.
27. Reinhold HS. A cell dispersion technique for use in quantitative transplantation studies with solid tumours. Eur J Cancer. 1965; 1:67–71.
28. van Bree C, Franken NA, Rodermond HM, Stalpers LJ, Haveman J. Repair of potentially lethal damage does not depend on functional TP53 in human glioblastoma cells. Radiation research. 2004; 161:511–516.
29. Bergs JW, Krawczyk PM, Borovski T, Ten Cate R, Rodermond HM, Stap J, Medema JP, Haveman J, Essers J, van Bree C, Stalpers LJ, Kanaar R, Aten JA, et al. Inhibition of homologous recombination by hyperthermia shunts early double strand break repair to non-homologous end-joining. DNA repair. 2013; 12:38–45.
30. Franken NA, Rodermond HM, Stap J, Haveman J and van Bree C. Clonogenic assay of cells in vitro. Nature protocols. 2006; 1:2315–2319.
31. Bergs JW, Franken NA, ten Cate R, van Bree C, Haveman J. Effects of cisplatin and gamma-irradiation on cell survival, the induction of chromosomal aberrations and apoptosis in SW-1573 cells. Mutation research. 2006; 594:148–154.
32. van Bree C, Franken NA, Snel FA, Haveman J, Bakker PJ. Wild-type p53-function is not required for hyperthermia-enhanced cytotoxicity of cisplatin. International journal of hyperthermia. 2001; 17:337–346.
33. Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nature protocols. 2006; 1:1458–1461.
34. van Bree C, Rietbroek RC, Schopman EM, Kipp JB, Bakker PJ. Local hyperthermia enhances the effect of cis-diamminedichloro-platinum(II) on nonirradiated and preirradiated rat solid tumors. International journal of radiation oncology, biology, physics. 1996; 36:135–140.